
Effects of Berberine against Pancreatitis and Pancreatic Cancer



Abstract
1. Introduction
1.1. Pancreatitis
1.1.1. Acute Pancreatitis
1.1.2. Chronic Pancreatitis
1.1.3. Pathophysiology of Pancreatitis
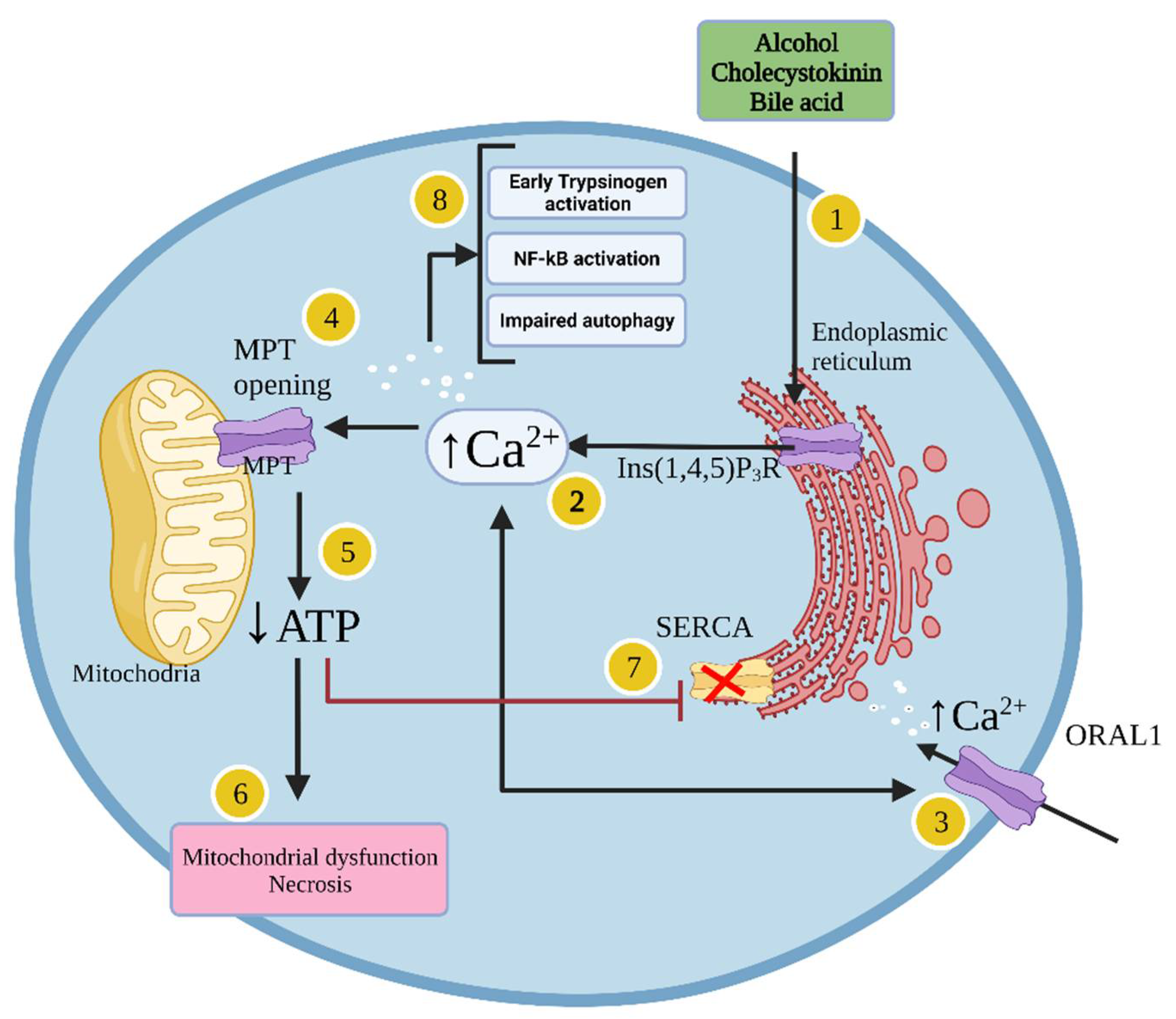
1.1.4. Cellular Mechanisms Leading to Acute Pancreatitis
1.2. Pancreatic Cancer
1.2.1. Pancreatic Cancer Risk Factors
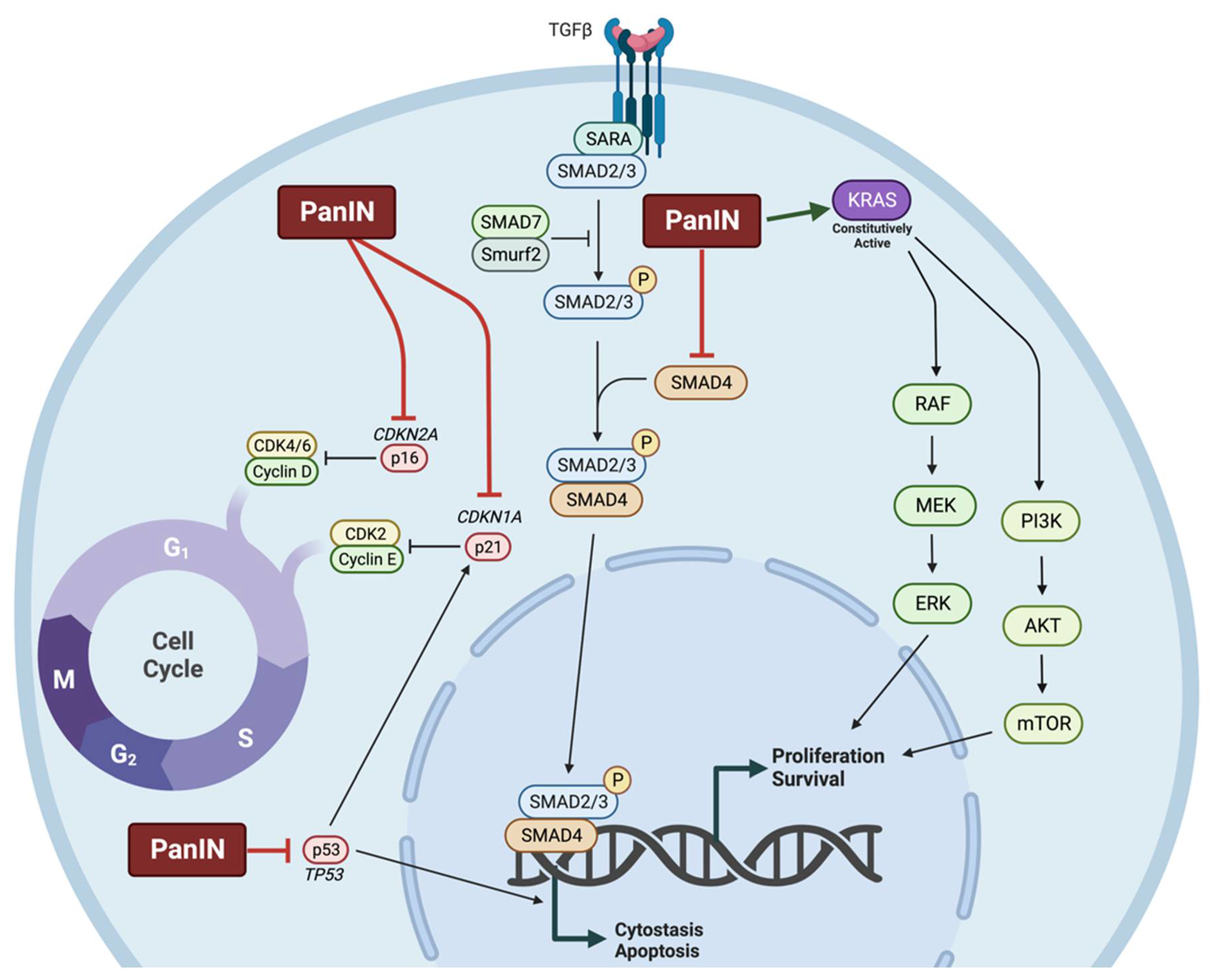
1.2.2. Molecular Characteristics of Pancreatic Cancer
1.2.3. Pancreatic Cancer Diagnosis and Treatment
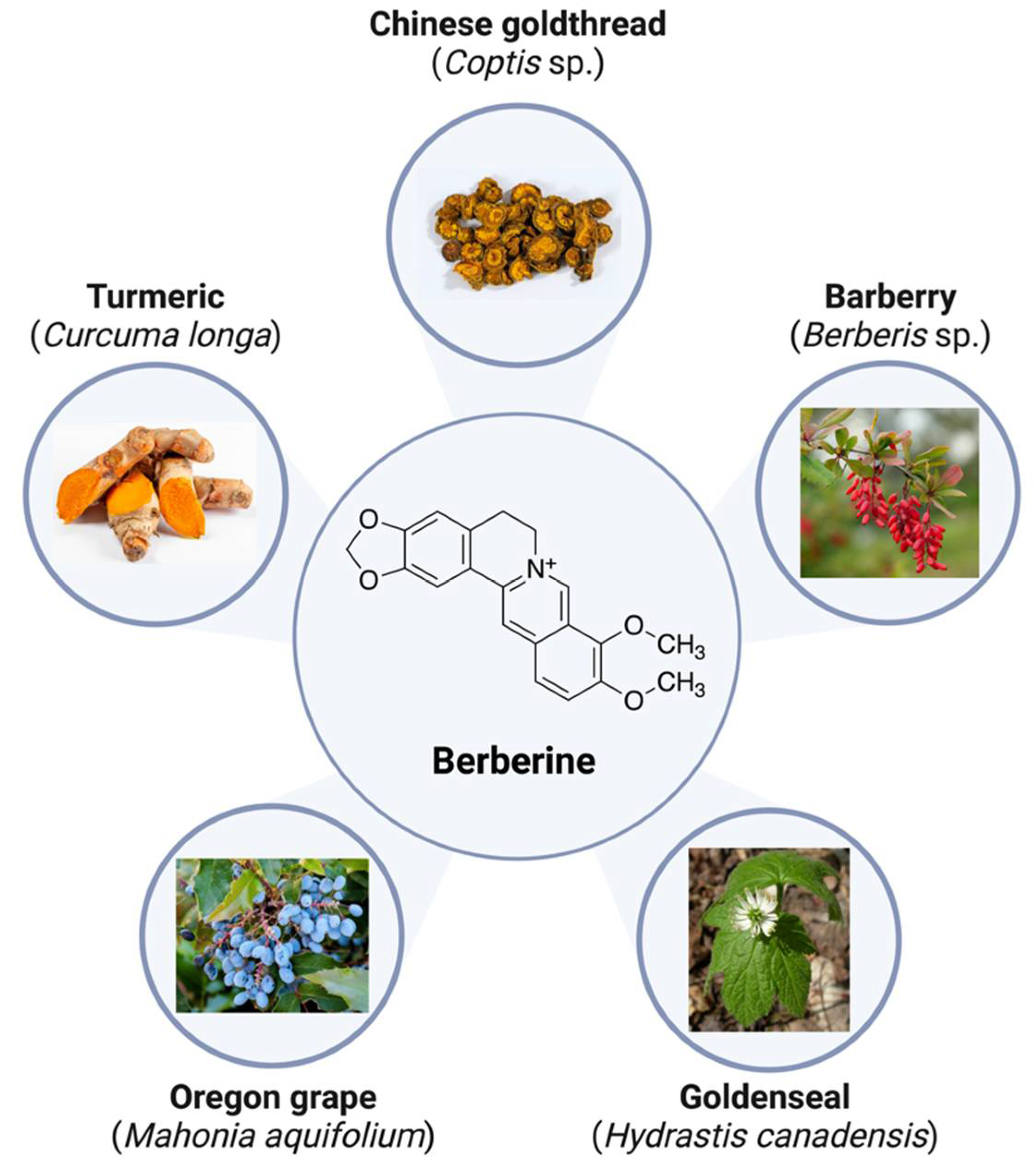
2. Berberine
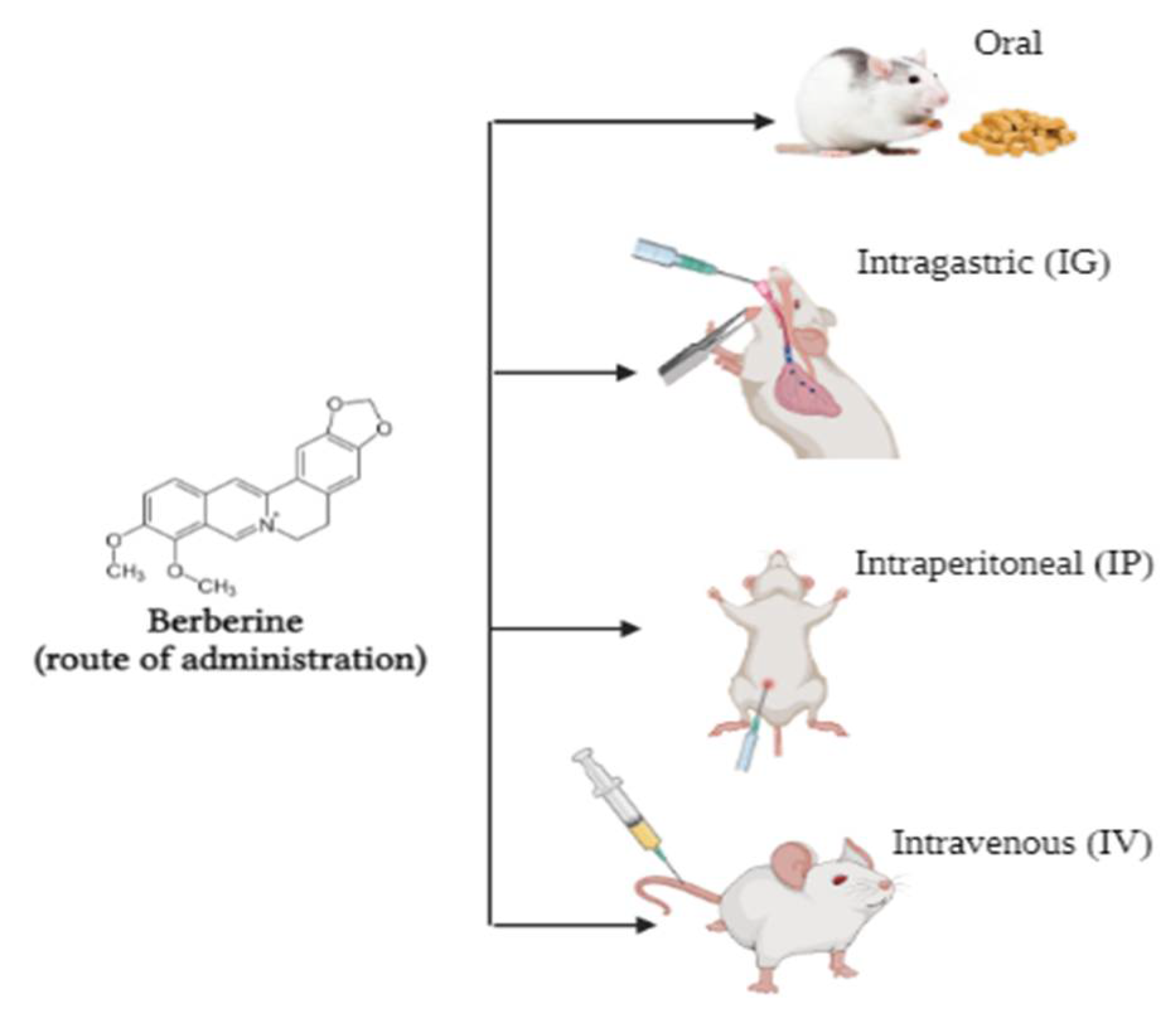
Bioavailability of Berberine
3. Berberine and Pancreatitis
3.1. Effects of Berberine on Pancreatitis: In Vitro Studies
3.2. Effects of Berberine against Pancreatitis: In Vivo Studies
4. Berberine and Pancreatic Cancer
4.1. Effects of Berberine against Pancreatic Cancer: In Vitro Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
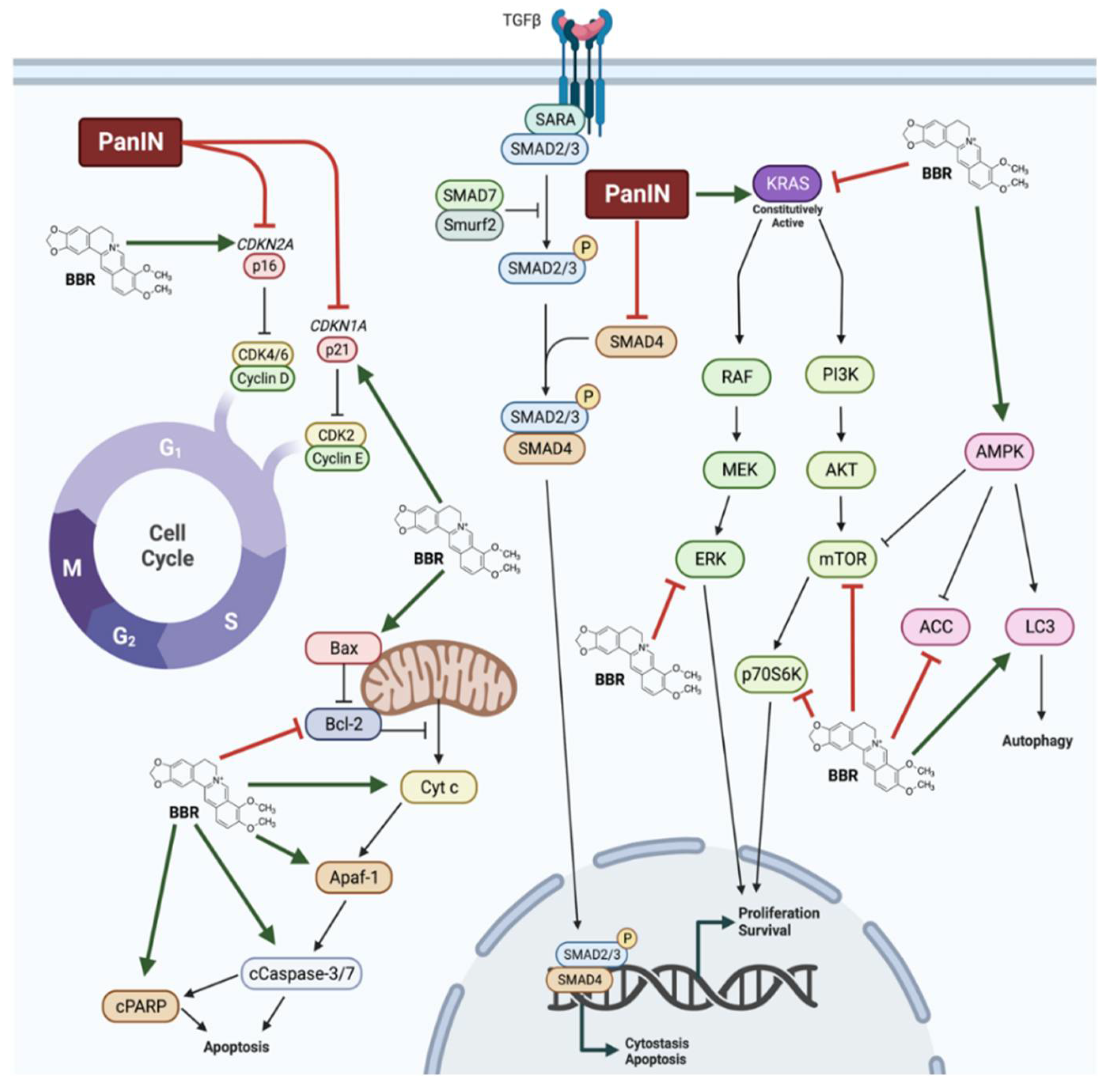{kind=link}
| Model | Treatment | Effects | Signalling | Ref. |
|---|---|---|---|---|
| BxPC-3 | 10–200 µM BBR 24–72 h | ↓ Proliferation | ↑ Caspase 3/7 | [108] |
| Panc02 | 0–10 µM BBR 0–0.12 µM Lovastatin 48 h | ↓ Cell viability | - | [109] |
| ↑ Sub-G1 and G1 phase populations | ||||
| PANC-1 MiaPaCa-2 | 0.3–6 µM BBR 17–72 h | ↓ DNA synthesis | ↑ pAMPK (Thr172) ↑ pACC (Ser79) ↓ mTORC1 ↓pp70 S6K (Thr389) ↓ pS6 (Ser240/244) ↓ pERK (Thr202/Tyr204) ↑ pRaptor (Ser792) | [110] |
| ↓ Proliferation | ||||
| ↑ G1-phase population | ||||
| ↓ S and G2/M-phase population | ||||
| ↓ Mitochondrial membrane potential | ||||
| ↓ ATP levels | ||||
| Mia-PaCa-2 PANC-1 | 15 µM BBR 72 h | ↓ CSC population | ↓ SOX2 ↓ OCT4 ↓ NANOG | [113] |
| PANC-1 Mia-PaCa-2 | 1–15 µM BBR 72 h | ↓ Cell viability | ↑ Caspase-3/7 activity | [114] |
| ↑ G1-phase population | ||||
| ↓ S-phase population | ||||
| ↑ Apoptosis | ||||
| ↑ ROS | ||||
| Mia-PaCa-2 | 10–50 µM BBR 1–48 h | ↓ Cell viability | ↑ p21 ↑ Caspase-3 activity ↑ LC3 ↑ DAP1 ↓ CXCR4 ↑DNMT1 ↑DNMT3A ↑ DNMT3B ↑ MGMT | [115] |
| BBR mitochondrial localisation | ||||
| ↓ Citrate synthase activity | ||||
| ↑ G1-phase population | ||||
| ↓ S and G2-phase population | ||||
| ↑ Senescence | ||||
| ↓ Migration | ||||
| ↓ Invasion | ||||
| Panc-1 | 1–60 µM BBR 48–72 h | ↓ Cell viability | ↓ TNFα ↓ CA242 ↓ K-Ras ↑ CDKN2A | [117] |
| ↑ Apoptosis | ||||
| ↓ Metastasis | ||||
| ↑ Glycolysis-associated metabolites | ||||
| ↑ Glutamine-associated metabolites | ||||
| ↓ Citric acid cycle-associated metabolites | ||||
| ↑ Mitochondrial damage | ||||
| ↓ Citrate metabolism | ||||
| MIA-PaCa-2 PANC-28 AsPC-1 | 1–2000 nM BBR 72 h–2 weeks | ↓ Cell viability | - | [119,121,122,123] |
| ↓ Colony formation | ||||
| PANC-1 AsPC-1 SW1990 | 0–30 µM BBR 24 h | ↓ Trans-endothelial migration | ↓ pSmad2 ↓ pSmad3 ↓ SNAIL1 ↓ SLUG | [124] |
| TGF-β-treated Primary acinar cells | ADM induction: 5 ng/mL TGF-β 2 Days 10 µM BBR 1–2 days | ↓ PanIN | ↓ CK19 ↓ LDHA ↓ALDOA ↓ PFKL ↓ PKM2 ↓ PDK1 ↑ pAMPK ↓ pmTOR ↓ HIF-1α | [127] |
| ↓ ADM | ||||
| ↓ Glycolysis | ||||
| MIN6 | 2.5–50 µM BBR 2–24 h | ↓ Cell viability | ↑ Cytochrome C ↑ AIF ↑ Apaf-1 ↑ Bax ↑ Cleaved Caspsase-3 ↑ Cleaved PARP ↓ Bcl-2 | [129] |
| ↑ Apoptosis | ||||
| ↑ DNA fragmentation |
4.2. Effects of Berberine against Pancreatic Cancer: In Vivo Studies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ćwik, G.; Gierbliński, I.W. Errors and Mistakes in the Ultrasound Diagnosis of the Pancreas. J. Ultrason. 2013, 13, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in Diagnosis of Pancreatic Cancer. World J. Gastroenterol. 2018, 24, 2047–2060. [Google Scholar] [CrossRef]
- Standring, S. Gray’s Anatomy E-Book: The Anatomical Basis of Clinical Practice; Elsevier Health Sciences: Amsterdam, The Netherlands, 2015; ISBN 978-0-7020-6851-5. [Google Scholar]
- Duin, S.; Schütz, K.; Ahlfeld, T.; Lehmann, S.; Lode, A.; Ludwig, B.; Gelinsky, M. 3D Bioprinting of Functional Islets of Langerhans in an Alginate/Methylcellulose Hydrogel Blend. Adv. Healthc. Mater. 2019, 8, e1801631. [Google Scholar] [CrossRef]
- Kim, J.; Kang, K.; Drogemuller, C.J.; Wallace, G.G.; Coates, P.T. Bioprinting an Artificial Pancreas for Type 1 Diabetes. Curr. Diab. Rep. 2019, 19, 53. [Google Scholar] [CrossRef]
- Berman, D.M.; O’Neil, J.J.; Coffey, L.C.K.; Chaffanjon, P.C.J.; Kenyon, N.M.; Ruiz, P.; Pileggi, A.; Ricordi, C.; Kenyon, N.S. Long-Term Survival of Nonhuman Primate Islets Implanted in an Omental Pouch on a Biodegradable Scaffold. Am. J. Transpl. 2009, 9, 91–104. [Google Scholar] [CrossRef]
- Di Piazza, E.; Pandolfi, E.; Cacciotti, I.; Del Fattore, A.; Tozzi, A.E.; Secinaro, A.; Borro, L. Bioprinting Technology in Skin, Heart, Pancreas and Cartilage Tissues: Progress and Challenges in Clinical Practice. Int. J. Environ. Res. Public Health 2021, 18, 10806. [Google Scholar] [CrossRef]
- Lankisch, P.G.; Apte, M.; Banks, P.A. Acute Pancreatitis. Lancet 2015, 386, 85–96. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Peery, A.F.; Crockett, S.D.; Murphy, C.C.; Lund, J.L.; Dellon, E.S.; Williams, J.L.; Jensen, E.T.; Shaheen, N.J.; Barritt, A.S.; Lieber, S.R.; et al. Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018. Gastroenterology 2019, 156, 254–272.e11. [Google Scholar] [CrossRef]
- Peery, A.F.; Dellon, E.S.; Lund, J.; Crockett, S.D.; McGowan, C.E.; Bulsiewicz, W.J.; Gangarosa, L.M.; Thiny, M.T.; Stizenberg, K.; Morgan, D.R.; et al. Burden of Gastrointestinal Disease in the United States: 2012 Update. Gastroenterology 2012, 143, 1179–1187.e3. [Google Scholar] [CrossRef] [PubMed]
- Forsmark, C.E.; Baillie, J. AGA Institute Technical Review on Acute Pancreatitis. Gastroenterology 2007, 132, 2022–2044. [Google Scholar] [CrossRef] [PubMed]
- Guda, N.M.; Trikudanathan, G.; Freeman, M.L. Idiopathic Recurrent Acute Pancreatitis. Lancet Gastroenterol. Hepatol. 2018, 3, 720–728. [Google Scholar] [CrossRef]
- Tenner, S.; Baillie, J.; DeWitt, J.; Vege, S.S.; American College of Gastroenterology. American College of Gastroenterology Guideline: Management of Acute Pancreatitis. Am. J. Gastroenterol. 2013, 108, 1400–1415. [Google Scholar] [CrossRef]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Tóth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Nakano, I.; Koyanagi, S.; Miyahara, T.; Migita, Y.; Ogoshi, K.; Sakai, H.; Matsunaga, S.; Yasuda, O.; Sumii, T.; et al. Autoimmune Pancreatitis as a New Clinical Entity (Three Cases of Autoimmune Pancreatitis with Effective Steroid Therapy). Dig. Dis. Sci. 1997, 42, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, S.J.S.; Sharma, A.; Chari, S.T. Autoimmune Pancreatitis. Am. J. Gastroenterol. 2018, 113, 1301. [Google Scholar] [CrossRef]
- Beyer, G.; Habtezion, A.; Werner, J.; Lerch, M.M.; Mayerle, J. Chronic Pancreatitis. Lancet 2020, 396, 499–512. [Google Scholar] [CrossRef]
- Lévy, P.; Domínguez-Muñoz, E.; Imrie, C.; Löhr, M.; Maisonneuve, P. Epidemiology of Chronic Pancreatitis: Burden of the Disease and Consequences. United Eur. Gastroenterol. J. 2014, 2, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Rizk, M.K.; Gerke, H. Utility of Endoscopic Ultrasound in Pancreatitis: A Review. World J. Gastroenterol. 2007, 13, 6321–6326. [Google Scholar] [CrossRef]
- Thaker, A.M.; Mosko, J.D.; Berzin, T.M. Post-Endoscopic Retrograde Cholangiopancreatography Pancreatitis. Gastroenterol. Rep. (Oxf) 2015, 3, 32–40. [Google Scholar] [CrossRef]
- Pförringer, S. Ueber die Selbstverdauung des Pankreas. Arch. Für Pathol. Anat. Und Physiol. Und Für Klin. Med. 1899, 158, 126–147. [Google Scholar] [CrossRef][Green Version]
- Rao, K.N.; Tuma, J.; Lombardi, B. Acute Hemorrhagic Pancreatic Necrosis in Mice. Intraparenchymal Activation of Zymogens, and Other Enzyme Changes in Pancreas and Serum. Gastroenterology 1976, 70, 720–726. [Google Scholar] [CrossRef]
- Bialek, R.; Willemer, S.; Arnold, R.; Adler, G. Evidence of Intracellular Activation of Serine Proteases in Acute Cerulein-Induced Pancreatitis in Rats. Scand. J. Gastroenterol. 1991, 26, 190–196. [Google Scholar] [CrossRef]
- Grady, T.; Saluja, A.; Kaiser, A.; Steer, M. Edema and Intrapancreatic Trypsinogen Activation Precede Glutathione Depletion during Caerulein Pancreatitis. Am. J. Physiol. 1996, 271, G20–G26. [Google Scholar] [CrossRef]
- Lee, P.J.; Papachristou, G.I. New Insights into Acute Pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 479–496. [Google Scholar] [CrossRef]
- Wen, L.; Voronina, S.; Javed, M.A.; Awais, M.; Szatmary, P.; Latawiec, D.; Chvanov, M.; Collier, D.; Huang, W.; Barrett, J.; et al. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 2015, 149, 481–492.e7. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Pandol, S.J.; Gukovsky, I. New Insights into the Pathways Initiating and Driving Pancreatitis. Curr. Opin. Gastroenterol. 2016, 32, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Mareninova, O.A.; Odinokova, I.V.; Huang, W.; Murphy, J.; Chvanov, M.; Javed, M.A.; Wen, L.; Booth, D.M.; Cane, M.C.; et al. Mechanism of Mitochondrial Permeability Transition Pore Induction and Damage in the Pancreas: Inhibition Prevents Acute Pancreatitis by Protecting Production of ATP. Gut 2016, 65, 1333–1346. [Google Scholar] [CrossRef]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Dawra, R.; Sah, R.P.; Dudeja, V.; Rishi, L.; Talukdar, R.; Garg, P.; Saluja, A.K. Intra-Acinar Trypsinogen Activation Mediates Early Stages of Pancreatic Injury but Not Inflammation in Mice with Acute Pancreatitis. Gastroenterology 2011, 141, 2210–2217.e2. [Google Scholar] [CrossRef]
- Aghdassi, A.A.; John, D.S.; Sendler, M.; Weiss, F.U.; Reinheckel, T.; Mayerle, J.; Lerch, M.M. Cathepsin D Regulates Cathepsin B Activation and Disease Severity Predominantly in Inflammatory Cells during Experimental Pancreatitis. J. Biol. Chem. 2018, 293, 1018–1029. [Google Scholar] [CrossRef]
- Sendler, M.; Weiss, F.-U.; Golchert, J.; Homuth, G.; van den Brandt, C.; Mahajan, U.M.; Partecke, L.-I.; Döring, P.; Gukovsky, I.; Gukovskaya, A.S.; et al. Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice. Gastroenterology 2018, 154, 704–718.e10. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, X.; Zhang, W.; Wu, K.; Ma, J. Hypertriglyceridemia Aggravates ER Stress and Pathogenesis of Acute Pancreatitis. Hepatogastroenterology 2012, 59, 2318–2326. [Google Scholar] [CrossRef]
- Sah, R.P.; Garg, S.K.; Dixit, A.K.; Dudeja, V.; Dawra, R.K.; Saluja, A.K. Endoplasmic Reticulum Stress Is Chronically Activated in Chronic Pancreatitis. J. Biol. Chem. 2014, 289, 27551–27561. [Google Scholar] [CrossRef] [PubMed]
- Lugea, A.; Tischler, D.; Nguyen, J.; Gong, J.; Gukovsky, I.; French, S.W.; Gorelick, F.S.; Pandol, S.J. Adaptive Unfolded Protein Response Attenuates Alcohol-Induced Pancreatic Damage. Gastroenterology 2011, 140, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal Autophagy Maintains Pancreatic Acinar Cell Homeostasis and Protein Synthesis and Prevents ER Stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef]
- Lugea, A.; Waldron, R.T.; Mareninova, O.A.; Shalbueva, N.; Deng, N.; Su, H.-Y.; Thomas, D.D.; Jones, E.K.; Messenger, S.W.; Yang, J.; et al. Human Pancreatic Acinar Cells: Proteomic Characterization, Physiologic Responses, and Organellar Disorders in Ex Vivo Pancreatitis. Am. J. Pathol. 2017, 187, 2726–2743. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S. Acute Pancreatitis Classification Working Group Classification of Acute Pancreatitis—2012: Revision of the Atlanta Classification and Definitions by International Consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Javed, M.A.; Wen, L.; Awais, M.; Latawiec, D.; Huang, W.; Chvanov, M.; Schaller, S.; Bordet, T.; Michaud, M.; Pruss, R.; et al. TRO40303 Ameliorates Alcohol-Induced Pancreatitis Through Reduction of Fatty Acid Ethyl Ester-Induced Mitochondrial Injury and Necrotic Cell Death. Pancreas 2018, 47, 18–24. [Google Scholar] [CrossRef]
- Lur, G.; Sherwood, M.W.; Ebisui, E.; Haynes, L.; Feske, S.; Sutton, R.; Burgoyne, R.D.; Mikoshiba, K.; Petersen, O.H.; Tepikin, A.V. InsP3 Receptors and Orai Channels in Pancreatic Acinar Cells: Co-Localization and Its Consequences. BioChem. J. 2011, 436, 231–239. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Gryshchenko, O.; Ferdek, P.E.; Stapleton, E.; Hébert, T.O.G.; Bychkova, S.; Peng, S.; Begg, M.; Gerasimenko, O.V.; Petersen, O.H. Ca2+ Release-Activated Ca2+ Channel Blockade as a Potential Tool in Antipancreatitis Therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 13186–13191. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.A.; Roberts, J.R.; Cartmell, M.T.; Demaine, A.G.; Kingsnorth, A.N. Heat Shock Factor-1 and Nuclear Factor-KappaB Are Systemically Activated in Human Acute Pancreatitis. JOP 2006, 7, 174–184. [Google Scholar]
- Gray, K.D.; Simovic, M.O.; Blackwell, T.S.; Christman, J.W.; May, A.K.; Parman, K.S.; Chapman, W.C.; Stain, S.C. Activation of Nuclear Factor Kappa B and Severe Hepatic Necrosis May Mediate Systemic Inflammation in Choline-Deficient/Ethionine-Supplemented Diet-Induced Pancreatitis. Pancreas 2006, 33, 260–267. [Google Scholar] [CrossRef]
- Watanabe, T.; Kudo, M.; Strober, W. Immunopathogenesis of Pancreatitis. Mucosal. Immunol. 2017, 10, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-B.; Liao, D.-H.; Nissen, T.D. Animal Models of Pancreatitis: Can It Be Translated to Human Pain Study? World J. Gastroenterol. 2013, 19, 7222–7230. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary Standards of Care and Recent Progress in Pancreatic Ductal Adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hao, L.; Zeng, X.-P.; Xin, L.; Wang, D.; Pan, J.; Bi, Y.-W.; Ji, J.-T.; Du, T.-T.; Lin, J.-H.; Zhang, D.; et al. Incidence of and Risk Factors for Pancreatic Cancer in Chronic Pancreatitis: A Cohort of 1656 Patients. Dig. Liver Dis. 2017, 49, 1249–1256. [Google Scholar] [CrossRef]
- McWilliams, R.R.; Maisonneuve, P.; Bamlmlet, W.R.; Petersen, G.M.; Li, D.; Risch, H.A.; Yu, H.; Fontham, E.T.H.; Luckett, B.; Bosetti, C.; et al. Risk Factors for Early-Onset and Very-Early-Onset Pancreatic Adenocarcinoma: A Pancreatic Cancer Case-Control Consortium (PanC4) Analysis. Pancreas 2016, 45, 311–316. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic Cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Rebours, V.; Gaujoux, S.; d’Assignies, G.; Sauvanet, A.; Ruszniewski, P.; Lévy, P.; Paradis, V.; Bedossa, P.; Couvelard, A. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN). Clin. Cancer Res. 2015, 21, 3522–3528. [Google Scholar] [CrossRef]
- Stolzenberg-Solomon, R.Z.; Schairer, C.; Moore, S.; Hollenbeck, A.; Silverman, D.T. Lifetime Adiposity and Risk of Pancreatic Cancer in the NIH-AARP Diet and Health Study Cohort. Am. J. Clin. Nutr. 2013, 98, 1057–1065. [Google Scholar] [CrossRef]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.P.; Oldfield, L.; Ney, A.; Hart, P.A.; Keane, M.G.; Pandol, S.J.; Li, D.; Greenhalf, W.; Jeon, C.Y.; Koay, E.J.; et al. Early Detection of Pancreatic Cancer. Lancet Gastroenterol. Hepatol. 2020, 5, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.; Parmigiani, G.; Kensler, T.W.; Wolfgang, C.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. Genetic Mutations Associated with Cigarette Smoking in Pancreatic Cancer. Cancer Res. 2009, 69, 3681–3688. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Das, S.; Brand, R.; Whitcomb, D.C. Inherited Pancreatic Cancer Syndromes. Cancer J. 2012, 18, 485–491. [Google Scholar] [CrossRef]
- Benzel, J.; Fendrich, V. Familial Pancreatic Cancer. ORT 2018, 41, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer Risk Estimates for BRCA1 Mutation Carriers Identified in a Risk Evaluation Program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef]
- Vasen, H.F.; Gruis, N.A.; Frants, R.R.; van Der Velden, P.A.; Hille, E.T.; Bergman, W. Risk of Developing Pancreatic Cancer in Families with Familial Atypical Multiple Mole Melanoma Associated with a Specific 19 Deletion of P16 (P16-Leiden). Int. J. Cancer 2000, 87, 809–811. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; Shelton, C.A.; Brand, R.E. Genetics and Genetic Testing in Pancreatic Cancer. Gastroenterology 2015, 149, 1252–1264.e4. [Google Scholar] [CrossRef]
- Tamura, K.; Yu, J.; Hata, T.; Suenaga, M.; Shindo, K.; Abe, T.; MacGregor-Das, A.; Borges, M.; Wolfgang, C.L.; Weiss, M.J.; et al. Mutations in the Pancreatic Secretory Enzymes CPA1 and CPB1 Are Associated with Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4767–4772. [Google Scholar] [CrossRef]
- van Heek, N.T.; Meeker, A.K.; Kern, S.E.; Yeo, C.J.; Lillemoe, K.D.; Cameron, J.L.; Offerhaus, G.J.A.; Hicks, J.L.; Wilentz, R.E.; Goggins, M.G.; et al. Telomere Shortening Is Nearly Universal in Pancreatic Intraepithelial Neoplasia. Am. J. Pathol. 2002, 161, 1541–1547. [Google Scholar] [CrossRef]
- Guo, J.; Xie, K.; Zheng, S. Molecular Biomarkers of Pancreatic Intraepithelial Neoplasia and Their Implications in Early Diagnosis and Therapeutic Intervention of Pancreatic Cancer. Int. J. Biol. Sci. 2016, 12, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Hansen, M.; Berendse, S.; Hamilton, W. Symptoms of Pancreatic Cancer in Primary Care: A Systematic Review. Pancreas 2016, 45, 814–818. [Google Scholar] [CrossRef]
- Al-Hawary, M.M.; Francis, I.R.; Chari, S.T.; Fishman, E.K.; Hough, D.M.; Lu, D.S.; Macari, M.; Megibow, A.J.; Miller, F.H.; Mortele, K.J.; et al. Pancreatic Ductal Adenocarcinoma Radiology Reporting Template: Consensus Statement of the Society of Abdominal Radiology and the American Pancreatic Association. Gastroenterology 2014, 146, 291–304.e1. [Google Scholar] [CrossRef]
- Valls, C.; Andía, E.; Sanchez, A.; Fabregat, J.; Pozuelo, O.; Quintero, J.C.; Serrano, T.; Garcia-Borobia, F.; Jorba, R. Dual-Phase Helical CT of Pancreatic Adenocarcinoma: Assessment of Resectability before Surgery. AJR Am. J. Roentgenol. 2002, 178, 821–826. [Google Scholar] [CrossRef]
- DeWitt, J.; Devereaux, B.; Chriswell, M.; McGreevy, K.; Howard, T.; Imperiale, T.F.; Ciaccia, D.; Lane, K.A.; Maglinte, D.; Kopecky, K.; et al. Comparison of Endoscopic Ultrasonography and Multidetector Computed Tomography for Detecting and Staging Pancreatic Cancer. Ann. Intern. Med. 2004, 141, 753–763. [Google Scholar] [CrossRef]
- Vachiranubhap, B.; Kim, Y.H.; Balci, N.C.; Semelka, R.C. Magnetic Resonance Imaging of Adenocarcinoma of the Pancreas. Top Magn. Reson. Imaging 2009, 20, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ballehaninna, U.K.; Chamberlain, R.S. The Clinical Utility of Serum CA 19-9 in the Diagnosis, Prognosis and Management of Pancreatic Adenocarcinoma: An Evidence Based Appraisal. J. Gastrointest Oncol. 2012, 3, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Isaji, S.; Mizuno, S.; Windsor, J.A.; Bassi, C.; Fernández-Del Castillo, C.; Hackert, T.; Hayasaki, A.; Katz, M.H.G.; Kim, S.-W.; Kishiwada, M.; et al. International Consensus on Definition and Criteria of Borderline Resectable Pancreatic Ductal Adenocarcinoma 2017. Pancreatology 2018, 18, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Millikan, K.W.; Deziel, D.J.; Silverstein, J.C.; Kanjo, T.M.; Christein, J.D.; Doolas, A.; Prinz, R.A. Prognostic Factors Associated with Resectable Adenocarcinoma of the Head of the Pancreas. Am. Surg. 1999, 65, 618–623, discussion 623–624. [Google Scholar] [CrossRef]
- Okasha, H.; Elkholy, S.; El-Sayed, R.; Wifi, M.-N.; El-Nady, M.; El-Nabawi, W.; El-Dayem, W.A.; Radwan, M.I.; Farag, A.; El-Sherif, Y.; et al. Real Time Endoscopic Ultrasound Elastography and Strain Ratio in the Diagnosis of Solid Pancreatic Lesions. World J. Gastroenterol. 2017, 23, 5962–5968. [Google Scholar] [CrossRef]
- Grycová, L.; Dostál, J.; Marek, R. Quaternary Protoberberine Alkaloids. Phytochemistry 2007, 68, 150–175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T. The Traditional Chinese Materia Medica: The Old and the New. Am. J. Chin. Med. 1987, 15, 35–38. [Google Scholar] [CrossRef]
- Sahibzada, M.U.K.; Sadiq, A.; Faidah, H.S.; Khurram, M.; Amin, M.U.; Haseeb, A.; Kakar, M. Berberine Nanoparticles with Enhanced in Vitro Bioavailability: Characterization and Antimicrobial Activity. Drug Des. Devel. Ther. 2018, 12, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yang, W.; Yang, X.; Mei, X.; Hu, T.; Liang, R.; Meng, D.; Yan, D. MgAl Monolayer Hydrotalcite Increases the Hypoglycemic Effect of Berberine by Enhancing Its Oral Bioavailability. Biomed. Pharmacother. 2020, 127, 110140. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Chi, C.-W.; Liu, T.-Y. The Anti-Inflammatory Potential of Berberine in Vitro and in Vivo. Cancer Lett. 2004, 203, 127–137. [Google Scholar] [CrossRef]
- Li, Z.; Geng, Y.-N.; Jiang, J.-D.; Kong, W.-J. Antioxidant and Anti-Inflammatory Activities of Berberine in the Treatment of Diabetes Mellitus. Evid. Based Complement AlterNat. Med. 2014, 2014, 289264. [Google Scholar] [CrossRef] [PubMed]
- Imenshahidi, M.; Hosseinzadeh, H. Chapter 14—Berberine Neuroprotection and Antioxidant Activity. In Oxidative Stress and Dietary Antioxidants in Neurological Diseases; Martin, C.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 199–216. ISBN 978-0-12-817780-8. [Google Scholar]
- Chang, W.; Chen, L.; Hatch, G.M. Berberine as a Therapy for Type 2 Diabetes and Its Complications: From Mechanism of Action to Clinical Studies. BioChem. Cell Biol. 2015, 93, 479–486. [Google Scholar] [CrossRef]
- Lan, J.; Zhao, Y.; Dong, F.; Yan, Z.; Zheng, W.; Fan, J.; Sun, G. Meta-Analysis of the Effect and Safety of Berberine in the Treatment of Type 2 Diabetes Mellitus, Hyperlipemia and Hypertension. J. Ethnopharmacol. 2015, 161, 69–81. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, X.; Yin, M.; Zhang, Y.; Huang, L.; Chen, R.; Ni, J. Effects of Berberine on Blood Glucose in Patients with Type 2 Diabetes Mellitus: A Systematic Literature Review and a Meta-Analysis. Endocr. J. 2019, 66, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xun, K.; Wang, Y.; Chen, X. A Systematic Review of the Anticancer Properties of Berberine, a Natural Product from Chinese Herbs. Anti-Cancer Drugs 2009, 20, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Du, X.; Ma, H.; Yao, J. The Anti-Cancer Mechanisms of Berberine: A Review. Cancer Manag. Res. 2020, 12, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Abu-Izneid, T.; Khalil, A.A.; Imran, M.; Shah, Z.A.; Emran, T.B.; Mitra, S.; Khan, Z.; Alhumaydhi, F.A.; Aljohani, A.S.M.; et al. Berberine as a Potential Anticancer Agent: A Comprehensive Review. Molecules 2021, 26, 7368. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Feng, X.; Chai, L.; Cao, S.; Qiu, F. The Metabolism of Berberine and Its Contribution to the Pharmacological Effects. Drug Metab Rev. 2017, 49, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, F.; Ma, X.; Cheng, X.; Zhou, H.; Klaassen, C.D. CYP2D Plays a Major Role in Berberine Metabolism in Liver of Mice and Humans. Xenobiotica 2011, 41, 996–1005. [Google Scholar] [CrossRef]
- Chen, W.; Miao, Y.-Q.; Fan, D.-J.; Yang, S.-S.; Lin, X.; Meng, L.-K.; Tang, X. Bioavailability Study of Berberine and the Enhancing Effects of TPGS on Intestinal Absorption in Rats. AAPS PharmSciTech 2011, 12, 705–711. [Google Scholar] [CrossRef]
- Gong, Z.; Chen, Y.; Zhang, R.; Wang, Y.; Guo, Y.; Yang, Q.; Zhang, H.; Dong, Y.; Weng, X.; Gao, S.; et al. Pharmacokinetic Comparison of Berberine in Rat Plasma after Oral Administration of Berberine Hydrochloride in Normal and Post Inflammation Irritable Bowel Syndrome Rats. Int. J. Mol. Sci. 2014, 15, 456–467. [Google Scholar] [CrossRef]
- Kheir, M.M.; Wang, Y.; Hua, L.; Hu, J.; Li, L.; Lei, F.; Du, L. Acute Toxicity of Berberine and Its Correlation with the Blood Concentration in Mice. Food Chem. Toxicol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Tan, X.-S.; Ma, J.-Y.; Feng, R.; Ma, C.; Chen, W.-J.; Sun, Y.-P.; Fu, J.; Huang, M.; He, C.-Y.; Shou, J.-W.; et al. Tissue Distribution of Berberine and Its Metabolites after Oral Administration in Rats. PLoS ONE 2013, 8, e77969. [Google Scholar] [CrossRef]
- Hua, W.; Ding, L.; Chen, Y.; Gong, B.; He, J.; Xu, G. Determination of Berberine in Human Plasma by Liquid Chromatography-Electrospray Ionization-Mass Spectrometry. J. Pharm. Biomed. Anal. 2007, 44, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-S.; Zheng, Y.-R.; Zhang, Y.-F.; Long, X.-Y. Research Progress on Berberine with a Special Focus on Its Oral Bioavailability. Fitoterapia 2016, 109, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Ochin, C.C.; Garelnabi, M. Berberine Encapsulated PLGA-PEG Nanoparticles Modulate PCSK-9 in HepG2 Cells. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, M.A.; Elnaggar, Y.S.R.; Hamdy, D.A.; Abdallah, O.Y. Novel Cremochylomicrons for Improved Oral Bioavailability of the Antineoplastic Phytomedicine Berberine Chloride: Optimization and Pharmacokinetics. Int. J. Pharm. 2018, 535, 316–324. [Google Scholar] [CrossRef]
- Pan, G.; Wang, G.-J.; Liu, X.-D.; Fawcett, J.P.; Xie, Y.-Y. The Involvement of P-Glycoprotein in Berberine Absorption. Pharmacol. Toxicol. 2002, 91, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jiang, S.-J.; Lu, F.-E.; Xu, L.-J.; Zou, X.; Wang, K.-F.; Dong, H. Effects of Berberine and Cinnamic Acid on Palmitic Acid-Induced Intracellular Triglyceride Accumulation in NIT-1 Pancreatic β Cells. Chin. J. Integr. Med. 2016, 22, 496–502. [Google Scholar] [CrossRef]
- Li, J.; Du, H.; Zhang, M.; Zhang, Z.; Teng, F.; Zhao, Y.; Zhang, W.; Yu, Y.; Feng, L.; Cui, X.; et al. Amorphous Solid Dispersion of Berberine Mitigates Apoptosis via IPLA2β/Cardiolipin/Opa1 Pathway in Db/Db Mice and in Palmitate-Treated MIN6 β-Cells. Int. J. Biol. Sci. 2019, 15, 1533–1545. [Google Scholar] [CrossRef]
- Bansod, S.; Doijad, N.; Godugu, C. Berberine Attenuates Severity of Chronic Pancreatitis and Fibrosis via AMPK-Mediated Inhibition of TGF-Β1/Smad Signaling and M2 Polarization. Toxicol. Appl. Pharmacol. 2020, 403, 115162. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, S.; Tang, J.; Zhang, K.; Guang, L.; Huang, Y.; Xu, Y.; Ying, Y.; Zhang, L.; Li, D. Protective Effect of Berberine on Beta Cells in Streptozotocin- and High-Carbohydrate/High-Fat Diet-Induced Diabetic Rats. Eur. J. Pharmacol. 2009, 606, 262–268. [Google Scholar] [CrossRef]
- Yang, X.; Yao, L.; Fu, X.; Mukherjee, R.; Xia, Q.; Jakubowska, M.A.; Ferdek, P.E.; Huang, W. Experimental Acute Pancreatitis Models: History, Current Status, and Role in Translational Research. Front. Physiol. 2020, 11, 614591. [Google Scholar] [CrossRef]
- Battu, S.K.; Repka, M.A.; Maddineni, S.; Chittiboyina, A.G.; Avery, M.A.; Majumdar, S. Physicochemical Characterization of Berberine Chloride: A Perspective in the Development of a Solution Dosage Form for Oral Delivery. AAPS PharmSciTech 2010, 11, 1466–1475. [Google Scholar] [CrossRef]
- Liang, H.-Y.; Chen, T.; Yan, H.-T.; Huang, Z.; Tang, L.-J. Berberine Ameliorates Severe Acute Pancreatitis-induced Intestinal Barrier Dysfunction via a Myosin Light Chain Phosphorylation-dependent Pathway. Mol. Med. Rep. 2014, 9, 1827–1833. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, S.-B.; Bae, G.-S.; Jo, I.-J.; Wang, S.; Song, H.-J.; Park, S.-J. Berberine Inhibits Inflammatory Mediators and Attenuates Acute Pancreatitis through Deactivation of JNK Signaling Pathways. Mol. Immunol. 2016, 74, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-B.; Bae, G.-S.; Jo, I.-J.; Song, H.-J.; Park, S.-J. Effects of Berberine on Acute Necrotizing Pancreatitis and Associated Lung Injury. Pancreas 2017, 46, 1046–1055. [Google Scholar] [CrossRef]
- Ou, X.; Hua, Y.; Liao, X.; Gong, C.; Kang, Y. Cognitive Impairments Induced by Severe Acute Pancreatitis Are Attenuated by Berberine Treatment in Rats. Mol. Med. Rep. 2018, 18, 3437–3444. [Google Scholar] [CrossRef]
- Pinto-Garcia, L.; Efferth, T.; Torres, A.; Hoheisel, J.D.; Youns, M. Berberine Inhibits Cell Growth and Mediates Caspase-Independent Cell Death in Human Pancreatic Cancer Cells. Planta Med. 2010, 76, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Issat, T.; Nowis, D.; Bil, J.; Winiarska, M.; Jakobisiak, M.; Golab, J. Antitumor Effects of the Combination of Cholesterol Reducing Drugs. Oncol. Rep. 2011, 26, 169–176. [Google Scholar] [CrossRef]
- Ming, M.; Sinnett-Smith, J.; Wang, J.; Soares, H.P.; Young, S.H.; Eibl, G.; Rozengurt, E. Dose-Dependent AMPK-Dependent and Independent Mechanisms of Berberine and Metformin Inhibition of MTORC1, ERK, DNA Synthesis and Proliferation in Pancreatic Cancer Cells. PLoS ONE 2014, 9, e114573. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef]
- Wu, C.; Alman, B.A. Side Population Cells in Human Cancers. Cancer Lett. 2008, 268, 1–9. [Google Scholar] [CrossRef]
- Park, S.H.; Sung, J.H.; Chung, N. Berberine Diminishes Side Population and Down-Regulates Stem Cell-Associated Genes in the Pancreatic Cancer Cell Lines PANC-1 and MIA PaCa-2. Mol. Cell BioChem. 2014, 394, 209–215. [Google Scholar] [CrossRef]
- Park, S.H.; Sung, J.H.; Kim, E.J.; Chung, N. Berberine Induces Apoptosis via ROS Generation in PANC-1 and MIA-PaCa2 Pancreatic Cell Lines. Braz. J. Med. Biol. Res. 2015, 48, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Agnarelli, A.; Natali, M.; Garcia-Gil, M.; Pesi, R.; Tozzi, M.G.; Ippolito, C.; Bernardini, N.; Vignali, R.; Batistoni, R.; Bianucci, A.M.; et al. Cell-Specific Pattern of Berberine Pleiotropic Effects on Different Human Cell Lines. Sci. Rep. 2018, 8, 10599. [Google Scholar] [CrossRef]
- Jin, B.; Robertson, K.D. DNA Methyltransferases, DNA Damage Repair, and Cancer. In Epigenetic Alterations in Oncogenesis; Karpf, A.R., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; pp. 3–29. ISBN 978-1-4419-9967-2. [Google Scholar]
- Liu, J.; Luo, X.; Guo, R.; Jing, W.; Lu, H. Cell Metabolomics Reveals Berberine-Inhibited Pancreatic Cancer Cell Viability and Metastasis by Regulating Citrate Metabolism. J. Proteome Res. 2020, 19, 3825–3836. [Google Scholar] [CrossRef]
- Dong, D.; Jia, L.; Zhang, L.; Ma, N.; Zhang, A.; Zhou, Y.; Ren, L. Periostin and CA242 as Potential Diagnostic Serum Biomarkers Complementing CA19.9 in Detecting Pancreatic Cancer. Cancer Sci. 2018, 109, 2841–2851. [Google Scholar] [CrossRef]
- Abrams, S.L.; Follo, M.Y.; Steelman, L.S.; Lertpiriyapong, K.; Cocco, L.; Ratti, S.; Martelli, A.M.; Candido, S.; Libra, M.; Murata, R.M.; et al. Abilities of Berberine and Chemically Modified Berberines to Inhibit Proliferation of Pancreatic Cancer Cells. Adv. Biol. Regul. 2019, 71, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.P.; Song, H.; Xu, Y. A Common Gain of Function of P53 Cancer Mutants in Inducing Genetic Instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Lertpiriyapong, K.; Yang, L.V.; Martelli, A.M.; Cocco, L.; Ratti, S.; Falasca, M.; Murata, R.M.; Rosalen, P.L.; Lombardi, P.; et al. Introduction of WT-TP53 into Pancreatic Cancer Cells Alters Sensitivity to Chemotherapeutic Drugs, Targeted Therapeutics and Nutraceuticals. Adv. Biol. Regul. 2018, 69, 16–34. [Google Scholar] [CrossRef]
- Abrams, S.L.; Akula, S.M.; Steelman, L.S.; Follo, M.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Libra, M.; Falzone, L.; Candido, S.; et al. Effects of the MDM2 Inhibitor Nutlin-3a on Sensitivity of Pancreatic Cancer Cells to Berberine and Modified Berberines in the Presence and Absence of WT-TP53. Adv. Biol. Regul. 2022, 83, 100840. [Google Scholar] [CrossRef]
- Abrams, S.L.; Akula, S.M.; Meher, A.K.; Steelman, L.S.; Gizak, A.; Duda, P.; Rakus, D.; Martelli, A.M.; Ratti, S.; Cocco, L.; et al. GSK-3β Can Regulate the Sensitivity of MIA-PaCa-2 Pancreatic and MCF-7 Breast Cancer Cells to Chemotherapeutic Drugs, Targeted Therapeutics and Nutraceuticals. Cells 2021, 10, 816. [Google Scholar] [CrossRef]
- Tian, W.; Hao, H.; Chu, M.; Gong, J.; Li, W.; Fang, Y.; Zhang, J.; Zhang, C.; Huang, Y.; Pei, F.; et al. Berberine Suppresses Lung Metastasis of Cancer via Inhibiting Endothelial Transforming Growth Factor Beta Receptor 1. Front. Pharmacol. 2022, 13, 917827. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Chen, X.; Wang, J.; Guo, L.; Wang, Q.; Gao, Z.; Kang, J.; Zhang, M.; Feng, J.; Guo, Q.; et al. Polypharmacology of Berberine Based on Multi-Target Binding Motifs. Front. Pharmacol. 2018, 9, 801. [Google Scholar] [CrossRef]
- Wagener, B.M.; Hu, M.; Zheng, A.; Zhao, X.; Che, P.; Brandon, A.; Anjum, N.; Snapper, S.; Creighton, J.; Guan, J.-L.; et al. Neuronal Wiskott-Aldrich Syndrome Protein Regulates TGF-Β1-Mediated Lung Vascular Permeability. FASEB J. 2016, 30, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Kang, W.; Liu, Y.; Tao, X.; Li, X.; Pan, Y. Berberine Inhibits Pancreatic Intraepithelial Neoplasia by Inhibiting Glycolysis via the Adenosine Monophosphate -Activated Protein Kinase Pathway. Eur. J. Pharmacol. 2022, 915, 174680. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Rachagani, S.; Chakraborty, S.; Singh, P.K.; Zhao, X.; Gurumurthy, C.B.; Anderson, J.M.; Lele, S.; Hollingsworth, M.A.; Band, V.; et al. Overexpression of Ecdysoneless in Pancreatic Cancer and Its Role in Oncogenesis by Regulating Glycolysis. Clin. Cancer Res. 2012, 18, 6188–6198. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Miao, X.-L.; Liu, J.-L.; Zhang, D.-W.; Wang, M.; Zhao, D.-D.; Mu, Q.-Q.; Yu, N.; Mo, F.-F.; Yin, H.-P.; et al. Berberine Induces Cell Apoptosis through Cytochrome C/Apoptotic Protease-Activating Factor 1/Caspase-3 and Apoptosis Inducing Factor Pathway in Mouse Insulinoma Cells. Chin. J. Integr. Med. 2019, 25, 853–860. [Google Scholar] [CrossRef]
- Tao, X.; Chen, Q.; Li, N.; Xiang, H.; Pan, Y.; Qu, Y.; Shang, D.; Go, V.L.W.; Xue, J.; Sun, Y.; et al. Serotonin-RhoA/ROCK Axis Promotes Acinar-to-Ductal Metaplasia in Caerulein-Induced Chronic Pancreatitis. Biomed. Pharmacother. 2020, 125, 109999. [Google Scholar] [CrossRef]





| Animal Model | Treatment | Effects | Signalling | Ref. |
|---|---|---|---|---|
| Taurocholate-induced SAP Sprague Dawley rats | Daily aqueous intragastric administration BBR 50 mg/kg for 5 days | ↓ Intestinal barrier dysfunction | ↓ phospho-MLC | [104] |
| ↓ Serum DAO activity | ||||
| ↓ Serum endotoxin levels | ||||
| ↓ Bacterial translocation | ||||
| Cerulein-induced AP and L-arginine-induced SAP C57BL/6 mice | Intraperitoneal injection BBR 1–20 mg/kg 1 h pre-treatment for 6 h | ↓ Pancreatic edema | ↓iNOS ↓NO ↓ TNF-α, IL-1β, and IL-6 ↓ phospho-JNK | [105] |
| ↓ Pancreatic inflammation | ||||
| ↓ Lung damage | ||||
| ↓Serum amylase and lipase | ||||
| ↓ Pancreatic myeloperoxidase | ||||
| ↓ pulmonary myeloperoxidase | ||||
| CDE diet-induced pancreatitis Female C57BL/6 mice | Intraperitoneal injection BBR 1, 5, 10 mg/kg/day For 3 days | ↓ histological damage | ↓ NFκB ↓ JNK ↓ p38 | [106] |
| ↓ plasma amylase and lipase | ||||
| ↓ myeloperoxidase activity | ||||
| ↓ TNF-α, IL-1β, and IL-6 | ||||
| ↓ mortality rate (60%) | ||||
| L-arginine-induced SAP Wistar rats | Intragastric administration BBR 100 mg/kg/day for 6 days | ↑ Survival | - | [107] |
| Protected from pancreatic encephalopathy | ||||
| ↓ pancreatic inflammation and necrosis | ||||
| ↓ amylase levels | ||||
| Prevented blood-brain barrier permeability | ||||
| Cerulein-induced CP Male Swiss albino mice | Intraperitoneal injection BBR 3 or 10 mg/kg/day for 21 days | ↑ pancreatic weight | ↓ α-SMA ↓ collagen 1a and 3a ↓ fibronectin ↑ AMPK ↓ TGF-β1/Smad2/3 ↑ Smad7 ↑ E-cadherin ↓ Slug and Snail ↓ CD206 | [100] |
| ↓ plasma amylase and lipase | ||||
| ↓ pancreatic MDA | ||||
| ↓ pancreatic nitrate | ||||
| ↑ GSH | ||||
| ↓ TNF-α, IL-6, IL-1β, and TGF-β1 | ||||
| ↓ collagen deposition | ||||
| ↓ inflammatory cell infiltration | ||||
| ↓ acinar cell atrophy | ||||
| ↓ exocrine vacuolization |
| Animal Model | Treatment | Effects | Ref. |
|---|---|---|---|
| Panc02 xenografted c57Bl/6 mice | Lovastatin: | ↓ Tumour volume Synergistic effect | [109] |
| Intraperitoneal injection 30 mg/kg/day | |||
| BBR: Oral administration 100 mg/kg/day for 14 days | |||
| MiaPaCa-2 xenografted nude mice | Intraperitoneal injection | ↓ Tumour weight ↓ Tumour volume | [110] |
| 5 mg/kg/day BBR | |||
| AsPC-1 intravenous injection (tail vein) in BALB/c nude mice | Oral Gavage | ↑ Survival ↓ Lung metastases ↓ Lung infiltration | [124] |
| 100–200 mg/kg/day BBR | |||
| Beginning 3 days prior to AsPC-1 injection | |||
| Cerulein-induced CP/CP-induced neoplasia C57BL/6 mice | Intragastric Administration | ↓ ADM ↓ PanIN ↓ Fibrosis ↓ CK19 ↑ Amylase | [127] |
| BBR 200 mg/kg | |||
| Daily, 3 days/week | |||
| 4–8 weeks |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlavcheski, F.; O’Neill, E.J.; Gagacev, F.; Tsiani, E. Effects of Berberine against Pancreatitis and Pancreatic Cancer. Molecules 2022, 27, 8630. https://doi.org/10.3390/molecules27238630
Vlavcheski F, O’Neill EJ, Gagacev F, Tsiani E. Effects of Berberine against Pancreatitis and Pancreatic Cancer. Molecules. 2022; 27(23):8630. https://doi.org/10.3390/molecules27238630
Chicago/Turabian StyleVlavcheski, Filip, Eric J. O’Neill, Filip Gagacev, and Evangelia Tsiani. 2022. "Effects of Berberine against Pancreatitis and Pancreatic Cancer" Molecules 27, no. 23: 8630. https://doi.org/10.3390/molecules27238630
APA StyleVlavcheski, F., O’Neill, E. J., Gagacev, F., & Tsiani, E. (2022). Effects of Berberine against Pancreatitis and Pancreatic Cancer. Molecules, 27(23), 8630. https://doi.org/10.3390/molecules27238630