
A Comparative Study of the Synthesis and Hydrolysis of sym-Triaminobenzene Homologues

Abstract
1. Introduction
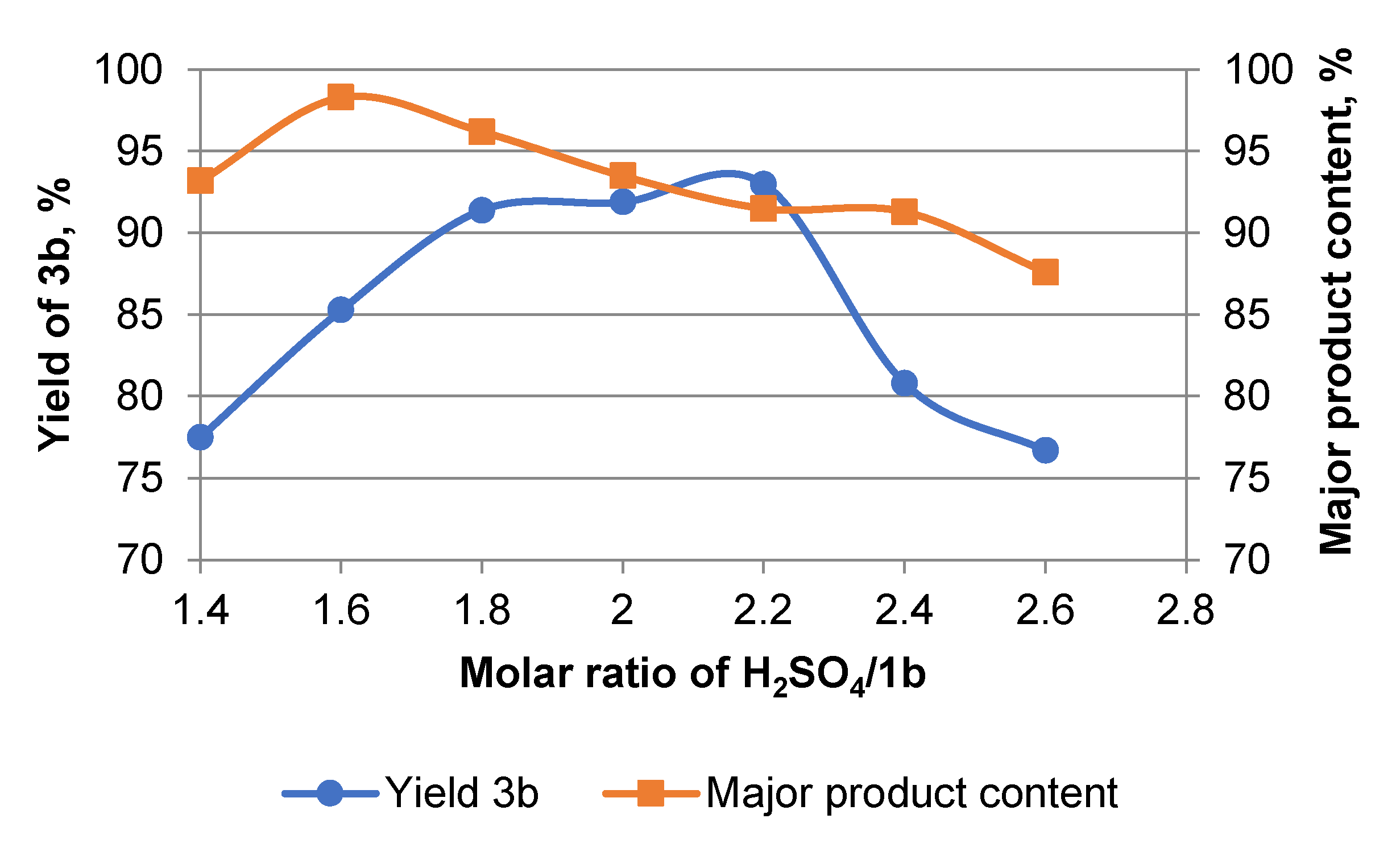
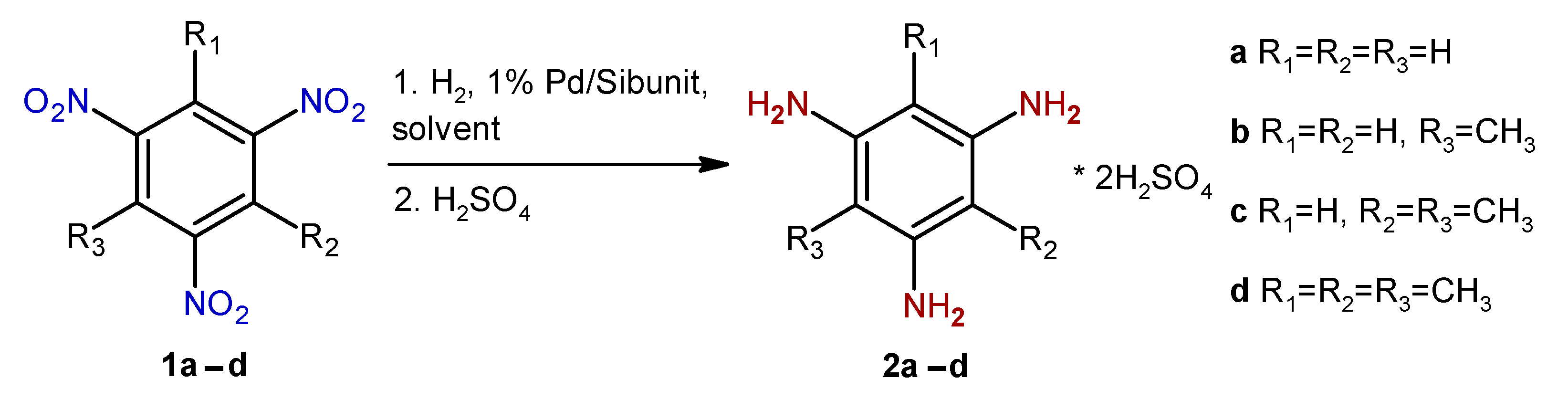
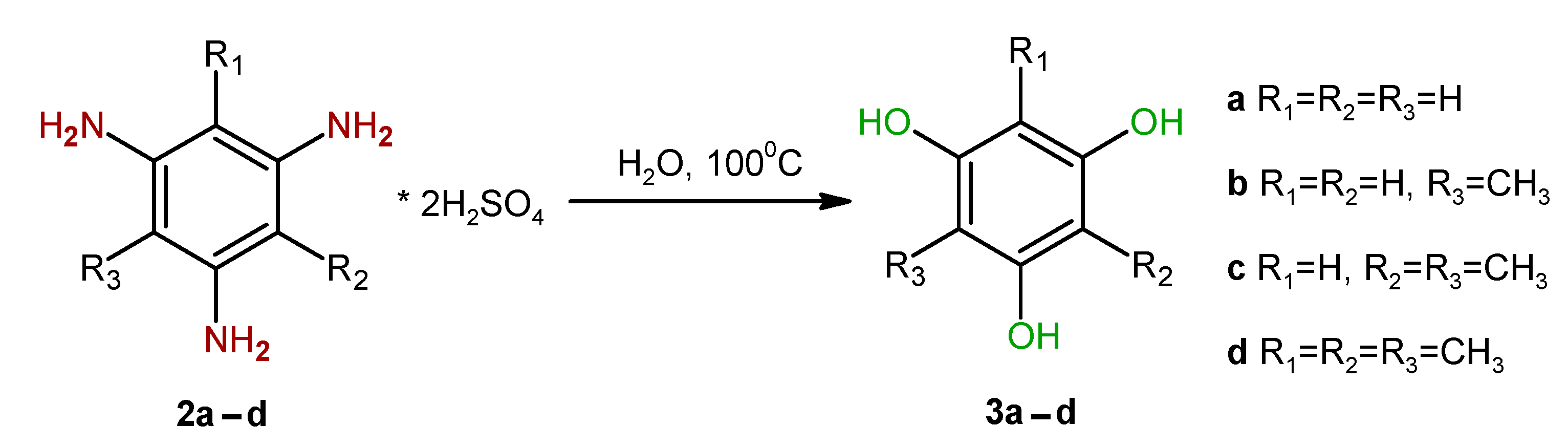
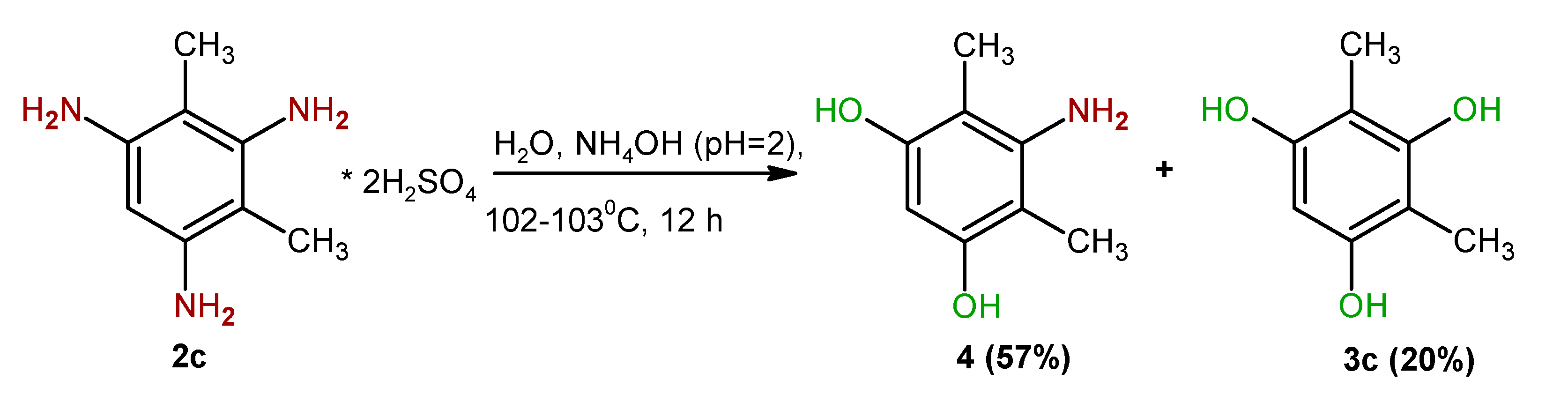
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Characterization Techniques
3.3. High-Performance Liquid Chromatography
3.4. Gas Chromatography
4. Experimental
4.1. General Procedure for Sulfuric Acid Salts of Triaminobenzene Derivatives 2b–d
- 2,4,6-Triaminotoluene disulfate (2b)
- 2,4,6-Triaminoxylene disulfate (2c)
- 2,4,6-Triaminomesitylene disulfate (2d)
4.2. General Procedure for the Hydrolysis of the Sulfuric-Acid Salts of Triaminobenzene Derivatives 2b–d
- 2-Methyl phloroglucinol (3b)
- 2,4-Dimethyl phloroglucinol (3c)
- 2,4,6-Trimethyl phloroglucinol (3d)
4.3. Synthesis of 2-Amino-4,6-dihydroxyxylene (4)
4.4. General Synthetic Procedure for Polyphenols 3b–d from Trinitrobenzenes 1b–d
- 2-Methyl phloroglucinol (3b)
- 2,4-Dimethyl phloroglucinol (3c)
- 2,4,6-Trimethyl phloroglucinol (3d)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alekseev, S.G.; Petrova, E.K.; Kuznetsov, D.N.; Kobrakov, K.I. A brief history of the chemical transformation of 2,4,6-trinitrotoluene. Butl. Commun. 2020, 62, 1–11. [Google Scholar] [CrossRef]
- Lee, H.; Park, R.Y.; Park, K. Total Syntheses of 4′,6′-dimethoxy-2′-hydroxy-3′,5′-dimethylchalcone derivatives. Bull. Korean Chem. Soc. 2021, 42, 66–71. [Google Scholar] [CrossRef]
- Teng, X.; Wang, Y.; Gu, J.; Shi, P.; Shen, Z.; Ye, L. Antifungal agents: Design, synthesis, antifungal activity and molecular docking of phloroglucinol derivatives. Molecules 2018, 23, 3116. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Tesfu, H.H.; Hogan, A.M.; Cardona, S.T.; Sorensen, J.L. Synthesis and antibiotic activity of novel acylated phloroglucinol compounds against methicillin-resistant Staphylococcus aureus. J. Antibiot. 2019, 72, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Verbanac, D.; Jain, S.C.; Jain, N.; Chand, M.; Paljetak, H.C.; Matijašic, M.; Peric, M.; Stepanic, V.; Saso, L. An efficient and convenient microwave-assisted chemical synthesis of (thio)xanthones with additional in vitro and in silico characterization. Bioorg. Med. Chem. 2012, 20, 3180–3185. [Google Scholar] [CrossRef]
- Yang, M.; Lai, W.; Li, J.; Ye, L. Design, Synthesis and Antifungal Activity of Phloroglucinol Derivatives. Pharm. Chem. J. 2022, 56, 356–360. [Google Scholar] [CrossRef]
- Meguellati, A.; Ahmed-Belkacem, A.; Nurisso, A.; Yi, W.; Brillet, R.; Berqouch, N.; Chavoutier, L.; Fortun, A.; Pawlotsky, J.-M.; Boumendjel, A.; et al. New pseudodimeric aurones as palm pocket inhibitors of Hepatitis C virus RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2016, 115, 217–229. [Google Scholar] [CrossRef]
- Shi, L.; Feng, X.E.; Cui, J.R.; Fang, L.H.; Du, G.H.; Li, Q.S. Synthesis and biological activity of flavanone derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 5466–5468. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Chen, H.-Y.; Chang, Y.-L.; Vasanthakumar, P.; Chen, S.-Y.; Kao, C.-L.; Wu, C.H.Y.; Hsu, S. Synthesis of triisocyanomesitylene b diketiminato copper(I) complexes and evaluation of isocyanide p-back bonding. Polyhedron 2020, 192, 114828. [Google Scholar] [CrossRef]
- Yuste, F.; Sánchez-Obregón, R.; Walls, F. The synthesis of grandinol. Tetrahedron Lett. 1978, 19, 4869–4870. [Google Scholar] [CrossRef]
- Belskaya, O.B.; Mironenko, R.M.; Talsi, V.P.; Rodionov, V.A.; Sysolyatin, S.V.; Likholobov, V.A. Transformation pathways of 2,4,6-trinitrobenzoic acid in the aqueous-phase hydrogenation over Pd/C catalyst. J. Mol. Catal. A Chem. 2016, 420, 190–199. [Google Scholar] [CrossRef]
- Belskaya, O.B.; Mironenko, R.M.; Talsi, V.P.; Rodionov, V.A.; Gulyaeva, T.I.; Sysolyatin, S.V.; Likholobov, V.A. The effect of preparation conditions of Pd/C catalyst on its activity and selectivity in the aqueous-phase hydrogenation of 2,4,6-trinitrobenzoic acid. Catal. Today 2018, 301, 258–265. [Google Scholar] [CrossRef]
- Kashaev, V.A.; Khisamutdinov, G.K.; Shevelev, S.A.; Shakhnes, A.K.; Bavrina, A.P. Preparation of 2,4,6-triaminotoluene and its salts with inorganic acids from 2,4,6-trinitrotoluene. Theor. Found. Chem. Eng. 2008, 42, 650–656. [Google Scholar] [CrossRef]
- Shchurova, I.A.; Alekseyeva, N.A.; Arbagozova, A.A.; Malykhin, V.V. Improved method for the preparation of 2,4,6-trihydroxytoluene. Uzno-Sib. Naucn. Vestn. 2019, 28, 221–225. (In Russian) [Google Scholar] [CrossRef]
- Ushkarov, V.I.; Kobrakov, K.I.; Alafinov, A.I.; Shevelev, S.A.; Shakhnes, A.K. Methylphloroglucinol As an Available Semiproduct for Azo Dye Synthesis. Theor. Found. Chem. Eng. 2007, 41, 671–674. [Google Scholar] [CrossRef]
- Parker, D.; Senanayake, K.; Vepsailainen, J.; Williams, S.; Batsanov, A.S.; Howard, J.A.K. Reaction chemistry of tri-substituted mesitylene derivatives and the synthesis of sterically buttressed 1,3,5-triaminocyclohexyl ligands. J. Chem. Soc. Perkin Trans. 2 1997, 8, 1445–1452. [Google Scholar] [CrossRef]
- Shchurova, I.A.; Alekseyeva, N.A.; Sysolyatin, S.V.; Malykhin, V.V. Method for the preparation of 2,4,6-trihydroxytoluene from 2,4,6-trinitrotoluene. Uzno-Sib. Naucn. Vestn. 2021, 40, 143–147. (In Russian) [Google Scholar] [CrossRef]
- Shchurova, I.A.; Alekseyeva, N.A.; Sysolyatin, S.V.; Malykhin, V.V. An improved method for the preparation of phloroglucinol from 1,3,5-trinitrobenzene. Uzno-Sib. Naucn. Vestn. 2021, 40, 148–154. (In Russian) [Google Scholar] [CrossRef]
- van Gelder, K.B.; Damhof, J.K.; Kroijenga, P.J.; Westerterp, K.R. Three-phase packed bed reactor with an evaporating solvent—I. Experimental: The hydrogenation of 2,4,6-trinitrotoluene in methanol. Chem. Eng. Sci. 1990, 45, 3159. [Google Scholar] [CrossRef]
- de Angelis, S.; Batsanov, A.; Norman, T.J.; Parker, D.; Senanayake, K.; Vepsalainen, J. Conformationally biased tri- and di-basic 1,3,5-triazacyclohexyl ligands. J. Chem. Soc. Chem. Commun. 1995, 22, 2361–2364. [Google Scholar] [CrossRef]
- Temme, O.; Dickner, T.; Laschat, S.; Fröhlich, R.; Kotila, S.; Bergander, K. Synthesis of Azapolycyclic Systems Based on the Indolizino [3,4-b]quinoline Skeleton—A Diastereoselective Entry to Potential Oligodentate Artificial Receptors. Eur. J. Org. Chem. 1998, 4, 651–659. [Google Scholar] [CrossRef]
- Mclean, A.; Tetlow, W.E.; Munro, J. Improvements in or Relating to the Production of Triamino-Monocyclic Aromatic Hydrocarbons. GB Patent 5,897,16-A, 27 June 1947. National Center for Biotechnology Information: Bethesda, MD, USA. [Google Scholar]
- Schuster, L. Process for the Preparation of Triaminotoluene. DE Patent 3,218,665, 16 December 1982. BASF AG: Ludwigshafen, Germany. [Google Scholar]
- Shchurova, I.A.; Alekseyeva, N.A.; Malykhin, V.V.; Sysolyatin, S.V. Development of the Technology of Phloroglucinol Production. Chem. Sustain. Dev. 2021, 29, 368–380. [Google Scholar] [CrossRef]
- Sumpter, B.G.; Meunier, V.; Valeev, E.F. A new class of supramolecular wires. J. Phys. Chem. C 2007, 111, 18912–18916. [Google Scholar] [CrossRef]
- Zielke, R.; Maegerlein, H. Process for the Preparation of Phloroglucinol Homologues. US Patent 4,296,260A, 20 October 1981. Akzona Inc.: Cnversion, DE, USA. [Google Scholar]
- Kashaev, V.A.; Bavrina, A.P.; Sedova, N.V.; Valeshnij, S.I.; Il’in, V.P.; Kolganov, E.V. Method for Preparing 2,4,6-Triaminotoluene as Sulfate or Phosphate. RU Patent 2,297,408C1, 20 April 2007. Russian Patent Office: Moscow, Russia. [Google Scholar]
- Kashaev, V.A.; Pechenev, Y.G.; Drozdova, T.S.; Bavrina, A.P.; Khisamutdinov, G.K. Investigation of the catalytic hydrogenation of trinitrobenzene. Vestn. Kazan. Tekhnol. Un-Ta 2014, 17, 82–85. (In Russian) [Google Scholar]
- Shchurova, I.A.; Arbagozova, A.A.; Alekseyeva, N.A.; Malykhin, V.V. Catalytic Hydrogenation of 1,3,5-Trinitrobenzene. Uzno-Sib. Naucn. Vestn. 2019, 28, 166–170. (In Russian) [Google Scholar] [CrossRef]
- Talsi, V.P.; Belskaya, O.B.; Yurpalov, V.L. The composition of transformation products of 2,4,6-trinitrobenzoic acid in the aqueous-phase hydrogenation over Pd/C catalysts. Magn. Reson. Chem. 2020, 58, 84. [Google Scholar] [CrossRef]
- Shevelev, S.A.; Shakhnes, A.K.; Vorob’ev, S.S. Method for Preparing 2,4,6-Trihydroxytoluene. RU Patent 2,292,329C1, 27 May 2007. Russian Patent Office: Moscow, Russia. [Google Scholar]
- Li, Y.; Wang, J.; Chen, J.; Wang, J.; Chai, X.; Li, M.; Fang, K.; Cao, D.; Zhao, L.; Chen, L. Method for Preparing TNT (Trinitrotoluene) by Taking Nitrotoluene as Raw Material Through One-Step Method. CN Patent 1,087,070,77A, 26 October 2018. Hubei Dongfang Chemical Industry Co Ltd.: Hubei, China. [Google Scholar]
- Xu, J.; Wei, J.; Li, F.; Ma, Q.; Peng, X. Conjugated Energetic Salts Based on 3,3′-((1E,1′E)-(2,4,6-trinitro-1,3-phenylene)-bis(ethene-2,1-diyl))bis(2,4,6-trinitrophenol). New J. Chem. 2014, 38, 5303–5311. [Google Scholar] [CrossRef]
- Risch, N. 1—Azaadamantane aus substituierten Phloroglucinen. Chem. Ber. 1985, 118, 4849–4856. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Solvent | Solvent/Substrate Ratio, mL/g | Reaction Time, h | Product | Yield,% |
|---|---|---|---|---|---|---|
| 1 | 1a 2 | Methanol | 7 | 2.0 | 2a | 83.9 |
| 2 | 1a2,3 | Methanol | 7 | 3.0 | 2a | 81.7 |
| 3 | 1b | Methanol | 7 | 1.8 | 2b | 98.5 |
| 4 | 1c | Methanol | 10 | 2.4 | 2c | 84.4 |
| 5 | 1c | Methanol | 25 | 2.5 | 2c | 75.2 |
| 6 | 1c | Methanol | 40 | 4.6 | 2c | 59.4 |
| 7 | 1c | Methanol/ toluene 4:1 (v/v) | 10 | 5.3 | 2c | 91.0 |
| 8 | 1d | Methanol | 15 | 4.0 | 2d | 97.2 |
| 9 | 1d | Methanol | 25 | 4.2 | 2d | 95.0 |
| 10 | 1d | Methanol | 50 | 2.3 | 2d | 91.5 |
| 11 | 1d 3 | Methanol | 50 | 7.0 | 2d | 79.1 |
| Entry | Substrate | Water/Salt Ratio, mL/g | Hydrolysis Time, h | Product | Yield, % |
|---|---|---|---|---|---|
| 1 | 2a1 | 10 | 13 | 3a | 86.0 |
| 2 | 2b | 10 | 16 | 3b | 83.7 |
| 3 | 2b | 10 | 24 | 3b | 90.3 |
| 4 | 2c | 10 | 13 | 3c | 54.2 |
| 5 | 2c | 10 | 35 | 3c | 76.6 |
| 6 | 2c2 | 10 | 12 | 3c | 20.0 |
| 7 | 2d | 10 | 14 | 3d | 82.3 |
| 8 | 2d | 5 | 14 | 3d | 84.8 |
| Entry | Substrate | Cycle | Hydrogenation Time, h | Hydrolysis Time, h | Product | Yield,% |
|---|---|---|---|---|---|---|
| 1 | 1a2 | 1 | 4.8 | 14 | 3a | 77.8 |
| 2 | 1a 2 | 10 | 5.5 | 14 | 3a | 75.1 |
| 3 | 1a 2 | 18 | 8.3 | 14 | 3a | 72.7 |
| 4 | 1a2 | 20 | 9.0 | 14 | 3a | 65.4 |
| 5 | 1b | 1 | 4.5 | 24 | 3b | 91.1 |
| 6 | 1b | 6 | 3.7 | 24 | 3b | 90.4 |
| 7 | 1b | 10 | 3.0 | 24 | 3b | 85.0 |
| 8 | 1c | 1 | 5.0 | 14 | 3c | 12.1 |
| 9 | 1d | 1 | 2.5 | 35 | 3d | 27.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shchurova, I.A.; Alekseyeva, N.A.; Sysolyatin, S.V.; Malykhin, V.V. A Comparative Study of the Synthesis and Hydrolysis of sym-Triaminobenzene Homologues. Molecules 2022, 27, 8595. https://doi.org/10.3390/molecules27238595
Shchurova IA, Alekseyeva NA, Sysolyatin SV, Malykhin VV. A Comparative Study of the Synthesis and Hydrolysis of sym-Triaminobenzene Homologues. Molecules. 2022; 27(23):8595. https://doi.org/10.3390/molecules27238595
Chicago/Turabian StyleShchurova, Irina A., Natalia A. Alekseyeva, Sergey V. Sysolyatin, and Valeriy V. Malykhin. 2022. "A Comparative Study of the Synthesis and Hydrolysis of sym-Triaminobenzene Homologues" Molecules 27, no. 23: 8595. https://doi.org/10.3390/molecules27238595
APA StyleShchurova, I. A., Alekseyeva, N. A., Sysolyatin, S. V., & Malykhin, V. V. (2022). A Comparative Study of the Synthesis and Hydrolysis of sym-Triaminobenzene Homologues. Molecules, 27(23), 8595. https://doi.org/10.3390/molecules27238595