
In Vitro Study of Cytotoxic Mechanisms of Alkylphospholipids and Alkyltriazoles in Acute Lymphoblastic Leukemia Models
 ,
,  , and
, and
Abstract

1. Introduction
2. Results
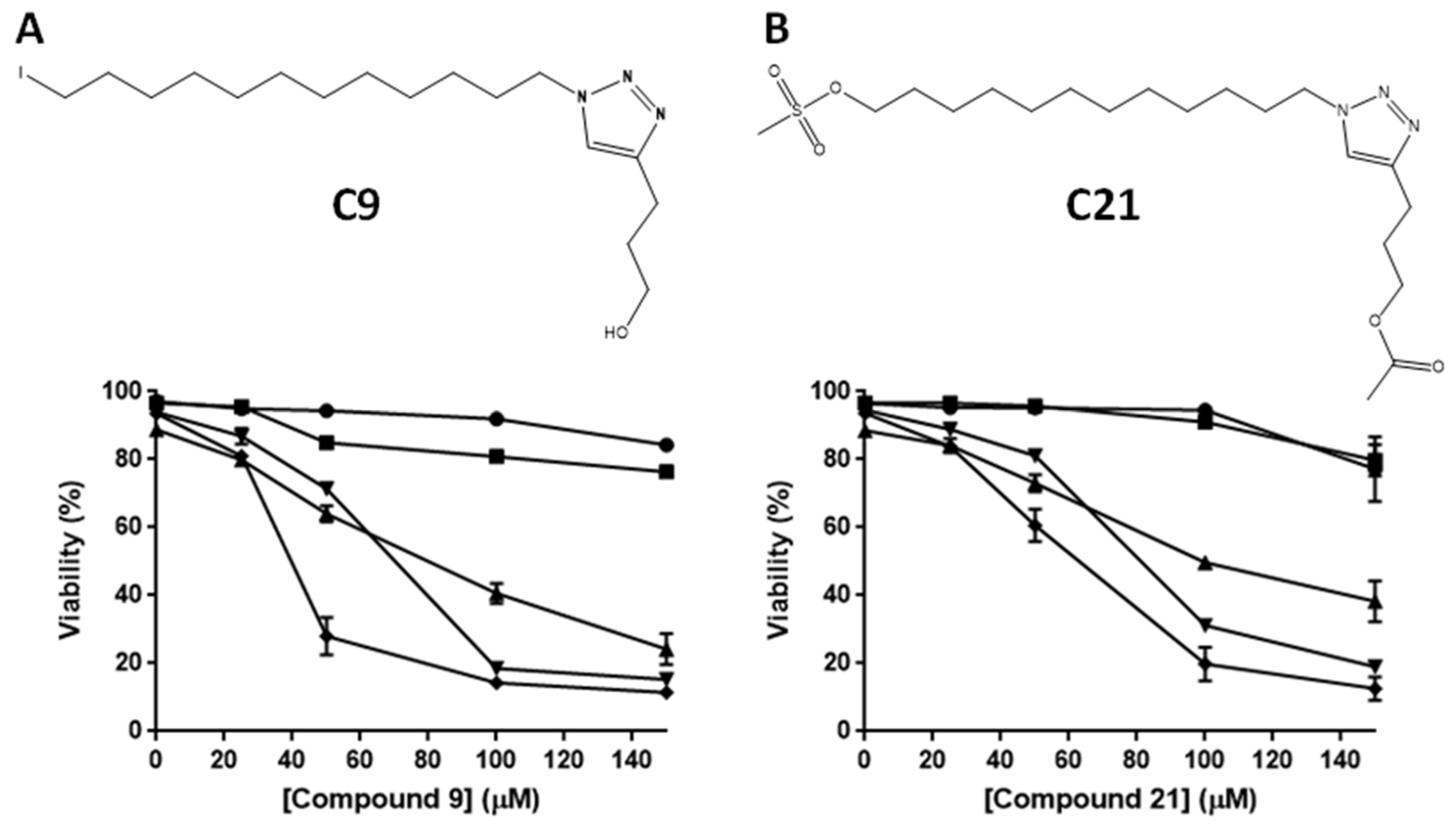
2.1. Cytotoxic Potential of Alkyltriazole and Alkylphospholipid Compounds in Different Leukemic Lines
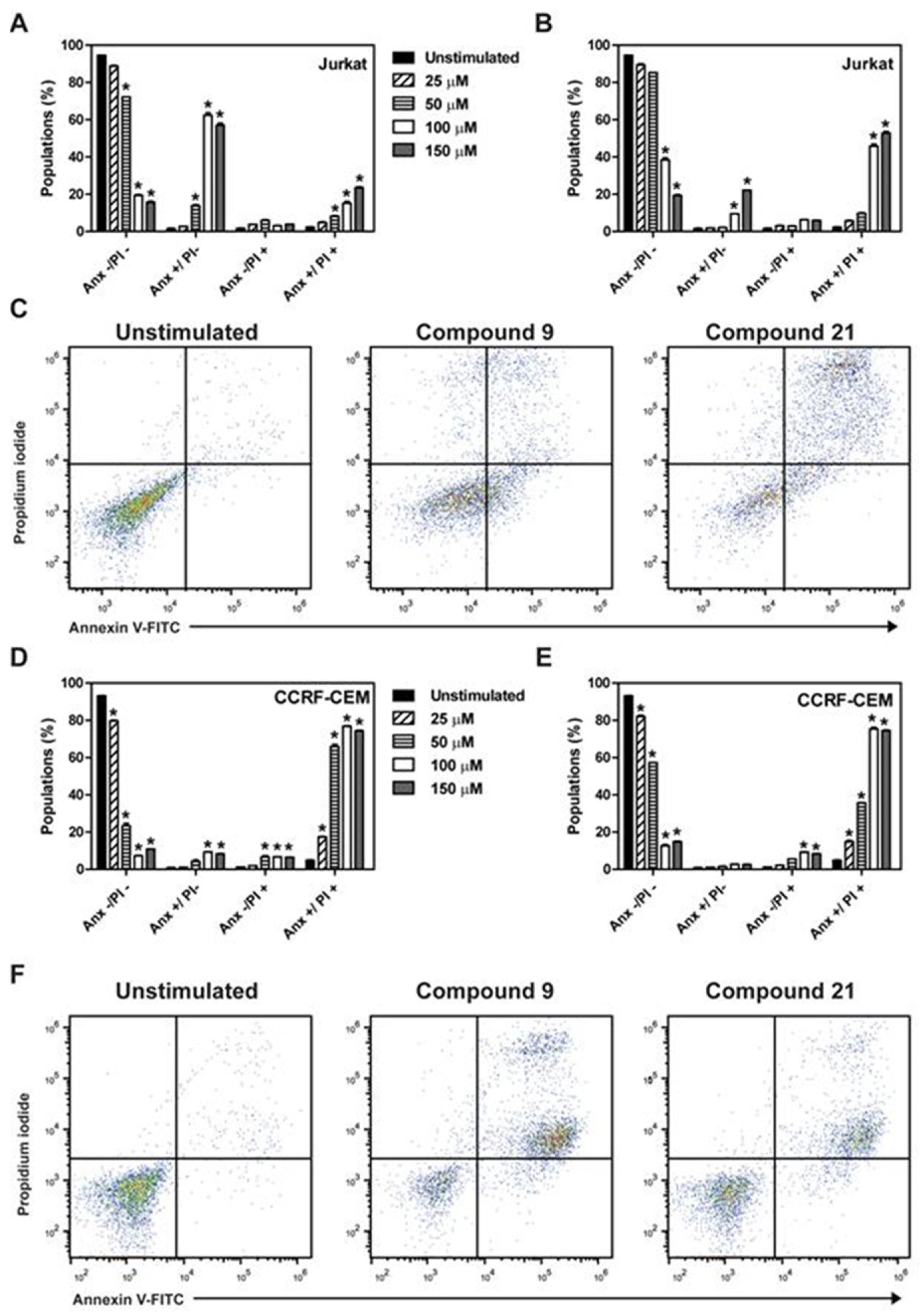
2.2. Determination of Induction of Cell Death by the Different Compounds
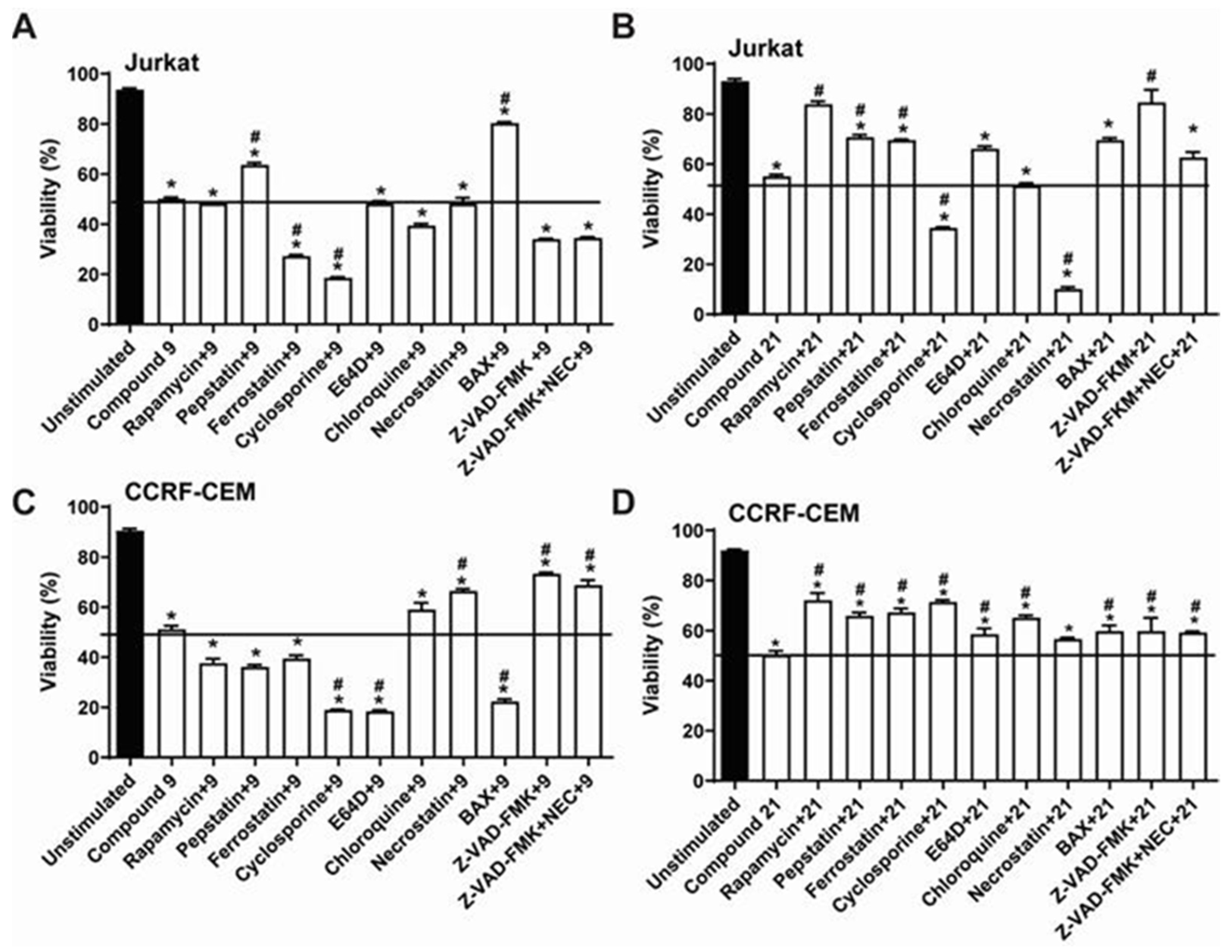
2.3. Comparative Analysis of Alkyltriazole Compounds with Inhibitors of Cell Death Pathways
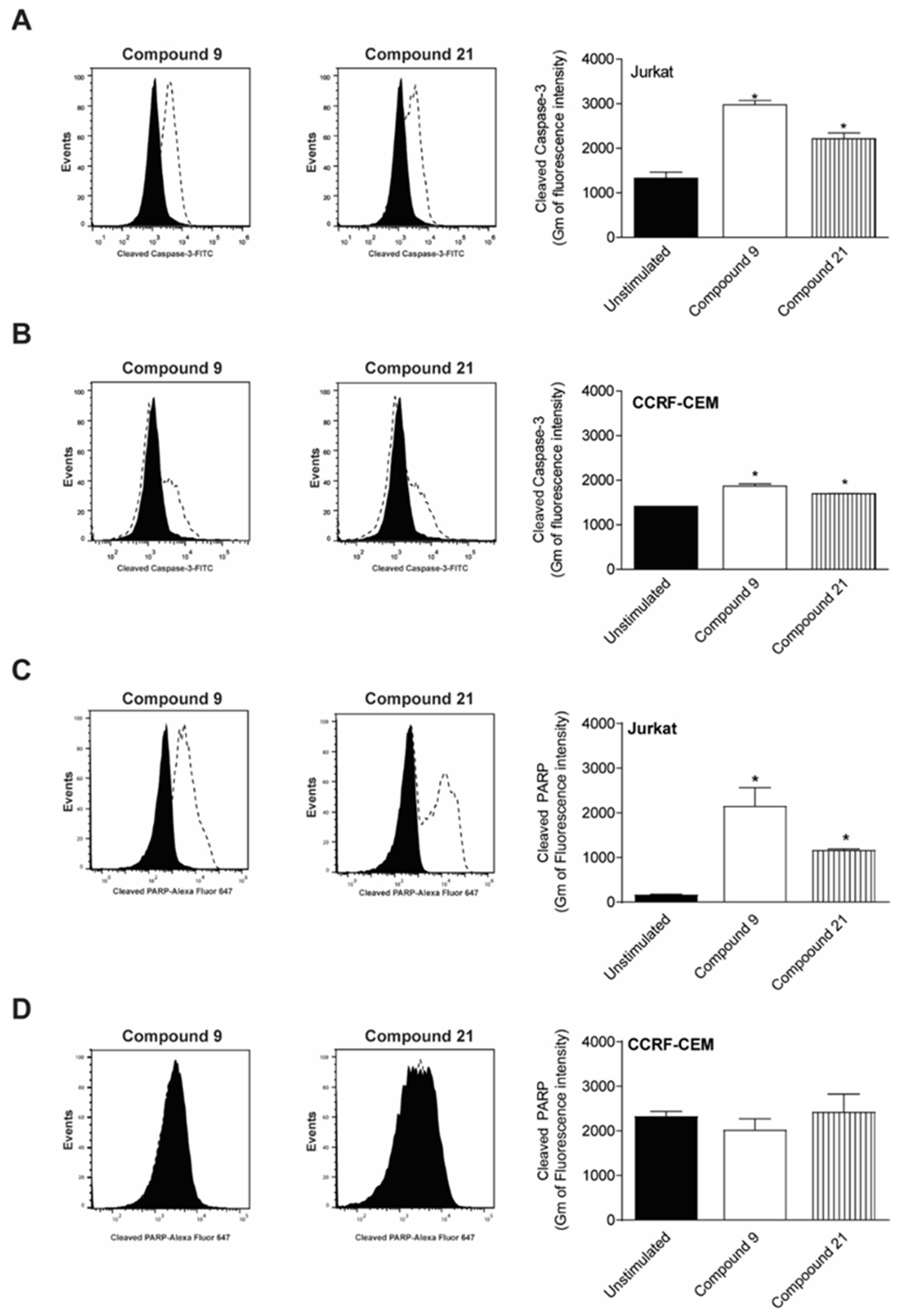
2.4. Caspase-3 and PARP Cleavage by Compounds
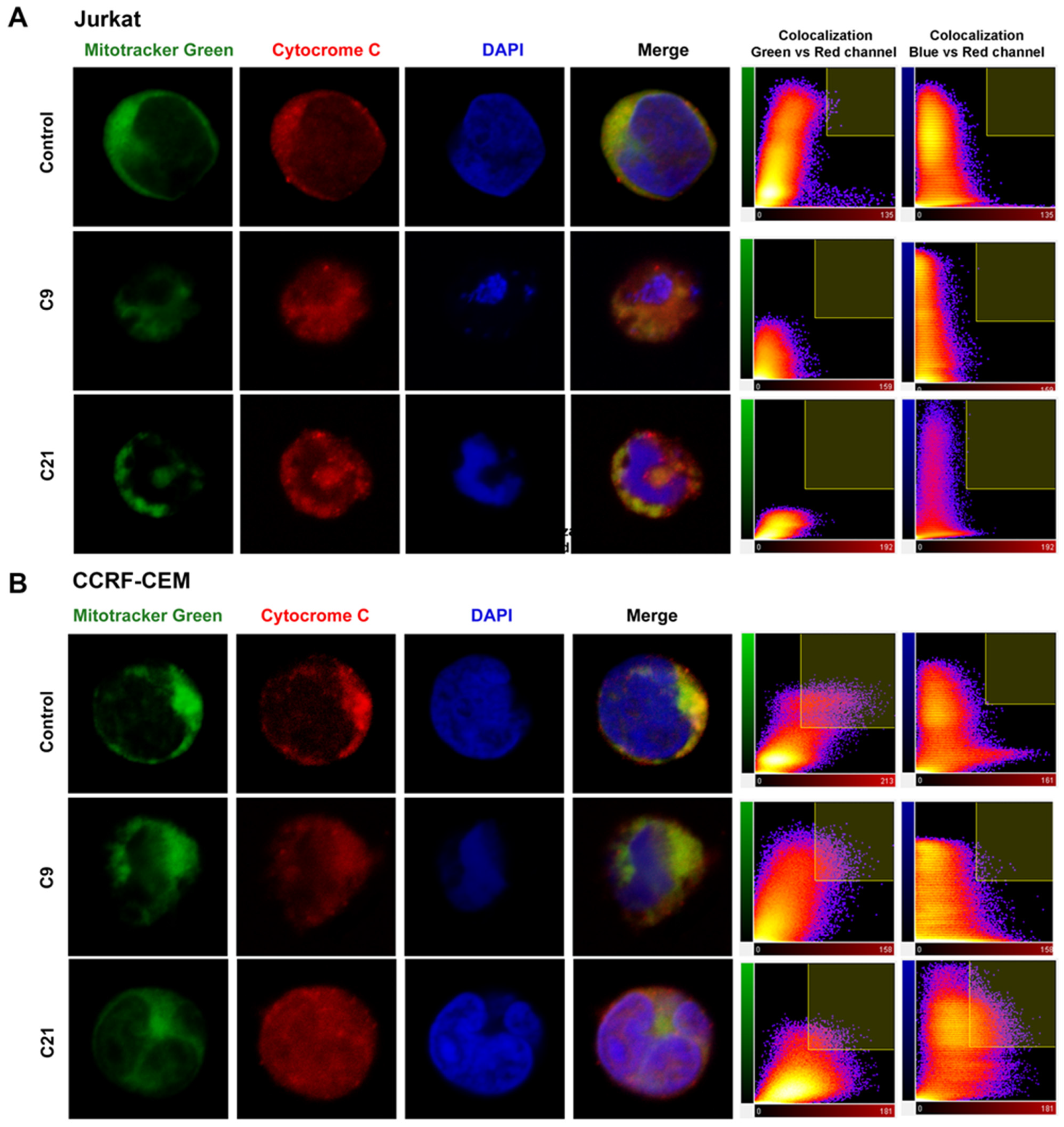
2.5. Release of Cytochrome c and Nuclear Changes
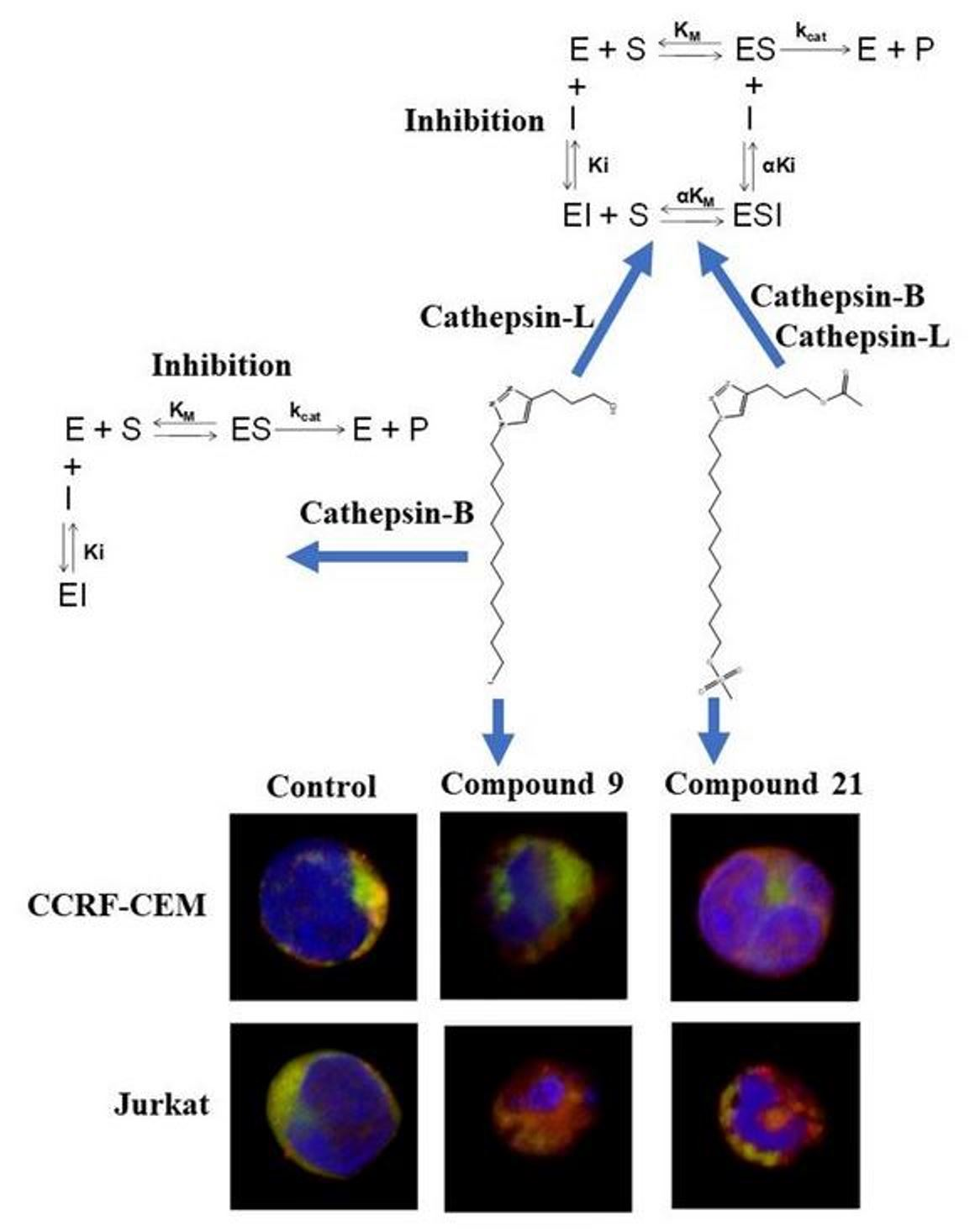
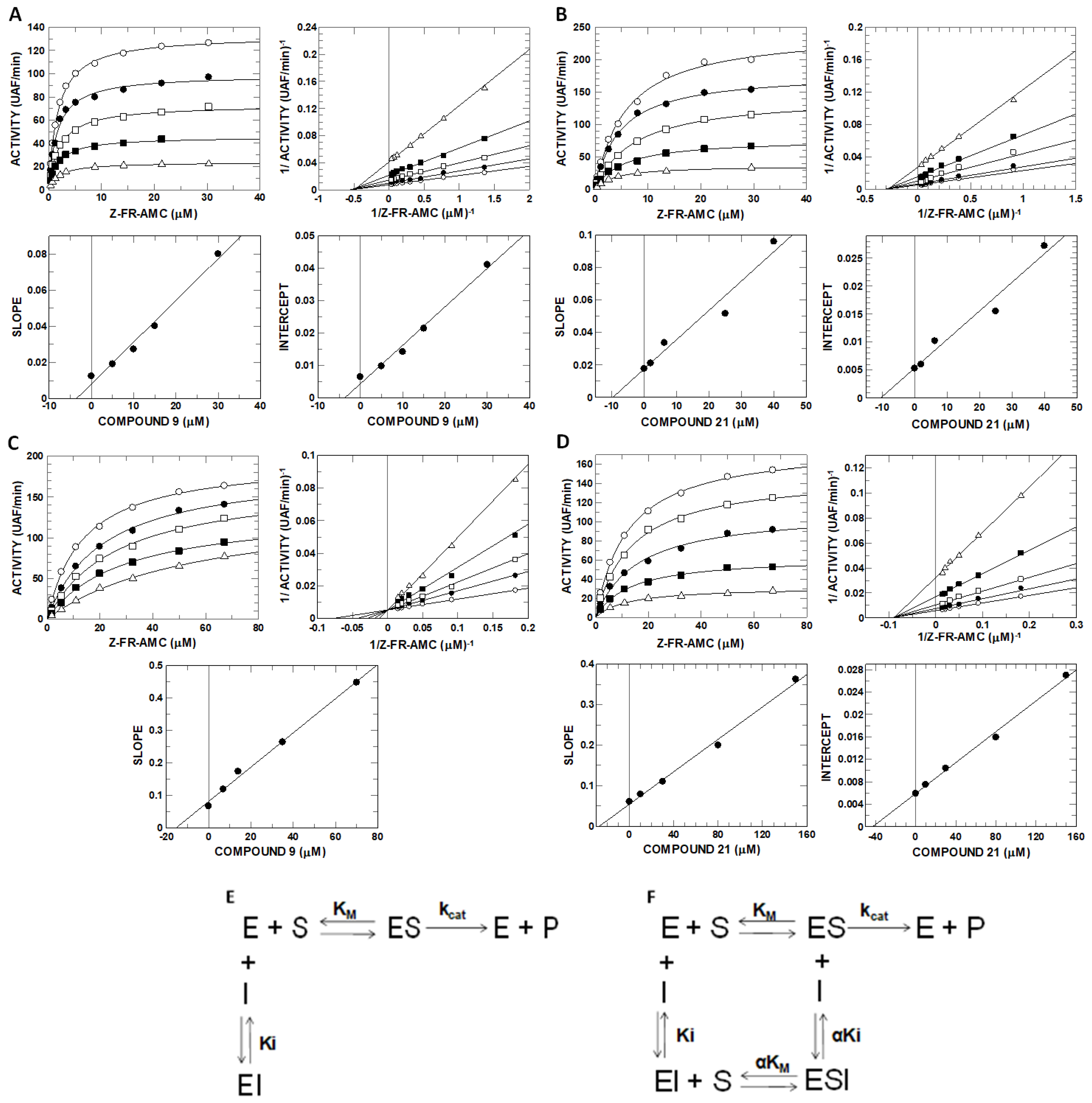
2.6. Inhibitory Potential (IC50) Evaluation and Mechanism Determination of Compounds with Cathepsins B and L
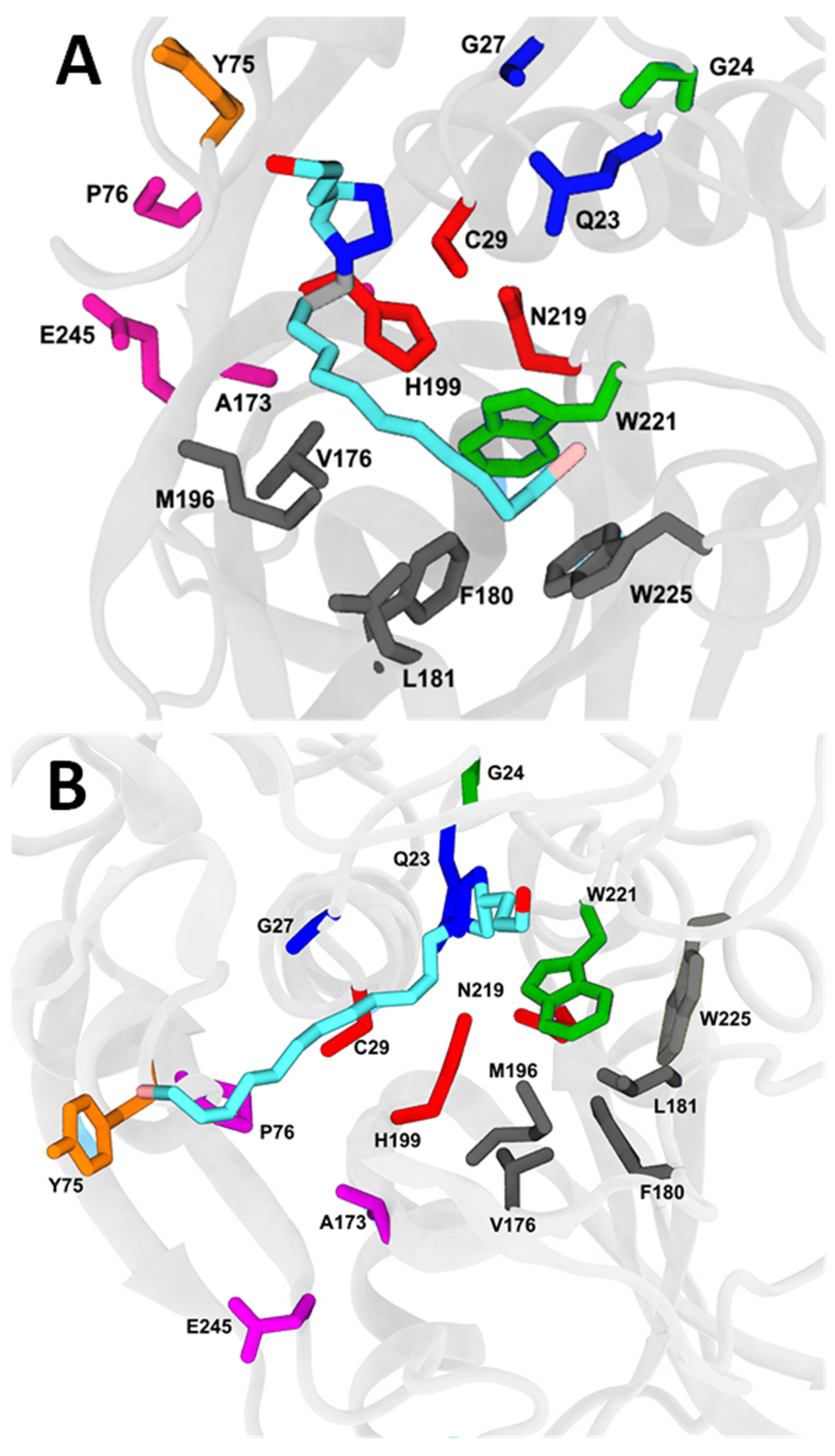
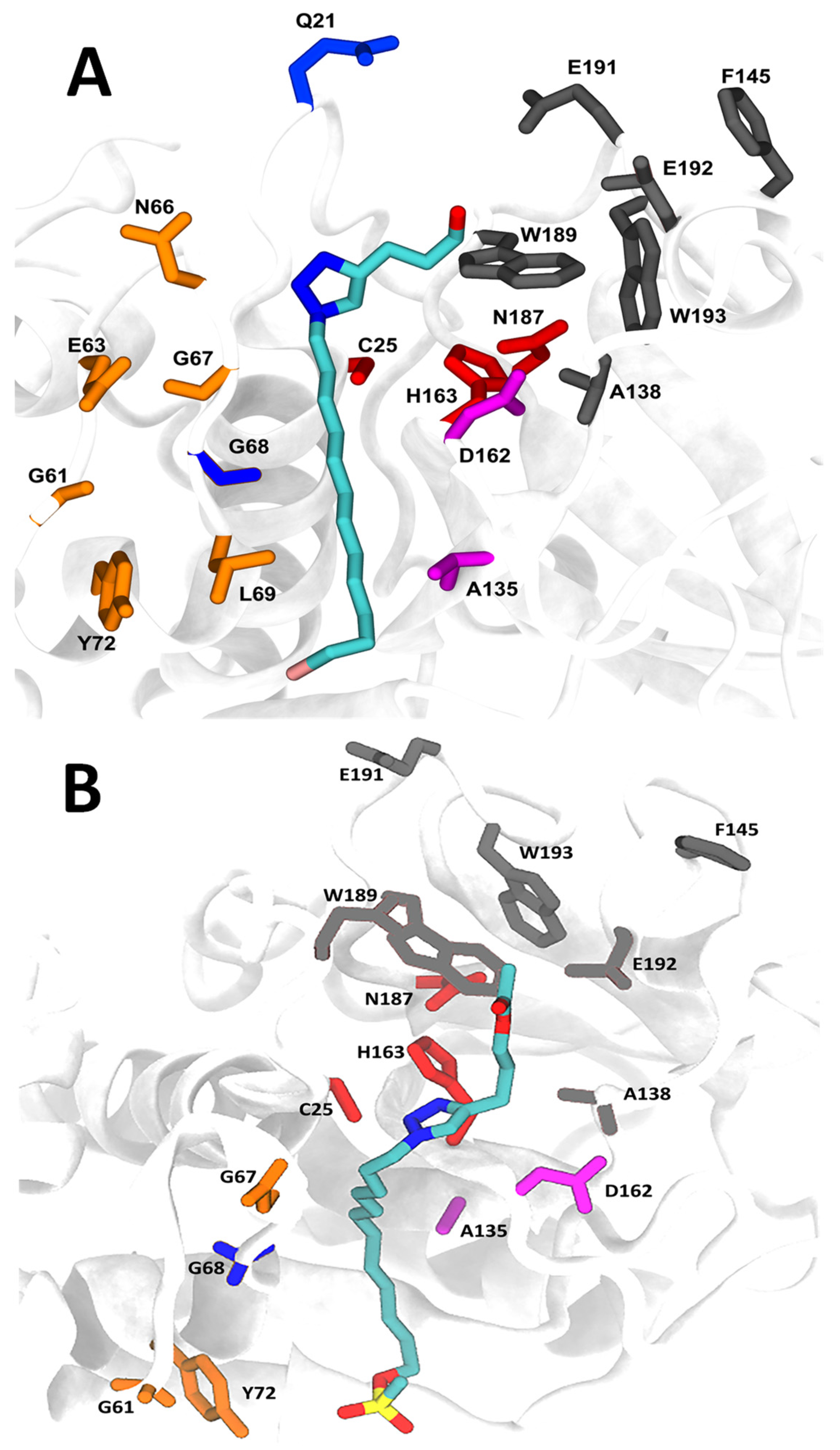
2.7. Molecular Docking and Molecular Dynamics
3. Discussion
4. Materials and Methods
4.1. Alkylphospholipid and Alkyltriazole Compounds
4.2. Cell Cultures
4.3. Peripheral Blood Mononuclear Cell Culture
4.4. Cellular Viability Using Calcein-AM and EthD-1 (Live/Dead Assay)
4.5. Annexin V/propidium Iodide Assay
4.6. Caspase- 3 and Poly(ADP-ribose) Polymerase-1(PARP) Cleavage
4.7. Cytochrome C Release and Nuclear Alteration Analyses
4.8. Determination of IC50 Values for Inhibitors
4.9. Enzyme Kinetics and Determination of the Mechanism of Inhibition
4.10. Molecular Docking Simulation
4.11. Molecular Dynamics
4.12. Free Energy Evaluation and Decomposition into Per-Residue Contributions
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Blitterswijk, W.J.; Verheij, M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim. Biophys. Acta 2013, 1831, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.P.; Jimenez-Lopez, J.M.; Segovia, J.L.; Marco, C. Hexadecylphosphocholine interferes with the intracellular transport of cholesterol in HepG2 cells. FEBS J. 2008, 275, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C.; Martin-Santamaria, S.; Gago, F. ET-18-OCH3 (edelfosine): A selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr. Med. Chem. 2004, 11, 3163–3184. [Google Scholar] [CrossRef] [PubMed]
- Busto, J.V.; Del Canto-Janez, E.; Goni, F.M.; Mollinedo, F.; Alonso, A. Combination of the anti-tumour cell ether lipid edelfosine with sterols abolishes haemolytic side effects of the drug. J. Chem. Biol. 2008, 1, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; de Frias, M.; Roue, G.; Gil, J.; Colomer, D.; Campanero, M.A.; et al. In Vitro and In Vivo selective antitumor activity of Edelfosine against mantle cell lymphoma and chronic lymphocytic leukemia involving lipid rafts. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2010, 16, 2046–2054. [Google Scholar] [CrossRef]
- Gajate, C.; Matos-da-Silva, M.; Dakir el, H.; Fonteriz, R.I.; Alvarez, J.; Mollinedo, F. Antitumor alkyl-lysophospholipid analog edelfosine induces apoptosis in pancreatic cancer by targeting endoplasmic reticulum. Oncogene 2012, 31, 2627–2639. [Google Scholar] [CrossRef]
- Hac-Wydro, K.; Dynarowicz-Latka, P. Effect of edelfosine on tumor and normal cells model membranes—A comparative study. Colloids Surf. B Biointerfaces 2010, 76, 366–369. [Google Scholar] [CrossRef]
- Khademvatan, S.; Gharavi, M.J.; Rahim, F.; Saki, J. Miltefosine-induced apoptotic cell death on Leishmania major and L. tropica strains. Korean J. Parasitol. 2011, 49, 17–23. [Google Scholar] [CrossRef]
- Nieto-Miguel, T.; Gajate, C.; Mollinedo, F. Differential targets and subcellular localization of antitumor alkyl-lysophospholipid in leukemic versus solid tumor cells. J. Biol. Chem. 2006, 281, 14833–14840. [Google Scholar] [CrossRef]
- Gajate, C.; Del Canto-Janez, E.; Acuna, A.U.; Amat-Guerri, F.; Geijo, E.; Santos-Beneit, A.M.; Veldman, R.J.; Mollinedo, F. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J. Exp. Med. 2004, 200, 353–365. [Google Scholar] [CrossRef]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Millard, N.E.; Modak, S.; Kushner, B.H.; Haque, S.; Spasojevic, I.; Trippett, T.M.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; et al. A phase I study of single-agent perifosine for recurrent or refractory pediatric CNS and solid tumors. PLoS ONE 2017, 12, e0178593. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Kagabu, M.; Mizuno, M.; Oda, K.; Aoki, D.; Mabuchi, S.; Kamiura, S.; Yamaguchi, S.; Aoki, Y.; Saito, T.; et al. Phase II basket trial of perifosine monotherapy for recurrent gynecologic cancer with or without PIK3CA mutations. Invest. New Drugs 2017, 35, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Bagley, R.G.; Kurtzberg, L.; Rouleau, C.; Yao, M.; Teicher, B.A. Erufosine, an alkylphosphocholine, with differential toxicity to human cancer cells and bone marrow cells. Cancer Chemother. Pharmacol. 2011, 68, 1537–1546. [Google Scholar] [CrossRef]
- Rios-Marco, P.; Marco, C.; Galvez, X.; Jimenez-Lopez, J.M.; Carrasco, M.P. Alkylphospholipids: An update on molecular mechanisms and clinical relevance. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1657–1667. [Google Scholar] [CrossRef]
- Pachioni Jde, A.; Magalhaes, J.G.; Lima, E.J.; Bueno Lde, M.; Barbosa, J.F.; de Sa, M.M.; Rangel-Yagui, C.O. Alkylphospholipids—A promising class of chemotherapeutic agents with a broad pharmacological spectrum. J. Pharm. Pharm. Sci. 2013, 16, 742–759. [Google Scholar] [CrossRef]
- Gontijo, V.S.; Oliveira, M.E.; Resende, R.J.; Fonseca, A.L.; Nunes, R.R.; Comar, M.; Taranto, A.G.; Torres, N.M.P.O.; Viana, G.H.R.; Silva, L.M.; et al. Long-chain alkyltriazoles as antitumor agents: Synthesis, physicochemical properties, and biological and computational evaluation. Med. Chem Res. 2015, 24, 430–441. [Google Scholar] [CrossRef]
- Gontijo, V.S.; Espuri, P.F.; Alves, R.B.; de Camargos, L.F.; Santos, F.V.; de Souza Judice, W.A.; Marques, M.J.; Freitas, R.P. Leishmanicidal, antiproteolytic, and mutagenic evaluation of alkyltriazoles and alkylphosphocholines. Eur. J. Med. Chem. 2015, 101, 24–33. [Google Scholar] [CrossRef]
- Xu, Z. 1,2,3-Triazole-containing hybrids with potential antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2020, 206, 112686. [Google Scholar] [CrossRef]
- Slavova, K.I.; Todorov, L.T.; Belskaya, N.P.; Palafox, M.A.; Kostova, I.P. Developments in the Application of 1,2,3-Triazoles in Cancer Treatment. Recent Pat. Anti-Cancer Drug Discov. 2020, 15, 92–112. [Google Scholar] [CrossRef]
- Huang, Q.; Xie, L.; Chen, X.; Yu, H.; Lv, Y.; Huang, X.; Ying, J.; Zheng, C.; Cheng, Y.; Huang, J. Synthesis and anticancer activity of novel rapamycin C-28 containing triazole moiety compounds. Arch. Der Pharm. 2018, 351, e1800123. [Google Scholar] [CrossRef]
- Alam, M.M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Der Pharm. 2022, 355, e2100158. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Saha, S.T.; Gu, L.; Palma, G.; Perumal, S.; Singh-Pillay, A.; Singh, P.; Anand, A.; Kaur, M.; Kumar, V. 1H-1,2,3-Triazole Tethered Nitroimidazole-Isatin Conjugates: Synthesis, Docking, and Anti-Proliferative Evaluation against Breast Cancer. ACS Omega 2018, 3, 12106–12113. [Google Scholar] [CrossRef]
- Sambasiva Rao, P.; Kurumurthy, C.; Veeraswamy, B.; Santhosh Kumar, G.; Shanthan Rao, P.; Pamanji, R.; Venkateswara Rao, J.; Narsaiah, B. Synthesis of novel 2-alkyl triazole-3-alkyl substituted quinoline derivatives and their cytotoxic activity. Bioorganic Med. Chem. Lett. 2013, 23, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Xiang, Z.J.; Yang, J.H.; Wang, W.J.; Xu, Z.C.; Xiang, R.L. Adverse Effects Associated with Currently Commonly Used Antifungal Agents: A Network Meta-Analysis and Systematic Review. Front. Pharm. 2021, 12, 697330. [Google Scholar] [CrossRef] [PubMed]
- Franklim, T.N.; Freire-de-Lima, L.; Chaves, O.A.; LaRocque-de-Freitas, I.F.; Silva-Trindade, J.D.d.; Netto-Ferreira, J.C.; Freire-de-Lima, C.G.; Decoté-Ricardo, D.; Previato, J.O.; Mendonça-Previato, L.; et al. Design, Synthesis, Trypanocidal Activity, and Studies on Human Album in Interaction of Novel S-Alkyl-1,2,4-triazoles %. J. Braz. Chem. Soc. 2019, 30, 1378–1394. [Google Scholar]
- NIH-NCI, Advances in Leukemia Research. U.S. Department of Health and Human Services, National Institutes of Health. National Cancer Institute. 2021. Available online: https://www.cancer.gov/types/leukemia/research (accessed on 22 November 2021).
- Portell, C.A.; Advani, A.S. Novel targeted therapies in acute lymphoblastic leukemia. Leuk. Lymphoma 2014, 55, 737–748. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef]
- Pandey, G.; Bakhshi, S.; Thakur, B.; Jain, P.; Chauhan, S.S. Prognostic significance of cathepsin L expression in pediatric acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Bakhshi, S.; Kumar, M.; Thakur, B.; Jain, P.; Kaur, P.; Chauhan, S.S. Prognostic and therapeutic relevance of cathepsin B in pediatric acute myeloid leukemia. Am. J. Cancer Res. 2019, 9, 2634–2649. [Google Scholar]
- Peng, S.; Yang, Q.; Li, H.; Pan, Y.; Wang, J.; Hu, P.; Zhang, N. CTSB Knockdown Inhibits Proliferation and Tumorigenesis in HL-60 Cells. Int. J. Med. Sci. 2021, 18, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Samaiya, M.; Bakhshi, S.; Shukla, A.A.; Kumar, L.; Chauhan, S.S. Epigenetic regulation of cathepsin L expression in chronic myeloid leukaemia. J. Cell Mol. Med. 2011, 15, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The Refined 2.4a X-Ray Crystal-Structure of Recombinant Human Stefin-B in Complex with the Cysteine Proteinase Papain—A Novel Type of Proteinase-Inhibitor Interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Ghasemi, M.; Brown, R.H. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Hojjat-Farsangi, M. Targeting non-receptor tyrosine kinases using small molecule inhibitors: An overview of recent advances. J. Drug Target. 2016, 24, 192–211. [Google Scholar] [CrossRef]
- Van Blitterswijk, W.J.; Verheij, M. Anticancer alkylphospholipids: Mechanisms of action, cellular sensitivity and resistance, and clinical prospects. Curr. Pharm. Des. 2008, 14, 2061–2074. [Google Scholar] [CrossRef]
- Yosifov, D.Y.; Kaloyanov, K.A.; Guenova, M.L.; Prisadashka, K.; Balabanova, M.B.; Berger, M.R.; Konstantinov, S.M. Alkylphosphocholines and curcumin induce programmed cell death in cutaneous T-cell lymphoma cell lines. Leuk. Res. 2014, 38, 49–56. [Google Scholar] [CrossRef]
- Leonard, R.; Hardy, J.; van Tienhoven, G.; Houston, S.; Simmonds, P.; David, M.; Mansi, J. Randomized, double-blind, placebo-controlled, multicenter trial of 6% miltefosine solution, a topical chemotherapy in cutaneous metastases from breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 4150–4159. [Google Scholar] [CrossRef]
- Chakrabandhu, K.; Huault, S.; Hueber, A.O. Distinctive molecular signaling in triple-negative breast cancer cell death triggered by hexadecylphosphocholine (miltefosine). FEBS Lett. 2008, 582, 4176–4184. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Del Sole, M.; Mongiorgi, S.; Gaboardi, G.C.; Cappellini, A.; Mantovani, I.; Follo, M.Y.; McCubrey, J.A.; Martelli, A.M. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia 2008, 22, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Vital, W.D.; Torquato, H.F.V.; Jesus, L.O.P.; Judice, W.A.S.; Silva, M.; Rodrigues, T.; Justo, G.Z.; Veiga, T.A.M.; Paredes-Gamero, E.J. 4-Deoxyraputindole C induces cell death and cell cycle arrest in tumor cell lines. J. Cell. Biochem. 2019, 120, 9608–9623. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gamero, E.J.; Casaes-Rodrigues, R.L.; Moura, G.E.; Domingues, T.M.; Buri, M.V.; Ferreira, V.H.; Trindade, E.S.; Moreno-Ortega, A.J.; Cano-Abad, M.F.; Nader, H.B.; et al. Cell-permeable gomesin peptide promotes cell death by intracellular Ca2+ overload. Mol. Pharm. 2012, 9, 2686–2697. [Google Scholar] [CrossRef] [PubMed]
- Buri, M.V.; Domingues, T.M.; Paredes-Gamero, E.J.; Casaes-Rodrigues, R.L.; Rodrigues, E.G.; Miranda, A. Resistance to degradation and cellular distribution are important features for the antitumor activity of gomesin. PLoS ONE 2013, 8, e80924. [Google Scholar] [CrossRef]
- De Ford, C.; Ulloa, J.L.; Catalan, C.A.N.; Grau, A.; Martino, V.S.; Muschietti, L.V.; Merfort, I. The sesquiterpene lactone polymatin B from Smallanthus sonchifolius induces different cell death mechanisms in three cancer cell lines. Phytochemistry 2015, 117, 332–339. [Google Scholar] [CrossRef]
- Vieira Torquato, H.F.; Ribeiro-Filho, A.C.; Buri, M.V.; Araujo Junior, R.T.; Pimenta, R.; de Oliveira, J.S.; Filho, V.C.; Macho, A.; Paredes-Gamero, E.J.; de Oliveira Martins, D.T. Canthin-6-one induces cell death, cell cycle arrest and differentiation in human myeloid leukemia cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 958–967. [Google Scholar] [CrossRef]
- Torquato, H.F.V.; Junior, M.T.R.; Lima, C.S.; Junior, R.T.A.; Talhati, F.; Dias, D.A.; Justo, G.Z.; Ferreira, A.T.; Pilli, R.A.; Paredes-Gamero, E.J. A canthin-6-one derivative induces cell death by apoptosis/necroptosis-like with DNA damage in acute myeloid cells. Biomed. Pharm. 2022, 145, 112439. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Y.Y.; Cheng, J.Y.; Chen, L.; Xu, H.; Li, Q.H.; Pang, T.X. Sam68 affects cell proliferation and apoptosis of human adult T-acute lymphoblastic leukemia cells via AKT/mTOR signal pathway. Leuk. Res. 2016, 46, 1–9. [Google Scholar] [CrossRef]
- Mu, Q.T.; Ma, Q.L.; Lu, S.S.; Zhan, T.; Yu, M.X.; Huang, X.; Chen, J.; Jin, J. 10058-F4, a c-Myc inhibitor, markedly increases valproic acid-induced cell death in Jurkat and CCRF-CEM T-lymphoblastic leukemia cells. Oncol. Lett. 2014, 8, 1355–1359. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Foa, R. T-cell acute lymphoblastic leukemia. Haematol-Hematol J. 2009, 94, 160–162. [Google Scholar] [CrossRef]
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Reif, S.; Hecht, K.; Pelka-Fleischer, R.; Pfister, K.; Schmetzer, H. High expression of urokinase plasminogen activator receptor (UPA-R) in acute myeloid leukemia (AML) is associated with worse prognosis. Am. J. Hematol. 2005, 79, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Bakhshi, S.; Shukla, A.A.; Chauhan, S.S. Cathepsins B and L in peripheral blood mononuclear cells of pediatric acute myeloid leukemia: Potential poor prognostic markers. Ann. Hematol. 2010, 89, 1223–1232. [Google Scholar] [CrossRef]
- Gocheva, V.; Zeng, W.; Ke, D.X.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef]
- Leatherbarrow, R.J. GraFit, Version 5.0.13; Erithacus Software Ltd.: Horley, UK, 2010. [Google Scholar]
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 2005, 46, 1–265. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Liu, Y.; Grimm, M.; Dai, W.T.; Hou, M.C.; Xiao, Z.X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharm. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Bell, E.W.; Yin, M.H.; Zhang, Y. EDock: Blind protein-ligand docking by replica-exchange monte carlo simulation. J. Cheminform. 2020, 12, 37. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; Schrödinger LLC.: New York, NY, USA, 2000. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.B.R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N. Amber 16; University of California: San Francisco, CA, USA, 2016; p. 923. [Google Scholar]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin Dynamics of Peptides - the Frictional Dependence of Isomerization Rates of N-Acetylalanyl-N’-Methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N.Log (N) method for Ewald Sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.P.L.; Berkowitz, M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.P.C.G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of N-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Genheden, S.K.O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell | Compound (EC50) µM | ||
|---|---|---|---|
| Miltefosine | 9 | 21 | |
| Kasumi | ND | >140 | >140 |
| K562 | ND | >140 | >140 |
| ARH-77 | ND | 71 ± 0.8 | 96 ± 1 |
| Jurkat | 186 ± 12 | 63 ± 1 | 77 ± 1.5 |
| CCRF-CEM | 120 ± 3 | 40 ± 1.2 | 60 ± 2 |
| Compound | IC50 (µM) | |
|---|---|---|
| Cat B | Cat L | |
| 1 | 7.5 ± 0.1 | 10.5 ± 0.2 |
| 2 | 23.7 ± 0.9 | 30.3 ± 3.3 |
| 3 | 20.6 ± 0.4 | 18.6 ± 0.3 |
| 4 | 11.3 ± 0.7 | 8.8 ± 0.3 |
| 5 | 9.9 ± 0.4 | 18.7 ± 0.5 |
| 6 | 23.4 ± 1.1 | 9.7 ± 0.4 |
| 7 | 10.8 ± 0.2 | 5.3 ± 0.1 |
| 8 | 10.9 ± 0.9 | 8.7 ± 0.3 |
| 9 | 3.5 ± 0.2 | 1.3 ± 0.1 |
| 10 | 61.9 ± 4.1 | 7.5 ± 0.2 |
| 11 | 12.3 ± 0.4 | 1.5 ± 0.1 |
| 12 | 4.6 ± 0.3 | 3.4 ± 0.1 |
| 13 | 6.2 ± 0.2 | 2.2 ± 0.1 |
| 14 | 10.5 ± 0.2 | 2.6 ± 0.1 |
| 15 | 11.4 ± 0.5 | 5.6 ± 0.2 |
| 16 | 8.8 ± 0.6 | 13.5 ± 0.2 |
| 17 | 22.7 ± 0.4 | 7.9 ± 0.2 |
| 18 | 13.2 ± 0.3 | 11.0 ± 0.2 |
| 19 | 8.1 ± 0.5 | 2.5 ± 0.1 |
| 20 | 4.4 ± 0.3 | 4.8 ± 0.1 |
| 21 | 3.9 ± 0.2 | 1.6 ± 0.1 |
| Enz. | Compound C9 | Compound C21 | ||||||
|---|---|---|---|---|---|---|---|---|
| Ki (µM) | αKi (µM) | α | Mec. | Ki (µM) | αKi (µM) | α | Mec. | |
| Cat L | 3.56 ± 0.26 | 3.66 ± 0.07 | 1 | SLNC | 9.72 ± 1.23 | 10.47 ± 1.17 | 1 | SLNC |
| Cat B | 15.3 ± 0.6 | -- | -- | SLC | 27.4 ± 1.2 | 43.9 ± 1.7 | 1.6 | SLNC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jesus, L.d.O.P.; de Souza, A.A.; Torquato, H.F.V.; Gontijo, V.S.; Pereira de Freitas, R.; Gesteira, T.F.; Coulson-Thomas, V.J.; Torquato, R.J.S.; Tanaka, A.S.; Paredes-Gamero, E.J.; et al. In Vitro Study of Cytotoxic Mechanisms of Alkylphospholipids and Alkyltriazoles in Acute Lymphoblastic Leukemia Models. Molecules 2022, 27, 8633. https://doi.org/10.3390/molecules27238633
Jesus LdOP, de Souza AA, Torquato HFV, Gontijo VS, Pereira de Freitas R, Gesteira TF, Coulson-Thomas VJ, Torquato RJS, Tanaka AS, Paredes-Gamero EJ, et al. In Vitro Study of Cytotoxic Mechanisms of Alkylphospholipids and Alkyltriazoles in Acute Lymphoblastic Leukemia Models. Molecules. 2022; 27(23):8633. https://doi.org/10.3390/molecules27238633
Chicago/Turabian StyleJesus, Larissa de Oliveira Passos, Aline Aparecida de Souza, Heron Fernandes Vieira Torquato, Vanessa Silva Gontijo, Rossimirian Pereira de Freitas, Tarsis Ferreira Gesteira, Vivien Jane Coulson-Thomas, Ricardo José Soares Torquato, Aparecida Sadae Tanaka, Edgar Julian Paredes-Gamero, and et al. 2022. "In Vitro Study of Cytotoxic Mechanisms of Alkylphospholipids and Alkyltriazoles in Acute Lymphoblastic Leukemia Models" Molecules 27, no. 23: 8633. https://doi.org/10.3390/molecules27238633
APA StyleJesus, L. d. O. P., de Souza, A. A., Torquato, H. F. V., Gontijo, V. S., Pereira de Freitas, R., Gesteira, T. F., Coulson-Thomas, V. J., Torquato, R. J. S., Tanaka, A. S., Paredes-Gamero, E. J., & Judice, W. A. d. S. (2022). In Vitro Study of Cytotoxic Mechanisms of Alkylphospholipids and Alkyltriazoles in Acute Lymphoblastic Leukemia Models. Molecules, 27(23), 8633. https://doi.org/10.3390/molecules27238633