Abstract
We developed a straightforward synthetic route to pharmacologically important 1,5-substituted pyrrolidin-2-ones from donor–acceptor cyclopropanes bearing an ester group as one of the acceptor substituents. This method includes a Lewis acid-catalyzed opening of the donor–acceptor cyclopropane with primary amines (anilines, benzylamines, etc.) to γ-amino esters, followed by in situ lactamization and dealkoxycarbonylation. The reaction has a broad scope of applicability; a variety of substituted anilines, benzylamines, and other primary amines as well as a wide range of donor–acceptor cyclopropanes bearing (hetero)aromatic or alkenyl donor groups and various acceptor substituents can be involved in this transformation. In this process, donor–acceptor cyclopropanes react as 1,4-C,C-dielectrophiles, and amines react as 1,1-dinucleophiles. The resulting di- and trisubstituted pyrrolidin-2-ones can be also used in subsequent chemistry to obtain various nitrogen-containing polycyclic compounds of interest to medicinal chemistry and pharmacology, such as benz[g]indolizidine derivatives.
1. Introduction
The γ-lactam skeleton is a component of many biologically active molecules, both natural and synthetic, including approved drugs [1,2]. In particular, 1,5-diarylpyrrolidin-2-ones or 5-aryl-1-benzylpyrrolidones have great potential in pharmacology and medicinal chemistry (Figure 1). Among 1,5-diarylpyrrolidin-2-ones, there are selective and effective inhibitors of histone deacetylases 5 and 6 [3,4,5], cannabinoid receptor 1 (CB1) [6,7], cyclin-dependent kinase CDK2 [8], tankyrase [9], etc. They are also capable of inhibiting glutaminyl cyclase [10] and the glucagon receptor [11]. In addition, 5-aryl-1-benzylpyrrolidones have been shown to antagonize the dual orexin receptor at the submicromolar level [12,13] and calcitonin gene-related peptide type I receptors at the subnanomolar level [14]. Therefore, the synthesis of these promising azaheterocycles is an urgent problem in synthetic organic and pharmaceutical chemistry.
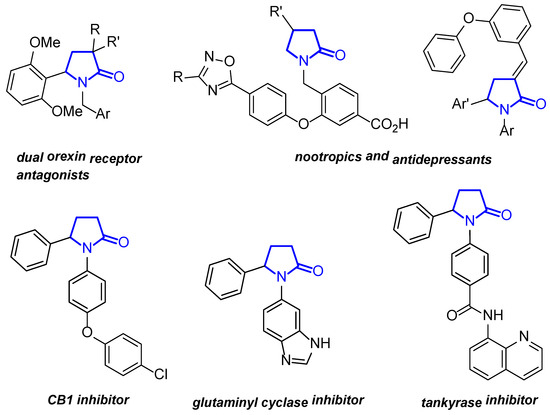
Figure 1.
Bioactive molecules containing 1,5-diarylpyrrolidin-2-one or 5-aryl-1-benzylpyrrolidin-2-one frameworks.
Although many methods for the γ-lactam synthesis are known [15,16,17,18], the development of new and simple strategies that also make it possible to introduce desired substituents into the resulting products remains an urgent task. Our interest in this problem is related to the possibility of solving it using the donor–acceptor (DA) cyclopropane [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33] reactivity, which has been the subject of our studies in recent years [27,28,34,35,36,37,38,39].
In general, two types of transformations of DA cyclopropanes can be used for the synthesis of 1,5-substituted pyrrolidin-2-ones. The first one is the (3 + 2)-cycloaddition of 2-aryl- or 2-alkenylcyclopropane-1,1-diesters with the appropriate isocyanates [40,41]. This process directly afforded the corresponding pyrrolidones (Scheme 1a); however, the resulting products contain two acceptor substituents at the C(3) atom; these groups must be removed to obtain the aforementioned bioactive compounds.
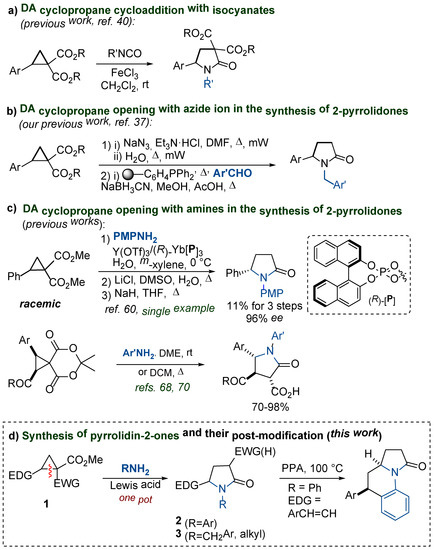
Scheme 1.
Synthesis of pyrrolidin-2-ones from DA cyclopropanes. (a–c) The reported methods for the synthesis of 1,5-substitutted pyrrolidine-2-ones from DA cyclopropanes. (d) Transformations reported in this work.
Alternatively, these DA cyclopropanes can undergo a small ring opening with N-nucleophiles followed by cyclization producing the target γ-lactams. For example, we have recently developed a method for the synthesis of 1,5-substituted pyrrolidin-2-ones, the key step of which is the opening of the DA cyclopropane ring with an azide ion (Scheme 1b) [37,38,39]. This method includes isolation and purification of the intermediate azides; a simpler and general approach to the synthesis of 1,5-disubstituted pyrrolidin-2-ones 2, 3 can be developed based on the reaction of DA cyclopropanes 1 with the corresponding primary amines, such as anilines, benzylamines, etc.
The reactions of DA cyclopropanes with primary amines affording both acyclic and various cyclic products, depending on the structure of the reagents and reaction conditions, have been well studied [29,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70]. However, only a few examples of the use of this reactivity for the synthesis of 1,5-functionalized pyrrolidin-2-ones have been described [59,60,61,62,63,64,65,66,67,68,69,70]. Usually, these examples were reported as postmodifications of the primary acyclic products [59,60,61,62,63,64] that provides not the principal advantage over other stepwise transformations. The one-step formation of the requisite pyrrolidones was achieved either on specific substrates [65,66,67,68,69,70] (Scheme 1c), i.e., has limited application, or proceeded under harsh conditions, giving pyrrolidones in moderate yields [62,64].
In this paper, we demonstrate that the transformation of DA cyclopropanes to 1,5-substituted pyrrolidones can be implemented as a one-pot process via a Lewis acid-initiated, three-membered ring opening with anilines, benzylamines, and other primary amines, followed by lactamization, as well as further modifications of the obtained pyrrolidones to polycyclic molecules such as benz[g]indolizidine derivatives (Scheme 1d).
2. Results and Discussion
We started our investigation with the study of the reaction of model cyclopropane 1a with aniline, leading to the acyclic product 4a (Table 1). To catalyze the reaction, we tested several available Lewis acids, which are commonly used for initiating reactions of DA cyclopropanes with N-nucleophiles. The reaction was carried out in dichloroethane (DCE) at room temperature for 1 h for all tested initiators. We found that Al(OTf)3 did not induce the target transformation (Table 1, entry 1). Conversely, in the presence of Fe(OTf)3, Sc(OTf)3, or Zn(OTf)2, cyclopropane 1a reacted with aniline affording acyclic product 4a in reasonable to good yields (Table 1, entries 2–5). The best results were achieved using 20 mol% Ni(ClO4)2∙6H2O or Y(OTf)3; with these catalysts, compound 4a was obtained in more than a 90% yield (Table 1, entries 6, 9). The decrease in nickel perchlorate loading led to a decrease in the product yield (Table 1, entries 7–9). The yield also decreased with increasing reaction time or when the reaction was carried out with heating; in both cases, the formation of byproducts was detected. When Brønsted acid, TfOH, was used, no reaction occurred at all presumably due to its neutralization with an excess of amine (Table 1, entry 10). With all the studied Lewis acids, only the acyclic product 4a was formed; its cyclization to pyrrolidin-2-one did not occur at room temperature.

Table 1.
Optimization of reaction conditions for the model cyclopropane 1a opening with aniline 1.
Then, the lactamization of γ-aminoester 4a was investigated. We found that the cyclization of compound 4a proceeded under the reflux of its toluene solution with acetic acid. Moreover, we showed that the crude reaction mixture obtained by a nickel perchlorate-induced reaction of cyclopropane 1a with aniline, when refluxing with 2 equiv. acetic acid in toluene efficiently produced the corresponding pyrrolidin-2-one in a one-vessel operation. This compound was obtained as a mixture of two diastereomers due to the presence of an ester group at the C(3) atom of the pyrrolidone ring. To obtain the target bioactive 1,5-diarylpyrrolidin-2-ones, this group must be removed by one of the known dealkoxycarbonylation methods. To further simplify the synthetic sequence and increase the practicality of this strategy, we realized this transformation in one pot using alkaline saponification of the ester group followed by thermolysis (Scheme 2). As a result, pyrrolidone 2a was synthesized by a four-step procedure, requiring chromatographic purification only at the last stage, with an overall yield of 70%.
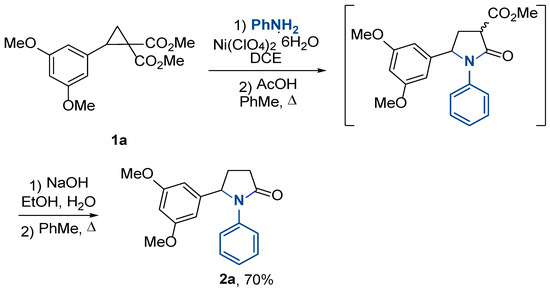
Scheme 2.
One-pot synthesis of pyrrolidone 2a.
With the optimized conditions in hand, we investigated the reaction scope using diversly substituted DA cyclopropanes and a range of anilines (Scheme 3). We found that this one pot transformation was efficient for a series of DA cyclopropanes where the electron-rich het(aryl) group or styryl group was the donor substituent. A broad variety of substituents on the aromatic moiety of both DA cyclopropanes and anilines, such as halogen, alkyl, and alkoxy, were well-tolerated in these transformations. The yields of the obtained pyrrolidones 2 varied considerably from moderate to good; however, it should be taken into account that these yields were given for four-step procedures. This is also reflected in the complex dependence of the obtained yields on the structure of the starting compounds. For example, DA cyclopropanes bearing electron-abundant aromatic substituents are typically more reactive than DA cyclopropanes with less electron-rich donors; i.e., the conversion time is shorter. However, their side reactions also proceed faster, and that can provide lower yields of the target products. For multistage processes, the overall effect of the substituent on the reaction yields is even more complex and cannot be followed by any simple model.
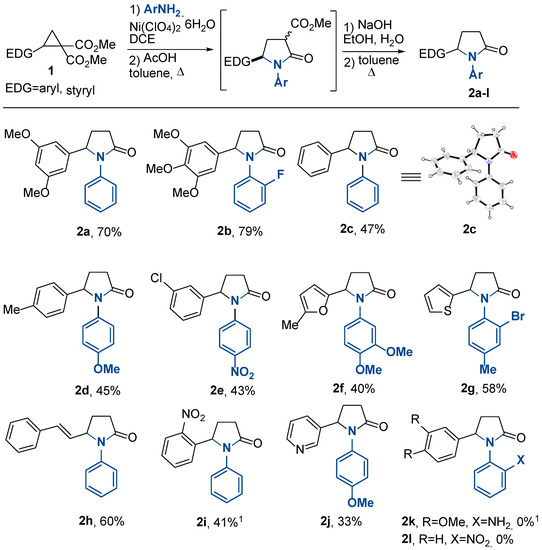
Scheme 3.
Synthesis of N-aryl and N-benzyl-substituted γ-lactams 2, 3. 1 Y(OTf)3 was used.
For example, the moderate yield of pyrrolidone 2f obtained from highly reactive furyl-substituted DA cyclopropane presumably resulted from the well-known tendency of the furan ring to undergo various acid-induced transformations [71,72]. In contrast, compound 2b was formed in a 79% yield. Other reactive DA cyclopropanes, thienyl- and styryl-derived, produced the corresponding pyrrolidones 2g,h in about a 60% yield. Less reactive 2-phenyl- and 2-(p-tolyl)cyclopropane-1,1-diesters produced the corresponding pyrrolidones 2c,d in 47 and 45% yields. The structure of the compound 2c was unambiguously proven by single-crystal X-ray data [73]. Cyclopropane-1,1-diesters containing the 2-nitrophenyl or 3-pyridyl groups at the C(2) atom of the small ring afforded the expected pyrrolidones 2i,j in low yields. However, the yield of 2i was improved by replacing the nickel perchlorate with 20 mol% Y(OTf)3. Anilines containing both electron-withdrawing and electron-donating substituents, including fluorine or bromine in the ortho position, reacted well. The exceptions were 4-nitroaniline, for which the first step occurred only when the reaction mixture was refluxed, and 2-nitroaniline and 1,2-phenylenediamine, which did not afford the desired products 2k,l at all. With these anilines, the process was stopped after the formation of the open-chain products 4b,c (see below); cyclization products were not detected in the reaction mixtures even in trace amounts.
To demonstrate the efficiency of this one pot process, we scaled up the synthesis of compound 2b using 1.00 g (3.08 mmol) of 3,4,5-trimethoxyphenyl-substituted cyclopropane 1b and 472 mg (3.08 mmol) of 2-fluoroaniline. With this loading, the yield of compound 2b was 841 mg (79%).
It is worth noting that, despite the potential ability of anilines to serve as ambident nucleophiles, in the studied reactions, they attacked the three-membered ring exclusively with the nitrogen atom, providing no isomeric products via the Friedel–Crafts alkylation of the electron-rich aromatic ring.
Benzylamines are known to be more nucleophilic than the corresponding anilines. The increased reactivity of benzylamines allowed us to synthesize pyrrolidones 3 in good yields by their direct reaction with DA cyclopropanes 1 by refluxing a dichloroethane solution in the presence of Ni(ClO4)2·6H2O without an additional lactamization step. Next, we used two methods of dealkoxycarbonylation of 3-substituted pyrrolidones. The first one included alkaline hydrolysis followed by decarboxylation according to the method developed for pyrrolidones 2 (method A, Scheme 4). Alternatively, dealkoxycarbonylation using a NaCl-promoted Krapcho reaction in wet DMSO at 160 °C under microwave (MW) irradiation provided pyrrolidones 3 in reasonable yields (method B, Scheme 4). For example, benzylamine and alkoxy-substituted benzylamines produced compounds 3a–d in up to 70% yields. In the reactions of DA cyclopropane with furfurylamine and (1H-indol-3-yl)methylamine, the corresponding pyrrolidones 3e and 3f were obtained in 32% and 42% yields. Given that these yields corresponded to a four-step sequence realized as a one pot procedure, these yields can be considered reasonable.
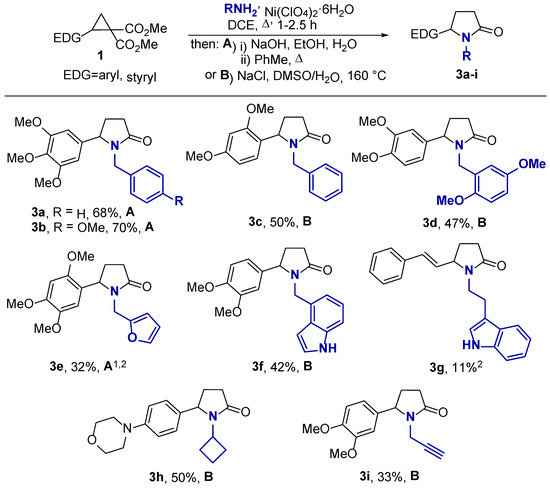
Scheme 4.
Synthesis of γ-lactams 3. 1 The reaction was performed at room temperature. 2 The reaction included a lactamization step under reflux in presence of AcOH in toluene.
Moreover, we applied the developed approach to the synthesis of 1-alkyl-5-arylpyrrolidones from DA cyclopropanes and some aliphatic amines (Scheme 4). Cyclobutylamine and propargylamine were found to participate quite efficiently in this transformation, affording the corresponding pyrrolidones 3h,i in yields close to those of 3c–f, although these substrates required a long reaction time (see Section 3). In contrast, tryptamine gave rise to the corresponding 3g product in only an 11% yield. A significant tarring of the reaction mixture was detected in this reaction. When simple primary aliphatic amines, such as methylamine or ethylamine, were reacted with cyclopropane 1a under the same reaction conditions, only unidentified byproducts were formed.
It was pointed out that above that, o-nitroaniline and 1,2-phenylenediamine did not produce the target products 2 under standard conditions. We tested their reactivity toward DA cyclopropanes 1 in the presence of the same catalysts in more detail (Scheme 5). We found that the full consumption of 2-phenylcyclopropane-1,1-dicarboxylate 1k in its reaction with o-nitroaniline catalyzed by Ni(ClO4)2·6H2O required 2 h of refluxing the solution in dichloroethane. Under these conditions, the expected acyclic product 4b was obtained in a 71% yield. The reaction of 3,4-dimethoxyphenyl-substituted cyclopropane 1j with 1,2-phenylenediamine under the same conditions produced the acyclic product 4c only in a low yield. Heating this reaction mixture at 100 °C in chlorobenzene resulted in a complex mixture of unidentified products. However, the acyclic compound 4c was obtained in a reasonable yield at room temperature using Y(OTf)3 as an initiator (Scheme 5). This product was unstable, and all attempts to cyclize it were unsuccessful.
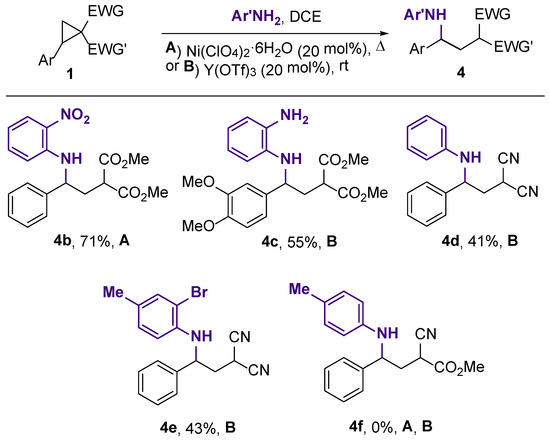
Scheme 5.
Synthesis of acyclic products 4b–f.
To test the generality of the DA cyclopropane ring opening with anilines, we tried to involve substrates with other acceptor groups in this reaction (Scheme 5). 2-Phenylcyclopropane-1,1-dicarbonitrile was found to undergo ring opening with aniline or 2-bromo-4-methylaniline under catalysis with 25 mol% Y(OTf)3 at room temperature for 4 days. The full conversion of these substrates required a significantly longer reaction time compared to the corresponding 1,1-diesters. Despite the mild reaction conditions, 4d,e were isolated only in 41% and 43% yields, presumably due to the competitive realization of the side processes resulting from the coexistence of the amino and cyano groups. 2-Phenyl-1-cyanocyclopropanecarboxylate turned out to be a less reactive substrate, which did not undergo conversion to amine 4f even at a high temperature. Other Lewis acids, such as Fe(OTf)3, Sc(OTf)3, and Ni(ClO4)2·6H2O, failed also to induce the reaction of this substrate with anilines.
Most biologically active compounds bearing chiral centers have different activities for different stereoisomers. This means that methods of their preparation in an optically pure form are highly desirable. Obviously, this also applies to bioactive-substituted pyrrolidones of types 2 and 3, which can be considered as cyclic analogs of GABA. We tested the possibility of using the developed procedure for the synthesis of chiral pyrrolidones 2 starting from optically active DA cyclopropane 1 as a substrate. We found that dimethyl (S)-2-(p-tolyl)cyclopropane-1,1-dicarboxylate (S)-1c was converted to the corresponding γ-lactam with a full inversion of the absolute configuration of the chiral center (Scheme 6). This result is consistent with previous investigations demonstrating that the nucleophilic ring opening of DA cyclopropanes under catalysis with moderately activating Lewis acids proceeds by an SN2-like mechanism, and subsequent stages (cyclization, saponification, and decarboxylation) do not affect the chiral center.
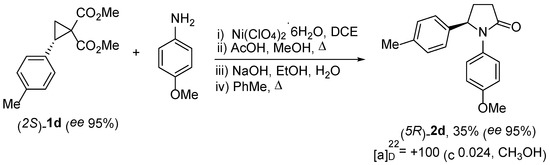
Scheme 6.
Synthesis of (R)-γ-lactam (2d).
The synthetic utility of the developed transformations can be significantly extended by diverse postmodifications of their multiple functionalities of the synthesized pyrrolidones 2,3, that allows for preparing complex azaheterocycles. In particular, the treatment of pyrrolidone 2h with PPA at 100 °C produced benz[g]indolizidine 5 in an acceptable yield (Scheme 7). Based on NOESY spectroscopy [73] and a comparison of its spectra with spectral data for the related compounds described earlier [74], the hydrogen atoms at the stereogenic centers in compound 5 have a cis arrangement.
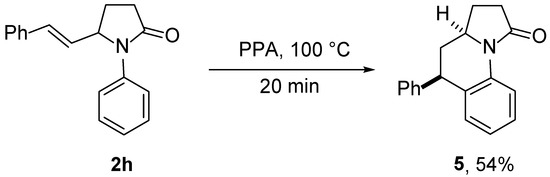
Scheme 7.
Synthesis of benz[g]indolizidine 5.
3. Experimental Section
3.1. General Information
The structures of synthesized compounds were elucidated with the aid of 1D NMR (1H, 13C) and 2D NMR (NOESY, HSQC and HMBC 1H-13C) spectroscopy. NMR spectra were acquired on Avance 600 and Avance 500 (Bruker, Billerica, MA, USA) and 400-MR (Agilent, Santa Clara, CA, USA) spectrometers at room temperature; the chemical shifts δ were measured in ppm with respect to solvent (1H: CDCl3, δ = 7.27 ppm; CD3OD, δ = 3.35 ppm; 13C: CDCl3, δ = 77.0 ppm; CD3OD: 13C: δ = 49.9 ppm). Splitting patterns were designated as s, singlet; d, doublet; m, multiplet; dd, double doublet; and br, broad. Coupling constants (J) were in Hertz. Infrared spectra were recorded on an FTIR spectrometer ALPHA II (Bruker, Billerica, MA, USA) in KBr for solid substances and as thin film for oils. High resolution and accurate mass measurements were carried out using a micrOTOF-QTM ESI-TOF (Electrospray Ionization/Time of Flight, Bruker, Billerica, MA, USA). Elemental analyses were performed with an EA-1108 CHNS elemental analyzer instrument (Fisons, Ipswich, UK). Melting points (mp) were measured using a Stuart® SMP3 melting point apparatus (Cole-Parmer, Stone, Staffordshire, UK). Microwave reactions were performed in a Monowave 200—Anton Paar microwave reactor in sealed reaction vessels. The temperature was monitored with installed IR detector. X-Ray analysis was performed on STOE STADI VARI PILATUS-100K diffractometer (Stoe & Sie, Darmstadt, Germany). Analytical thin-layer chromatography (TLC) was carried out with silica gel plates (silica gel 60, F254, supported on aluminum); visualization was performed using a UV lamp (365 nm). Column chromatography was performed on silica gel 60 (230–400 mesh, Merck, Darmstadt, Germany). Enantiomeric purity of the optically active compounds was determined by chiral HPLC with a Hitachi LaChrome Elite-2000 chromatograph (Hitachi Hugh-Tech Corp, Toranomonon Minato-Ku, Japan) using a Daicel (Daicel Corp, Osaka, Japan) Chiralcel OD-H column (0.46 × 25 cm) at room temperature. The column was eluted with heptane/i-PrOH = 70:30 at a flow rate of 1 mL/min, and peak detection was accomplished using a UV detector at 219 nm. Optical rotation was measured on a Krüss P8000 polarimeter (A. Krüss Optronic GmbH, Hamburg, Germany). All reactions were carried out using freshly distilled and dry solvents. Cyclopropanes 1 were prepared by Knoevenagel/Corey–Chaykovsky reaction sequences from the corresponding aldehydes [75,76]. Compounds 2c,j, 3b,c, and (2S)-1d were described previously [15,16,17,39,77,78]. Commercial reagents employed in the synthesis were analytical-grade, obtained from Sigma-Aldrich (St. Louis, MO, USA) or Alfa Aesar (Ward Hill, MA, USA). The 1H NMR and 13C NMR for synthesized compounds as well as 2D (HSQC and HMBC) NMR spectra for selected compounds are available in the Supplementary Materials.
3.2. Synthesis of Pyrrolidin-2-ones 2,3 from Anilines and Benzylamines
3.2.1. General Procedure 1
To a 0.2 M solution of aniline or benzylamine (1.0–1.2 equiv.) in DCE in the presence of molecular sieves, 4 Å Ni(ClO4)2∙6H2O or Y(OTf)3 (0.2 equiv.) was added under Ar atmosphere; then, cyclopropane 1 (1–4 mmol, 1.0 equiv.) was added. The resulting mixture was stirred at room temperature for 1–3 h, diluted with dichloromethane (DCM), and filtered through a short pad of silica gel using EtOAc as the eluent. The filtrate was concentrated under vacuum; the residue was dissolved in toluene (0.13 M). Next, acetic acid (2.0 equiv.) was added, and the reaction mixture was stirred under reflux for 7 h. Then, solvent was removed under vacuum, and residue was dissolved in ethanol (0.17 M); 1M aq. solution of sodium hydroxide (2.0 equiv.) was added in one portion. The reaction mixture was stirred at room temperature for 2 h, and after that, ethanol was removed under vacuum. The residue was diluted with water, and 1M HCl was added until pH 1. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). Combined organic layers were dried with Na2SO4 and concentrated in vacuo. The residue was dissolved in toluene (0.07 M) and was refluxed for 7 h. The solvent was removed under vacuum; the pure product was isolated by silica gel column chromatography.
3.2.2. General Procedure 2
To a 2 M solution of cyclopropane 1 (1 equiv.) and benzylamine (1.2 equiv.) in DCM or DCE in the presence of molecular sieves, 4 Å Ni(ClO4)2∙6H2O (0.1 equiv.) was added under Ar atmosphere The reaction mixture was placed into oil bath, which was preheated to 45 °C, and stirred at the same temperature for 1–2.5 h, and after, it was cooled to room temperature and diluted with DCM. The resulting solution was passed through a plug of silica using 1:1 petroleum ether:EtOAc system as an eluent. Concentration under reduced pressure gave a residue, which was dissolved in DMSO/H2O mixture (3:1, 0.16 M). To this solution, NaCl (1.5 equiv.) was added; the resulting mixture was heated at 160 °C under MW irradiation for 4–6 h. Then, the reaction mixture was diluted with H2O and extracted with EtOAc three times. Combined organic layers were washed with brine, dried with Na2SO4, and concentrated in vacuo. Pure product was isolated by silica gel column chromatography.
5-(3,5-Dimethoxyphenyl)-1-phenylpyrrolidin-2-one (2a) was obtained from dimethyl 2-(3,5-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (300 mg, 1.02 mmol), aniline (0.1 mL, 1.10 mmol), Ni(ClO4)2∙6H2O (75 mg, 0.2 mmol), DCE (5.1 mL), AcOH (120 μL), toluene (8.0 mL), NaOH (84 mg, 2.1 mmol), ethanol (5.9 mL), and water (2.0 mL) according to the general procedure 1. Yield: 210 mg (69%); white solid, Rf = 0.34 (ethyl acetate:petroleum ether; 1:1). 1H NMR (CDCl3, 600 MHz): δ 7.44 (d, 3J = 7.9 Hz, 2H, Ph), 7.27–7.25 (m, 2H, Ph), 7.08–7.06 (m, 1H, Ph), 6.37 (d, 4J = 2.0 Hz, 2H, Ar), 6.33–6.32 (m, 1H, Ar), 5.17 (dd, 3J = 7.2 Hz, 3J = 4.2 Hz, 1H, CH), 3.73 (s, 6H, 2 × CH3O), 2.80–2.74 (m, 1H, CH2), 2.64–2.56 (m, 2H, CH2), 2.03–1.97 (m, 1H, CH2). 13C{1H} NMR (CDCl3, 150 MHz): δ 174.8 (CO), 161.3 (2 × C), 144.0 (C), 138.3 (C), 128.7 (2 × CH), 124.9 (CH), 122.0 (2 × CH), 103.9 (2 × CH), 99.2 (CH), 63.9 (CH), 55.3 (2 × CH3O), 31.3 (CH2), 29.3 (CH2). IR (KBr, cm−1) 2996, 2980, 2941, 2904, 2874, 2840, 1693, 1654, 1613, 1594, 1538, 1499, 1490, 1482, 1461, 1443, 1429, 1370, 1352, 1318, 1284, 1250, 1227, 1201, 1162, 1145, 1117, 1066, 1030, 1011, 978. HRMS ESI-TOF: m/z = 298.1435 [M + H]+ (298.1438 calcd. for C18H20NO3+).
5-(3,4,5-Trimethoxyphenyl)-1-(2-fluorophenyl)pyrrolidin-2-one (2b) was obtained from dimethyl 2-(3,4,5-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (1.00 g, 3.08 mmol), 2-fluoroaniline (472 mg, 3.08 mmol), Ni(ClO4)2∙6H2O (226 mg, 0.62 mmol), DCE (15.4 mL), AcOH (355 μL), toluene (26.7 mL), NaOH (247 mg, 6.18 mmol), ethanol (17.8 mL), and water (3.1 mL) according to the general procedure 1. Yield: 841 mg (79%); yellowish viscous oil, Rf = 0.42 (ethyl acetate:petroleum ether; 1:2). 1H NMR (CDCl3, 400 MHz): δ 7.19–7.10 (m, 2H, Ar), 7.04–7.00 (m, 2H, Ar), 6.43 (s, 2H, Ar), 5.14–5.11 (m, 1H, CH), 3.75 (s, 6H, 2 × CH3O), 3.74 (s, 3H, CH3O), 2.78–2.58 (m, 3H), 2.09–2.00 (m, 1H). 13C{1H} NMR (CDCl3, 100 MHz): δ 174.7 (CO), 157.2 (d, 1JCF = 250 Hz), 153.2 (2 × C), 137.1 (C), 136.3 (C), 128.4 (d, 3JCF = 7 Hz, CH), 127.9 (CH), 125.0 (d, 2JCF = 12 Hz, C), 124.1 (CH), 116.4 (d, 2JCF = 20 Hz, CH), 103.0 (2 × CH), 64.7 (CH), 60.5 (CH3O), 55.8 (2 × CH3O), 30.5 (CH2), 29.6 (CH2). IR (film, cm−1): 2840, 2827, 1703, 1593, 1504, 1237, 1131. HRMS (ESI/TOF): m/z = 346.1449 [M + H]+ (346.1449 calcd. for C19H21FNO4+).
1,5-Diphenylpyrrolidin-2-one (2c) [17] was obtained from dimethyl 2-phenylcyclopropane-1,1-dicarboxylate (100 mg, 0.4 mmol), aniline (38.6 μL, 0.4 mmol), Ni(ClO4)2∙6H2O (30.8 mg, 0.08 mmol), DCE (2 mL), AcOH (46 μL), toluene (2.7 mL), NaOH (35 mg, 0.8 mmol), ethanol (2.5 mL), and water (0.4 mL) according to the general procedure. Yield: 47 mg (47%); yellow crystals; mp = 109–112 °C (dec.); lit. 100 °C [17]; 106– 108 [16]; Rf = 0.25 (ethyl acetate:petroleum ether; 1:3). 1H NMR (CDCl3, 600 MHz): δ 7.43 (d, 3J = 8.0 Hz, 2H, Ph), 7.32–7.30 (m, 2H, Ph), 7.26–7.22 (m, 5H, Ph), 7.07 (m, 1H, Ph), 5.27–5.25 (m, 1H), 2.80–2.74 (m, 1H), 2.67–2.59 (m, 2H), 2.04–1.98 (m, 1H). 13C{1H} NMR (CDCl3, 150 MHz MHz): δ 175.0, 141.4, 138.3, 129.1 (2 × C), 128.8 (2 × C), 127.8, 126.0 (2 × C), 125.0, 122.2 (2 × C), 64.0, 31.3, 29.3. The spectral data were in accordance with the literature [17]. IR (KBr, cm−1): 3379, 3107, 3095, 3083, 3042, 3028, 2992, 2969, 2952, 2895, 1709, 1596, 1497, 1480, 1456, 1415, 1367, 1356, 1324, 1308, 1281, 1238, 1224, 1194, 1179, 1194, 1114, 1077, 1052, 1031, and 1002. HRMS (ESI/TOF): m/z = 238.1226 [M + H]+ (238.1228 calcd. for C16H16NO+).
1-(4-Methoxyphenyl)-5-(p-tolyl)pyrrolidin-2-one (2d) was obtained from dimethyl 2-(4-methylphenyl)cyclopropane-1,1-dicarboxylate (182 mg, 0.73 mmol), p-anisidine (91 mg, 0.73 mmol), Ni(ClO4)2∙6H2O (55 mg, 0.15 mmol), DCE (5.0 mL), AcOH (90 μL), toluene (4.9 mL), NaOH (55.6 mg, 1.39 mmol), ethanol (3.9 mL), and water (1.4 mL) according to the general procedure 1. Yield: 92 mg (45%); ivory solid; mp 112–114 °C; Rf = 0.47 (ethyl acetate:petroleum ether; 2:1). 1H NMR (CDCl3, 600 MHz): δ 7.28 (d, 3J = 8.9 Hz, 2H, Ar), 7.10 (s, 4H, Ar), 6.76 (d, 3J = 8.9 Hz, 2H, Ar), 5.14 (dd, 3J = 7.4 Hz, 3J = 4.6 Hz, 1H, CH), 3.63 (s, 3H, CH3O), 2.77–2.70 (m, 1H, CH2), 2.62–2.55 (m, 2H, CH2), 2.29 (s, 3H, CH3), 2.00–1.95 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 174.6 (CO), 156.8 (C), 138.4 (C), 137.4 (C), 131.2 (C), 129.5 (2 × CH), 126.0 (2 × CH), 124.2 (2 × CH), 113.9 (2 × CH), 64.1 (CH), 55.2 (CH3O), 31.1 (CH2), 29.1 (CH2), 21.0 (CH3). IR (KBr, cm−1): 2835, 1694, 1512, 1248, 1035. HRMS (ESI/TOF): m/z = 282.1492 [M + H]+ (282.1489 calcd. for C18H20NO2+). Anal. calcd. for C18H19NO2: C, 76.84; H, 6.81, N, 4.95. Found: C, 76.63; H, 6.85; N, 4.98.
(R)-1-(4-Methoxyphenyl)-5-(p-tolyl)pyrrolidin-2-one (5R)-2d) was obtained from dimethyl (S)-2-(p-tolyl)cyclopropane-1,1-dicarboxylate [79] (80 mg, 0.32 mmol, ee 95%, [α]D20 –130° (c 1.0, CHCl3)), 4-methoxyaniline (40 mg, 0.32 mmol), Ni(ClO4)2∙6H2O (24 mg, 0.066 mmol), DCE (1.6 mL), AcOH (37 μL), toluene (2.1 mL), NaOH (27 mg, 0.68 mmol), ethanol (1.9 mL), and water (0.65 mL) according to the general procedure. Yield 32 mg (35%); ivory solid; mp 112–114 °C; [α]D22 = +100 (c 0.024, CH3OH); Rf = 0.47 (ethyl acetate:petroleum ether; 2:1). Spectral data were identical to those of 2d.
5-(3-Chlorophenyl)-1-(4-nitrophenyl)pyrrolidin-2-one (2e) was obtained from dimethyl 2-(3-chlorophenyl)cyclopropane-1,1-dicarboxylate (300 mg, 1.11 mmol), 4-nitroaniline (170 mg, 1.22 mmol), and Ni(ClO4)2∙6H2O (82 mg, 0.223 mmol) in DCE (5.6 mL) according to the modified general procedure 1 (2.5 h under reflux for the first step and 12 h for the second step). Yield: 151 mg (43%); yellowish solid, mp 64–66 °C, Rf = 0.56 (ethyl acetate:petroleum ether; 1:1). 1H NMR (CDCl3, 600 MHz): δ 8.09 (d, 3J = 9.6 Hz, 2H, Ar), 7.66 (d, 3J = 9.6 Hz, 2H, Ar), 7.30–7.22 (m, 2H, Ar), 7.20 (s, 1H, Ar), 7.07 (dt, 3J = 7.2 Hz, 4J = 1.8 Hz, 1H, Ar), 5.30 (dd, 3J = 7.9 Hz, 3J = 4.2 Hz, 1H, C(5)H), 2.83–2.60 (m, 3H, C(3)H2, C(4)H2), 2.07–1.99 (m, 1H, C(4)H2). 13C NMR (CDCl3, 150 MHz): δ 175.2 (CO), 143.7 (C), 143.6 (C), 142.4 (C), 135.4 (C), 130.7 (CH), 128.5 (CH), 125.8 (CH), 124.5 (2 × CH), 123.6 (CH), 120.7 (2 × CH), 62.8 (C(5)H), 31.0 (C(3)H2), 28.8 (C(4)H2). IR (KBr, cm−1): 1708, 1592, 1510, 1496, 1335, 1323, 1296, 1286, 1219, 1194, 112, 848. HRMS ESI-TOF: m/z = 317.0687 [M + H]+ (317.0687 calcd. for C16H14N2O3+).
1-(3,4-Dimethoxyphenyl)-5-(5-methylfuran-2-yl)pyrrolidin-2-one (2f) was obtained from dimethyl 2-(5-methylfuran-2-yl)cyclopropane-1,1-dicarboxylate (300 mg, 1.26 mmol), 3,4-dimethoxyaniline (193 mg, 1.26 mmol), Ni(ClO4)2∙6H2O (92.0 mg, 0.25 mmol), DCE (5.6 mL), AcOH (144 μL), toluene (8 mL), NaOH (104 mg, 2.6 mmol), ethanol (7.3 mL), and water (2.5 mL) according to the general procedure 1 (at 0 °C for the first step). Yield: 150 mg (40%); yellowish oil, Rf = 0.39 (ethyl acetate:petroleum ether; 2:1). 1H NMR (CDCl3, 600 MHz): δ 6.87 (d, 4J = 2.0 Hz, 1H, Ar), 6.78 (d, 3J = 8.6 Hz, 1H, Ar), 6.73 (dd, 3J = 8.6 Hz, 4J = 2.0 Hz, 1H, Ar), 6.00 (d, 3J = 2.7 Hz, 1H, Fu), 5.83 (d, 3J = 2.7 Hz, 1H, Fu), 5.05–5.00 (m, 1H, CH), 3.83 (s, 3H, CH3O), 3.79 (s, 3H, CH3O), 2.87–2.81 (m, 1H, CH2), 2.62–2.56 (m, 1H, CH2), 2.53–2.46 (m, 1H, CH2), 2.33–2.28 (m, 1H, CH2), 2.25 (s, 3H, CH3). 13C NMR (CDCl3, 150 MHz): δ 174.4 (CO), 152.0 (C), 151.0 (C), 148.6 (C), 147.0 (C), 130.9 (C), 116.3 (CH), 110.8 (CH), 108.8 (CH), 108.4 (CH), 106.1 (CH), 58.7 (CH), 55.8 (CH3O), 55.6 (CH3O), 30.9 (CH2), 25.2 (CH2), 13.4 (CH3). IR (film, cm−1): 2837, 1674, 1565, 1511, 1239, 1022, 911, 538. HRMS ESI-TOF: m/z = 302.1389 [M + H]+ (302.1387 calcd. for C17H20NO4+).
1-(2-Bromo-4-methylphenyl)-5-(thiophen-2-yl)pyrrolidin-2-one (2g) was obtained from dimethyl 2-(thiophen-2-yl)cyclopropane-1,1-dicarboxylate (200 mg, 0.79 mmol), 2-bromo-4-methylaniline (147 mg, 0.78 mmol), Ni(ClO4)2∙6H2O (61.8 mg, 0.17 mmol), DCE (4.2 mL), AcOH (96 μL), toluene (5.3 mL), NaOH (63 mg, 1.57 mmol), ethanol (4.8 mL), and water (1.7 mL) according to the general procedure 1. Yield: 162 mg (58%); colorless solid; mp 127–129 °C; Rf = 0.54 (ethyl acetate). 1H NMR (CDCl3, 600 MHz): δ 7.26 (dd, 3J = 8.2 Hz, 4J = 2.0 Hz, 1H, Ar), 7.22 (d, 3J = 5.0 Hz, 1H, Ar), 7.15 (br. s, 1H, Ar), 7.05 (d, 3J = 8.2 Hz, 1H, Ar), 6.87–6.84 (m, 1H, Ar), 6.83–6.81 (m, 1H, Ar), 5.32–5.26 (m, 1H, CH), 2.87–2.81 (m, 1H, CH2), 2.77–2.71 (m, 1H, CH2), 2.70–2.64 (m, 1H, CH2), 2.41–2.34 (m, 1H, CH2), 2.11 (s, 3H, CH3). 13C NMR (CDCl3, 150 MHz): δ 173.7 (CO), 143.5 (C), 137.3 (C), 135.7 (C), 132.4 (CH), 130.9 (CH), 130.0 (C), 126.9 (CH), 126.8 (CH), 125.8 (CH), 119.1 (CH), 60.7 (CH), 30.9 (CH2), 29.6 (CH2), 18.0 (CH3). IR (KBr, cm−1): 3100, 2853, 1694, 1528, 716, 525. HRMS ESI-TOF: m/z = 336.0052 [M + H]+ (336.0052 calcd. for C15H15 Br79NOS+).
(E)-1-Phenyl-5-styrylpyrrolidin-2-one (2h) was obtained from dimethyl 2-(E)-styrylcyclopropane-1,1-dicarboxylate (300 mg, 1.15 mmol), aniline (107 mg, 1.15 mmol), Ni(ClO4)2∙6H2O (84 mg, 0.23 mmol), DCE (5.7 mL), AcOH (140 μL), toluene (8 mL), NaOH (99 mg, 2.48 mmol), ethanol (6.9 mL), and water (2.4 mL) according to the general procedure 1. Yield: 182 mg (60%); yellow solid; mp 71–73 °C; Rf = 0.38 (ethyl acetate:petroleum ether; 1:1). 1H NMR (CDCl3, 600 MHz): δ 7.50 (d, 3J = 8.6 Hz, 2H, Ar), 7.36–7.23 (m, 7H, Ar), 7.16–7.13 (m, 1H, Ar), 6.52 (d, 3J = 15.8 Hz, 1H, CH=), 6.14 (dd, 3J = 15.8 Hz, 3J = 7.4 Hz, 1H, CH=), 4.86–4.82 (m, 1H), 2.76–2.70 (m, 1H), 2.62–2.56 (m, 1H), 2.50–2.44 (m, 1H), 2.04–1.99 (m, 1H). 13C NMR (CDCl3, 150 MHz): δ 174.5, 138.2, 136.1, 132.2, 129.0, 128.9 (2 × C), 128.7 (2 × C), 128.1, 126.6 (2 × C), 125.4, 123.0 (2 × C), 62.4, 31.3, 26.7. IR (KBr, cm−1): 3059, 3026, 2978, 2943, 2873, 1699, 1695, 1598, 1529, 1497, 1456, 1449, 1382, 1294, 1219, 1210, 1154, 1115, 1072, 1042, 1029, 968. HRMS (ESI/TOF): m/z = 286.1202 [M+Na]+ (286.1202 calcd. for C18H17NNaO+).
5-(2-Nitrophenyl)-1-phenylpyrrolidin-2-one (2i) was obtained from dimethyl 2-(2-nitrophenyl)cyclopropane-1,1-dicarboxylate (300 mg, 1.07 mmol), aniline (120 mg, 1.29 mmol), Y(OTf)3 (118 mg, 0.22 mmol), DCE (5.4 mL), AcOH (123 μL), toluene (8 mL), NaOH (87 mg, 2.18 mmol), ethanol (6.2 mL), and water (2 mL) according to the general procedure 1. Yield: 124 mg (41%); thick dark brown oil, Rf = 0.27 (ethyl acetate:petroleum ether; 1:3). 1H NMR (CDCl3, 600 MHz): δ 8.09–8.07 (m, 1H, Ar), 7.56–7.54 (m, 1H, Ar), 7.44–7.42 (m, 3H, Ar), 7.40–7.38 (m, 1H, Ar), 7.28–7.26 (m, 2H, Ar), 7.10–7.07 (m, 1H, Ar), 5.99 (dd, 3J = 8.7 Hz, 3J = 3.4 Hz, 1H, CH), 2.92–2.85 (m, 1H, CH2), 2.75–2.64 (m, 2H, CH2), 2.07–2.02 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 175.1 (CO), 137.0 (C), 134.2 (CH), 129.1 (2 × CH), 128.8 (CH), 128.5 (C), 127.3 (CH), 126.8 (C), 125.8 (CH), 125.3 (CH), 121.5 (2 × CH), 59.7 (CH), 30.8 (CH2), 28.1 (CH2). IR (film, cm−1): 3308, 3199, 3136, 3103, 3066, 3043, 2953, 2924, 2853, 1699, 1598, 1579, 1526, 1498, 1457, 1444, 1382, 1348, 1295, 1251, 1225, 1162, 1118, 1073, 1040. HRMS ESI-TOF: m/z = 283.1077 [M + H]+ (283.1077 calcd. for C16H15N2O3+).
1-(4-Methoxyphenyl)-5-(pyridin-3-yl)pyrrolidin-2-one (2j) [15] was obtained from dimethyl 2-(pyridin-3-yl)cyclopropane-1,1-dicarboxylate (250 mg, 1.06 mmol), 4-methoxyaniline (131 mg, 1.06 mmol), Ni(ClO4)2∙6H2O (78 mg, 0.21 mmol), DCE (5.3 mL), AcOH (122 μL), toluene (6.7 mL), NaOH (77 mg, 1.9 mmol), ethanol (5.4 mL), and water (1.9 mL) according to the general procedure 1 but the product of hydrolysis was refluxed in water (14 mL, 0.065 M) for 14 h. Yield: 95 mg (33%); brown solid; mp 72–73 °C; lit. 73–75 °C [17]; Rf = 0.14 (ethyl acetate). 1H NMR (CDCl3, 600 MHz): δ 8.51–8.50 (m, 1H, Ar), 8.48–8.47 (m, 1H, Ar), 7.52–7.50 (m, 1H, Ar), 7.23–7.20 (m, 3H, Ar), 6.77 (d, 3J = 9.1 Hz, 2H, Ar), 5.22–5.20 (m, 1H, CH), 3.70 (s, 3H, CH3O), 2.50–2.64 (m, 3H, CH2), 2.03–1.97 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 174.6 (CO), 157.2 (C), 149.4 (CH), 148.4 (CH), 136.9 (C), 133.8 (CH), 130.4 (C), 124.6 (2 × CH), 123.9 (CH), 114.2 (2 × CH), 62.0 (CH), 55.4 (CH3O), 31.0 (CH2), 28.9 (CH2). IR (KBr, cm−1): 3309, 3128, 3041, 3000, 2953, 2934, 2911, 2834, 1672, 1607, 1547, 1509, 1463, 1441, 1431, 1409, 1385, 1296, 1243, 1178, 1107, 1028. HRMS ESI-TOF: m/z = 269.1285 [M + H]+ (269.1285 calcd. for C16H17N2O2+). Spectral data are consistent with the reported ones [15].
1-Benzyl-5-(3,4,5-trimethoxyphenyl)pyrrolidin-2-one (3a) To a solution of dimethyl 2-(3,4,5-trimethoxyphenyl)cyclopropane-1,1-dicarboxylate (550 mg, 1.70 mmol) in DCE (8.5 mL) in the presence of molecular sieves, 4 Å Ni(ClO4)2∙6H2O (63.4 mg, 0.17 mmol) was added; then, benzylamine (222 μL, 2.03 mmol) was added. The resulting mixture was refluxed for 1.5 h, diluted with DCM, and filtered through a small pad of silica gel using EtOAc as the eluent. Then, solvent was removed under vacuum; residue was dissolved in ethanol (9.8 mL), and aq. solution of NaOH (136 mg, 3.40 mmol; 3.5 mL) was added in one portion. The reaction mixture was stirred at room temperature for 2 h, and after that, ethanol was removed under vacuum. The residue was diluted with water, and 1 M HCl was added until pH 1. Then, mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried with Na2SO4 and concentrated in vacuo. The residue was dissolved in toluene (26.1 mL) and refluxed for 7 h. The solvent was removed under vacuum; pure product was isolated by column chromatography on silica gel. Yield: 393 mg (68%); yellowish solid; mp 99–102 °C; Rf = 0.69 (ethyl acetate). 1H NMR (CDCl3, 600 MHz): δ 7.27–7.21 (m, 3H, Ar), 7.08–7.07 (m, 2H, Ar), 6.29 (s, 2H, Ar), 5.01 (d, 2J = 14.5 Hz, 1H, CH2), 4.32 (dd, 3J = 7.9 Hz, 3J = 6.1 Hz, 1H, CH), 3.84 (s, 3H, CH3O), 3.79 (s, 6H, 2 × CH3O), 3.62 (d, 2J = 14.5 Hz, 1H, CH2), 2.65–2.60 (m, 1H, CH2), 2.51–2.45 (m, 1H, CH2), 2.41–2.35 (m, 1H, CH2), 1.91–1.85 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 175.4 (CO), 153.7 (2 × C), 137.6 (C), 136.5 (C), 136.4 (C), 128.6 (2 × CH), 128.5 (2 × CH), 127.5 (CH), 103.5 (2 × CH), 61.9 (CH), 60.9 (CH3O), 56.2 (2 × CH3O), 44.6 (CH2), 30.4 (CH2), 28.3 (CH2). IR (KBr, cm−1): 2926, 1672, 1594, 1247, 1117, 1008, 700. HRMS ESI-TOF: m/z = 342.1700 [M + H]+ (342.1700 calcd. for C20H24NO4+).
1-(4-Methoxybenzyl)-5-(3,4,5-trimethoxyphenyl)pyrrolidin-2-one (3b) [39] was obtained from dimethyl 2-(3,4,5-trimethoxyphenyl)cyclopropane-1,1-dicarboxylate (550 mg, 1.70 mmol), (4-methoxyphenyl)methanamine (285 μL, 2.18 mmol), Ni(ClO4)2∙6H2O (63.0 mg, 0.17 mmol), DCE (8.5 mL), NaOH (128 mg, 3.20 mmol), ethanol (9.0 mL), and water (3.2 mL) according to the general procedure 2b. Yield: 440 mg (70%); light yellow oil; Rf = 0.66 (ethyl acetate).1H NMR (CDCl3, 600 MHz): δ 6.96 (d, 3J = 8.6 Hz, 2H, Ar), 6.75 (d, 3J = 8.6 Hz, 2H, Ar), 6.27 (s, 2H, Ar), 4.92 (d, 2J = 14.5 Hz, 1H, CH2), 4.27 (dd, 3J = 8.2 Hz, 3J = 6.4 Hz, 1H, CH), 3.81 (s, 3H, CH3O), 3.77 (s, 6H, 2 × CH3O), 3.72 (s, 3H, CH3O), 3.52 (d, 2J = 14.5 Hz, 1H, CH2), 2.60–2.55 (m, 1H, CH2), 2.45–2.40 (m, 1H, CH2), 2.36–2.30 (m, 1H, CH2), 1.86–1.80 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 175.3 (CO), 158.9 (C), 153.6 (2 × C), 137.5 (C), 136.4 (C), 129.8 (2 × CH), 128.4 (C), 113.7 (2 × CH), 103.5 (2 × CH), 61.8 (CH), 60.8 (CH3O), 56.1 (2 × CH3O), 55.2 (CH3O), 43.9 (CH2), 30.4 (CH2), 28.2 (CH2). Spectral data are consistent with the reported ones [39].
1-Benzyl-5-(2,4-dimethoxyphenyl)pyrrolidin-2-one (3c) was obtained from dimethyl 2-(2,4-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (125 mg, 0.425 mmol), benzylamine (0.056 mL, 0.51 mmol), Ni(ClO4)2∙6H2O (16 mg, 0.044 mmol), and NaCl (37 mg, 0.64 mmol) in DCM (0.5 mL) according to the general procedure 2 (1 h for the first step and 4 h for the second step). Yield: 66 mg (50%); yellowish oil, Rf = 0.47 (ethyl acetate:petroleum ether; 2:1). 1H NMR (CDCl3, 400 MHz): δ 7.22–7.29 (m, 3H, Ar), 7.08–7.13 (m, 2H, Ar), 6.91–6.95 (m, 1H, Ar), 6.43–6.49 (m, 2H, Ar), 5.05 (d, 2J = 14.7 Hz, 1H, CH2), 4.73 (dd, 3J = 8.5 Hz, 3J = 4.2 Hz, 1H, CH), 3.82 (s, 3H, CH3O), 3.71 (s, 3H, CH3O), 3.54 (d, 2J = 14.7 Hz, 1H, CH2), 2.54–2.64 (m, 1H, CH2), 2.40–2.49 (m, 1H, CH2), 2.27–2.38 (m, 1H, CH2), 1.83–1.94 (m, 1H, CH2). 13C NMR (CDCl3, 100 MHz): δ 175.5 (CO), 160.5 (C), 158.2 (C), 136.7 (C), 128.3 (4 × CH), 128.0 (CH), 127.2 (CH), 120.6 (C), 104.0 (CH), 98.8 (CH), 56.1 (CH), 55.32 (CH3O), 55.21 (CH3O), 44.2 (CH2Ph), 30.2 (C(4)H2), 26.3 (C(3)H2). IR (film, cm−1): 3086, 3064, 3030, 3001, 2940, 2837, 1739, 1685, 1612, 1588, 1507, 1456, 1441, 1416, 1358, 1292, 1277, 1262, 1209, 1158, 1119, 1033. HRMS ESI-TOF: m/z = 312.1599 [M + H]+ (312.1594 calcd. for C19H22NO3+). Spectral data are consistent with the reported ones [77].
1-(2,5-Dimethoxybenzyl)-5-(3,4-dimethoxyphenyl)pyrrolidin-2-on (3d) was obtained from dimethyl 2-(3,4-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (300 mg, 1.02 mmol), 2,5-dimethoxybenzylamine (0.184 mL, 1.22 mmol), Ni(ClO4)2∙6H2O (37 mg, 0.101 mmol), and NaCl (85 mg, 1.5 mmol) in DCM (0.5 mL) according to the general procedure 2 (1 h for the first step and 6 h for the second step). Yield: 178 mg (47%); yellow oil, Rf = 0.49 (ethyl acetate:petroleum ether; 3:1). 1H NMR (CDCl3, 500 MHz): δ 6.81 (d, 3J = 8.2 Hz, 1H, Ar), 6.73 (dd, 3J = 8.8 Hz, 4J = 2.9 Hz, 1H, Ar), 6.70 (d, 3J = 8.8 Hz, 1H, Ar), 6.66 (dd, 3J = 8.2 Hz, 4J = 2.0 Hz, 1H, Ar), 6.63 (d, 4J = 2.9 Hz, 1H, Ar), 6.57 (d, 4J = 2.0 Hz, 1H, Ar), 4.85 (d, 2J = 14.8 Hz, 1H, CH2), 4.42 (dd, 3J = 8.1 Hz, 3J = 5.8 Hz, 1H, CH), 3.86 (s, 3H, CH3O), 3.81 (s, 3H, CH3O), 3.78 (d, 2J = 14.8 Hz, 1H, CH2), 3.70 (s, 3H, CH3O), 3.61 (s, 3H, CH3O), 2.57–2.64 (m, 1H, CH2), 2.42–2.50 (m, 1H, CH2), 2.35–2.45 (m, 1H, CH2), 1.82–1.90 (m, 1H, CH2). 13C NMR (CDCl3, 125 MHz): δ 175.3 (CO), 153.3 (C), 151.6 (C), 149.2 (C), 148.5 (C), 133.7 (C), 125.5 (C), 118.8 (CH), 115.8 (CH), 112.8 (CH), 111.1 (CH), 111.0 (CH), 109.3 (CH), 61.7 (CH), 55.8 (CH3O), 55.7 (CH3O), 55.59 (CH3O), 55.53 (CH3O), 39.4 (CH2Ar), 30.1 (C(4)H2), 28.3 (C(3)H2). IR (film, cm−1): 3074, 2998, 2940, 2912, 2835, 2251, 2063, 1693, 1608, 1593, 1518, 1500, 1465, 1412, 1357, 1315, 1303, 1278, 1260, 1218, 1154, 1138, 1120, 1046, 1026. HRMS ESI-TOF: m/z = 372.1817 [M + H]+ (372.1805 calcd. for C21H26NO5+).
1-(Furan-2-ylmethyl)-5-(2,4,5-trimethoxyphenyl)pyrrolidin-2-one (3e) was obtained from dimethyl 2-(2,4,5-trimethoxyphenyl)cyclopropane-1,1-dicarboxylate (400 mg, 1.23 mmol), furfurylamine (114 μL, 1.29 mmol), Ni(ClO4)2∙6H2O (90.2 mg, 0.25 mmol), DCE (6.2 mL), AcOH (140 μL), toluene (10.7 mL), NaOH (41.3 mg, 1.03 mmol), ethanol (3 mL), and water (1 mL) according to the general procedure 1. Yield: 132 mg (32%); light yellow thick oil, Rf = 0.18 (ethyl acetate:petroleum ether; 1:1). 1H NMR (CDCl3, 600 MHz) δ 7.28–7.26 (m, 1H, Fu), 6.53 (s, 1H, Ar), 6.51 (s, 1H, Ar), 6.22 (dd, 3J = 3.0 Hz, 3J = 1.7 Hz, 1H, Fu), 6.01 (br. d, 3J = 3.0 Hz, 1H, Fu), 4.86–4.83 (m, 2H, CH + CH2), 3.87 (s, 3H, CH3O), 3.77 (s, 3H, CH3O), 3.74 (s, 3H, CH3O), 3.69 (d, 2J = 15.4 Hz, 1H, CH2), 2.57–2.52 (m, 1H, CH2), 2.44–2.32 (m, 2H, CH2), 1.87–1.81 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 175.5 (CO), 151.7 (C), 150.3 (C), 149.4 (C), 143.3 (C), 142.1 (CH), 119.9 (C), 111.2 (CH), 110.2 (CH), 108.3 (CH), 98.0 (CH), 56.7 (CH3O), 56.4 (CH + CH3O), 56.2 (CH3O), 37.4 (CH2), 30.3 (CH2), 26.7 (CH2). IR (film, cm−1): 2992, 2935, 2836, 1687, 1684, 1611, 1513, 1463, 1456, 1440, 1411, 1399, 1348, 1317, 1276, 1208, 1163, 1116, 1079, 1032, 1012. HRMS (ESI/TOF): m/z = 331.1420 [M]+ (331.1414 calcd. for C18H21NO5+).
1-[(1H-Indol-4-yl)methyl]-5-(3,4-dimethoxyphenyl)pyrrolidin-2-one (3f) was obtained from dimethyl 2-(2,4-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (100 mg, 0.34 mmol), 4-aminomethylindole (60 mg, 0.41 mmol), Ni(ClO4)2∙6H2O (13 mg, 0.036 mmol) in DCM (0.2 mL), and NaCl (30 mg, 0.51 mmol) according to the general procedure 2 (1 h for the first step and 4 h for the second step). Yield: 50 mg (42%); yellow oil, Rf = 0.35 (ethyl acetate:petroleum ether; 4:1). 1H NMR (CDCl3, 500 MHz): δ 8.56 (br. s, 1H, NH), 7.34–7.37 (m, 1H, Ar), 7.18–7.21 (m, 1H, Ar), 7.05–7.09 (m, 1H, Ar), 6.86–6.90 (m, 1H, Ar), 6.68–6.72 (m, 2H, Ar), 6.56–6.59 (m, 2H, Ar), 5.45 (d, 2J = 14.3 Hz, 1H, CH2), 4.25 (dd, 3J = 8.0 Hz, 3J = 5.8 Hz, 1H, CH), 3.93 (s, 3H, CH3O), 3.83 (s, 3H, CH3O), 3.81 (d, 2J = 14.3 Hz, 1H, CH2), 2.68 (ddd, 2J = 17.2 Hz, 3J = 9.9 Hz, 3J = 5.8 Hz, 1H, CH2), 2.51 (ddd, 2J = 17.2 Hz, 3J = 9.9, 3J = 7.2 Hz, 1H, CH2), 2.26–2.35 (m, 1H, CH2), 1.82–1.90 (m, 1H, CH2). 13C NMR (CDCl3, 125 MHz): δ 175.0 (CO), 149.3 (C), 148.6 (C), 135.9 (C), 135.6 (C), 127.8 (C), 127.0 (C), 124.3 (CH), 121.4 (CH), 120.7 (CH), 119.1 (CH), 111.2 (CH), 110.8 (CH), 109.7 (CH), 101.3 (CH), 61.1 (CH), 55.91 (CH3O), 55.85 (CH3O), 42.8 (CH2Ar), 30.5 (C(4)H2), 28.1 (C(3)H2). IR (film, cm−1): 3465, 3396, 3371, 3308, 3293, 3272, 3118, 2998, 2932, 2875, 2836, 2384, 1730, 1665, 1611, 1595, 1516, 1462, 1443, 1415, 1347, 1317, 1305, 1259, 1236, 1173, 1151, 1137, 1086, 1025. HRMS ESI-TOF: m/z = 351.1696 [M + H]+ (351.1703 calcd. for C21H23N2O3+).
(E)-1-[2-(1H-Indol-3-yl)ethyl]-5-styrylpyrrolidin-2-one (3g) was obtained from dimethyl (E)-2-styrylcyclopropane-1,1-dicarboxylate (200 mg, 0.77 mmol), tryptamine (123 mg, 0.77 mmol), Ni(ClO4)2∙6H2O (56 mg, 0.155 mmol), DCE (3.8 mL), AcOH (90 μL), toluene (5.3 mL), NaOH (63 mg, 1.58 mmol), ethanol (4.4 mL), and water (0.8 mL) according to the general procedure 1. Yield: 28 mg (11%); brownish thick oil, Rf = 0.31 (ethyl acetate). 1H NMR (CDCl3, 600 MHz): 1H NMR (CDCl3, 600 MHz): δ 8.23 (br. s, 1H, NH), 7.54 (d, 3J = 7.9 Hz, 1H, Ar), 7.37–7.32 (m, 5H, Ar), 7.30–7.26 (m, 1H, Ar), 7.19–7.16 (m, 1H, Ar), 7.03–7.00 (m, 2H, Ar), 6.34 (d, 3J = 15.6 Hz, 1H, CH=), 5.91 (dd, 3J = 15.6 Hz, 3J = 8.6 Hz, 1H, CH=), 4.01–3.97 (m, 1H, CH), 3.89–3.85 (m, 1H, CH2), 3.36–3.31 (m, 1H, CH2), 3.11–3.06 (m, 1H, CH2), 2.99–2.94 (m, 1H, CH2), 2.52–2.46 (m, 1H, CH2), 2.42–2.36 (m, 1H, CH2), 2.21–2.15 (m, 1H, CH2), 1.80–1.74 (m, 1H, CH2). 13C NMR (CDCl3, 150 MHz): δ 175.1 (CO), 136.3 (C), 135.9 (C), 133.3 (CH), 128.9 (CH), 128.7 (2 × CH), 128.2 (CH), 127.4 (C), 126.6 (2 × CH), 122.1 (CH), 122.0 (CH), 119.3 (CH), 118.8 (CH), 113.0 (C), 111.2 (CH), 61.8 (CH), 41.4 (CH2), 30.4 (CH2) 25.9 (CH2), 23.5 (CH2). IR (film, cm−1): 3210, 3180, 3110, 3079, 3057, 3028, 2962, 2929, 2878, 2853, 2723, 2601, 2545, 2385, 2350, 2253, 1956, 1883, 1804, 1666, 1548, 1493, 1452, 1417, 1368, 1340, 1312, 1274, 1264, 1230, 1181, 1160, 1143, 1126, 1102, 1071, 1052, 1029, 1008, 979, 905. HRMS ESI-TOF: m/z = 331.1805 [M + H]+ (331.1805 calcd. for C22H23N2O+).
1-Cyclobutyl-5-(4-morpholinophenyl)pyrrolidin-2-one (3h) was obtained from dimethyl 2-(4-morpholinophenyl)cyclopropane-1,1-dicarboxylate (300 mg, 0.94 mmol), cyclobutylamine (84 μL, 0.99 mmol) in DCE (0.5 mL), and NaCl (75 mg, 1.28 mmol) according to the general procedure 2 (2.5 h for the first step and 4 h for the second step). Yield: 142 mg (50%); yellow oil, Rf = 0.71 (chloroform:methanol, 10:1). 1H NMR (CDCl3, 500 MHz): δ 7.09 (d, 3J = 8.6 Hz, 2H, Ar), 6.96–6.88 (m, 2H, Ar), 4.73 (dd, 3J = 8.3 Hz, 3J = 3.4 Hz, 1H, CH), 4.32–4.23 (m, 1H, CH), 3.90–3.83 (m, 4H, 2 × CH2), 3.20–3.13 (m, 4H, 2 × CH2), 2.58–2.59 (m, 1H, CH2), 2.46–2.39 (m, 1H, CH2), 2.38–2.33 (m, 1H, CH2), 2.32–2.25 (m, 1H, CH2), 2.15–2.07 (m, 1H, CH2), 1.97–1.88 (m, 1H, CH2), 1.82–1.74 (m, 2H, CH2), 1.56–1.45 (m, 2H, CH2). 13C NMR (CDCl3, 125 MHz): δ 175.3 (CO), 126.6 (2 × CH), 115.8 (2 × CH), 66.6 (2 × CH2), 60.49 (CHAr), 49.2 (2 × CH2), 47.7 (CHN), 30.2 (CH2), 29.0 (CH2), 28.18 (CH2), 28.14 (CH2), 15.4 (CH2), quaternary aromatic carbons not observed. HRMS ESI-TOF: m/z = 301.1904 [M + H]+ (301.1911 calcd. for C18H25N2O2+).
5-(3,4-Dimethoxyphenyl)-1-(prop-2-yn-1-yl)pyrrolidin-2-one (3i) was obtained from dimethyl 2-(3,4-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (300 mg, 1.02 mmol), propargylamine (78 μL, 1.22 mmol) in DCE (0.5 mL), and NaCl (70 mg, 1.2 mmol) according to the general procedure 2 (2.5 h for the first step and 4 h for the second step). Yield: 86 mg (33%); yellow oil, Rf = 0.43 (ethyl acetate). 1H NMR (CDCl3, 400 MHz): δ 6.83 (d, 3J = 8.3 Hz, 1H, Ar), 6.77 (dd, 3J = 8.3 Hz, 4J = 1.9 Hz, 1H, Ar), 6.69 (d, 4J = 1.9 Hz, 1H, Ar), 4.73–4.69 (m, 1H, CH), 4.52 (dd, 2J = 17.4 Hz, 4J = 2.5 Hz, 1H, CH2), 3.85 (s, 3H, CH3O), 3.84 (s, 3H, CH3O), 3.22 (dd, 2J = 17.4 Hz, 4J = 1.7 Hz, 1H, CH2), 2.61–2.52 (m, 1H, CH2), 2.50–2.42 (m, 2H, CH2), 2.13 (dd, 4J = 2.5 Hz, 4J = 1.7 Hz, 1H, CH), 1.95–1.86 (m, 1H, CH2). 13C NMR (CDCl3, 100 MHz): δ 174.8 (CO), 149.4 (C), 148.9 (C), 132.5 (C), 119.3 (CH), 111.2 (CH), 109.3 (CH), 77.6 (C), 71.7 (CH), 61.2 (CH), 55.84 (CH3O), 55.80 (CH3O), 30.3 (CH2), 30.0 (CH2), 28.0 (CH2). IR (film, cm−1): 3250, 3073, 2998, 2957, 2939, 2837, 2585, 2468, 2280, 2117, 2030, 2846, 1693, 1607, 1594, 1519, 1465, 1465, 1422, 1371, 1347, 1306, 1237, 1179, 1139, 1066, 1026. HRMS ESI-TOF: m/z = 260.1292 [M + H]+ (260.1281 calcd. for C15H18NO3+).
Dimethyl 2-[2-(3,5-dimethoxyphenyl)-2-(phenylamino)ethyl]malonate (4a). To a solution of dimethyl 2-(3,5-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (300 mg, 1.02 mmol) in DCE (5.1 mL) in the presence of molecular sieves, 4 Å Ni(ClO4)2∙6H2O (75 mg, 0.206 mmol) was added under Ar atmosphere; then, aniline (0.1 mL, 1.10 mmol) was added. The resulting mixture was stirred at room temperature for 1 h, diluted with DCM, and filtered. The filtrate was concentrated under vacuum; pure product was isolated by column chromatography on silica gel. Yield: 363 mg (92%); yellow viscous oil; Rf = 0.50 (ethyl acetate:petroleum ether; 1:3). 1H NMR (CDCl3, 600 MHz): δ 7.13–7.10 (m, 2H, Ar), 6.69–6.67 (m, 1H, Ar), 6.56 (d, 3J = 7.8 Hz, 2H, Ar), 6.52 (d, 4J = 2.2 Hz, 2H, Ar), 6.36–6.35 (m, 1H, Ar), 4.40–4.37 (m, 1H, CH), 4.23 (br.s, 1H, NH), 3.77 (s, 6H, 2 × CH3O), 3.76 (s, 3H, CH3O), 3.73 (s, 3H, CH3O), 3.60–3.58 (m, 1H, CH), 2.43–2.41 (m, 2H, CH2). 13C NMR (CDCl3, 150 MHz): δ 170.1 (CO2Me), 169.7 (CO2Me), 161.2 (2 × C), 146.9 (C), 145.3 (C), 129.2 (2 × CH), 117.7 (CH), 113.5 (2 × CH), 104.4 (2 × CH), 99.1 (CH), 56.6 (CH), 55.3 (2× CH3O), 52.8 (CH3O), 52.8 (CH3O), 49.3 (CH), 37.0 (CH2). IR (film, cm−1) 3386, 3088, 3051, 3003, 2953, 2839, 1748, 1732, 1602, 1506, 1460, 1433, 1347, 1313, 1290, 1277, 1260, 1234, 1205, 1156, 1120, 1064, 1020, 993. HRMS ESI-TOF: m/z = 410.1574 [M + Na]+ (410.1562 calcd. for C21H25NNaO6+).
Dimethyl 2-{2-[(2-nitrophenyl)amino]-2-phenylethyl}malonate (4b). To a solution of dimethyl 2-phenylcyclopropane-1,1-dicarboxylate (200 mg, 0.85 mmol) in DCE (4.3 mL) in the presence of molecular sieves, 4 Å Ni(ClO4)2∙6H2O (62 mg, 0.17 mmol) was added under Ar atmosphere; then, 2-nitroaniline (118 mg, 0.85 mmol) was added. The resulting mixture was refluxed for 2 h, diluted with DCM, and filtered. The filtrate was concentrated under vacuum; the pure product was isolated by column chromatography on silica gel. Yield: 226 mg (71%); yellow oil; Rf = 0.47 (ethyl acetate:petroleum ether; 1:3). 1H NMR (CDCl3, 600 MHz): δ 8.45 (d, 3J = 6.9 Hz, 1H, NH), 8.17–8.16 (m, 1H, Ar), 7.38–7.29 (m, 6H, Ar), 6.73 (d, 3J = 8.7 Hz, 1H, Ar), 6.65–6.63 (m, 1H, Ar), 4.69–4.65 (m, 1H, CH), 3.77(s, 3H, CH3O), 3.71 (s, 3H, CH3O), 3.50 (t, 3J = 7.2 Hz, 1H, CH), 2.59–2.47 (m, 2H, CH2). 13C NMR (CDCl3, 150 MHz): δ 169.4 (CO2Me), 169.3 (CO2Me), 144.4 (C), 140.9 (C), 136.3 (CH), 132.7 (C), 129.3 (2 × CH), 128.2 (CH), 126.9 (CH), 126.4 (2 × CH), 116.2 (CH), 115.0 (CH), 55.8 (CH), 53.0 (CH3O), 53.0 (CH3O), 49.0 (CH), 37.0 (CH2). IR (film, cm−1): 3373, 3084, 3032, 3007, 2959, 2921, 2886, 2851, 1751, 1727, 1619, 1584, 1575, 1512, 1504, 1451, 1440, 1418, 1359, 1339, 1317, 1281, 1262, 1232, 1205, 1169, 1159, 1121, 1096, 1062, 1041, 1028, 1011, 985, 961. HRMS ESI-TOF: m/z = 373.1394 [M + H]+ (373.1394 calcd. for C19H21N2O6+).
Dimethyl 2-{2-[(2-aminophenyl)amino]-2-(3,4-dimethoxyphenyl)ethyl}malonate (4c). To a solution of dimethyl 2-(3,4-dimethoxyphenyl)cyclopropane-1,1-dicarboxylate (200 mg, 0.68 mmol) and o-phenylenediamine (74 mg, 0.68 mmol) in DCE (3.4 mL) in the presence of molecular sieves, 4 Å Y(OTf)3 (74 mg, 0.14 mmol) was added under Ar atmosphere. The resulting mixture was stirred at room temperature for 2.25 h, diluted with DCM, and filtered. The filtrate was concentrated under vacuum; pure product was isolated by column chromatography on silica gel. Yield: 150 mg (55%); viscous yellowish oil; Rf = 0.52 (ethyl acetate:petroleum ether; 2:1). 1H NMR (CDCl3, 400 MHz): δ 6.87–6.83 (m, 2H, Ar), 6.80 (d, 3J = 8.1 Hz, 1H, Ar), 6.71–6.62 (m, 3H, Ar), 6.45–6.43 (m, 1H, Ar), 3.56 (dd, 3J = 8.0 Hz, 1H, 3J = 6.4 Hz, 1H, 1H, CH), 3.85 (s, 3H, CH3O), 3.84 (s, 3H, CH3O), 3.75 (s, 3H, CH3O), 3.71 (s, 3H, CH3O), 3.56 (dd, 3J = 7.4 Hz, 3J = 6.6 Hz, 1H, CH), 2.54–2.38 (m, 2H, CH2). Signals of NH2 groups were not observed. 13C NMR (CDCl3, 100 MHz): δ 170.1 (CO2Me), 169.7 (CO2Me), 149.1 (C), 148.2 (C), 136.0 (C), 134.7 (C), 134.4 (C), 120.4 (CH), 119.0 (CH), 118.3 (CH), 116.5 (CH), 113.6 (CH), 111.2 (CH), 109.4 (CH), 56.3 (CH), 55.8 (2 × CH3O), 52.7 (2 × CH3O), 49.3 (CH), 37.1 (CH2). IR (film, cm−1): 3400, 3350, 3002, 2953, 2837, 2254, 1738, 1729, 1598, 1512, 1453, 1437, 1343, 1263, 1237, 1142, 1053, 912. HRMS ESI-TOF: m/z = 403.1855 [M + H]+ (403.1864 calcd. for C21H27N2O6+).
2-[2-Phenyl-2-(phenylamino)ethyl]malononitrile (4d) To a solution of 2-phenylcyclopropane-1,1-dicarbonitrile (417 mg, 2.48 mmol) in DCE (12 mL) and aniline (0.26 mL, 2.88 mmol) in the presence of molecular sieves, 4Å Y(OTf)3 (265 mg, 0.49 mmol, 20 mol%) was added under Ar atmosphere. The reaction mixture was stirred at room temperature for 4 days. Then, the resulting mixture was poured into saturated aq. solution of NaHCO3 (12 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic fractions were washed with saturated aq. solution of NaHCO3 (2 × 10 mL) and water (1 × 10 mL), dried with Na2SO4, and concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel. Yield: 266 mg (41%); white solid; m.p. 149–151 °C; Rf = 0.68 (ethyl acetate:petroleum ether; 1:3). 1H NMR (CDCl3, 600 MHz): δ 7.41–7.38 (m, 2H, Ph), 7.34–7.32 (m, 3H, Ph), 7.19–7.15 (m, 2H, Ph), 6.80–6.76 (m, 1H, Ph), 6.67 (d, 3J = 8.1 Hz, 2H, Ph), 4.72–4.68 (m, 1H, CH), 3.98–3.93 (m, 2H, CH, NH), 2.51–2.47 (m, 2H, CH2). 13C NMR (CDCl3, 150 MHz): δ 145.8 (C), 139.9 (C), 129.5 (2 × CH), 129.4 (2 × CH), 128.6 (CH), 126.1 (2 × CH), 119.3 (CH), 114.4 (2 × CH), 112.5 (2 × CN), 55.3 (CH), 38.4 (CH), 20.2 (CH2). IR (KBr, cm−1) 3379, 3057, 2882, 2257, 1601, 1506, 1453, 1428, 1311, 1258, 769, 754, 704. HRMS ESI-TOF: m/z = 262.1339 [M + H] (262.1339 calcd. for C17H16N3).
2-{2-[(2-Bromo-4-methylphenyl)amino]-2-phenylethyl}malononitrile (4e). To a solution of 2-phenylcyclopropane-1,1-dicarbonitrile (150 mg, 0.89 mmol) and 2-bromo-4-methylaniline (200 mg, 1.07 mmol) in DCE (4.5 mL) in the presence of molecular sieves, 4 Å Y(OTf)3 (96 mg, 0.18 mmol) was added under Ar atmosphere. The reaction mixture was stirred at room temperature for 3 days, poured into saturated aq. solution of NaHCO3, and extracted with CH2Cl2 (3 × 10 mL). The combined organic fractions were washed with saturated aq. solution of NaHCO3 (2 × 10 mL) and water (1 × 10 mL), dried with Na2SO4, and concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel. Yield: 136 mg (43%); yellow viscous oil; Rf = 0.40 (ethyl acetate:petroleum ether; 1:4). 1H NMR (CD3OD, 600 MHz): δ 7.34–7.29 (m, 4H, Ar) 7.24–7.21 (m, 1H, Ar), 6.84 (d, 3J = 7.9 Hz, 1H, Ar), 6.65 (dd, 3J = 7.9 Hz, 4J = 1.8 Hz, 1H, Ar), 6.59 (d, 4J = 1.8 Hz, 1H, Ar), 4.68 (dd, 3J = 10.6 Hz, 3J = 4.4 Hz, 1H, CH), 2.57 (dd, 2J = 14.2 Hz, 3J = 10.6 Hz, 1H, CH2), 2.45 (dd, 2J = 14.2 Hz, 3J = 4.4 Hz, 1H, CH2), 2.21 (s, 3H, CH3). Signals of CH and NH groups were not observed. 13C NMR (CD3OD, 150 MHz): δ 147.2 (CN), 142.2 (CN), 132.4 (CH), 129.9 (2 × CH), 128.9 (CH), 127.2 (2 × CH), 123.6 (C), 121.2 (CH), 121.0 (C), 115.2 (CH), 114.7 (C), 114.6 (C), 56.1 (CH), 38.5 (CH2), 17.6 (CH3). Signal of CH group was not observed. IR (film, cm−1) 3391, 2898, 2524, 2257, 1597, 1490, 1408, 1266, 1071, 835, 701. HRMS ESI-TOF: m/z = 354.0584 [M + H+] (354.0600 calcd. for C18H17Br79 N3+).
(3aRS,5RS)-5-Phenyl-3,3a,4,5-tetrahydropyrrolo [1,2-a]quinolin-1(2H)-one (5). (E)-1-Phenyl-5-styrylpyrrolidin-2-one (2h) (50 mg, 0.19 mmol) was dissolved in polyphosphoric acid (400 mg) in triple evacuated/N2-filled vial. The obtained mixture was stirred at 100 °C for 20 min. Then, the reaction mixture was cooled and quenched with saturated aq. NaHCO3 solution. The resulted mixture was extracted with ethyl acetate (3 × 7 mL). The combined organic phases were dried with Na2SO4. The solvent was removed under vacuum; the pure product was isolated by column chromatography on silica gel. Yield: 27 mg (54%); dark ivory solid; m.p. 174–176 °C; Rf = 0.69 (ethyl acetate:petroleum ether; 1:1). 1H NMR (CDCl3, 600 MHz): δ 8.71 (d, 3J = 8.3 Hz, 1H, C(9)H), 7.35–7.32 (m, 2H, C(3′)H, C(5′)H), 7.28–7.26 (m, 1H, C(4′)H), 7.23–7.21 (m, 1H, C(8)H), 7.16 (br. d, 3J = 7.2 Hz, 2H, C(2′)H, C(6′)H), 6.91–6.87 (m, 1H, C(7)H), 6.77 (d, 3J = 7.9 Hz, 1H, C(6)H), 4.17 (dd, 3J = 12.4 Hz, 3J = 5.8 Hz, 1H, C(5)H), 4.14–4.10 (m, 1H, C(3a)H), 2.63 (ddd, 2J = 17.1 Hz, 3J = 10.8 Hz, 3J = 9.9 Hz, 1H, C(2)H2), 2.55 (ddd, 2J = 17.1 Hz, 3J = 9.7 Hz, 3J = 2.3 Hz, 1H, C(2)H2), 2.39 (ddd, 2J = 13.3 Hz, 3J = 5.8 Hz, 3J = 2.3 Hz, 1H, C(4)H2), 2.35–2.29 (m, 1H, C(3)H2), 1.97–1.93 (m, 1H, C(4)H2), 1.80–1.73 (m, 1H, C(3)H2). 13C NMR (CDCl3, 150 MHz): δ 173.9 (C(1)), 145.0 (C(1′)), 136.7 (C(9a)), 130.0 (C(6)H), 129.7 (C(5a)), 128.8 (C(2′)H, C(6′)H), 128.6 (C(3′)H, C(5′)H), 127.2 (C(8)H), 126.9 (C(4′)H), 123.9 (C(7)H), 119.1 (C(9)H), 57.9 (C(3a)H), 45.0 (C(5)H), 40.2 (C(4)H2), 32.1 (C(2)H2), 25.0 (C(3)H2). IR (KBr, cm−1): 3080, 3064, 3021, 2985, 2934, 2916, 2849, 1682, 1645, 1598, 1580, 1487, 1449, 1439, 1385, 1372, 1320, 1307, 1294, 1271, 1240, 1227, 1202, 1184, 1166, 1147, 1124, 1116, 1066, 1042, 1032, 1024. HRMS ESI-TOF: m/z = 264.1390 [M + H]+ (264.1383 calcd. for C18H18NO+).
4. Conclusions
In summary, we developed a convenient general method for the synthesis of substituted γ-lactams based on Lewis acid-catalyzed DA cyclopropane ring opening with primary amines as the key step. Various 1,5-disubstituted γ-lactams were synthesized in moderate to good yields in three or four steps, requiring only a single purification procedure. We also demonstrated the potential of our method in the synthesis of an optically pure γ-lactam derivative from optically active DA cyclopropane. Additionally, the presence of reactive functionalities at the C(1) and C(5) atoms of γ-lactams ensured the possibility for postmodifications of the obtained products to convert them into more complex azaheterocycles, such as benz[g]indolizidine derivatives.
Supplementary Materials
Copies of NMR spectra for novel compounds are available online. The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27238468/s1.
Author Contributions
Conceptualization, O.A.I. and I.V.T.; methodology, O.A.I. and M.A.B.; investigation, M.A.B., A.Y.P., V.V.S., D.S.L., A.V.F., S.S.Z., E.A.T. and V.B.R.; resources, O.A.I. and I.V.T.; formal analysis, S.S.Z. and O.A.I.; writing, S.S.Z., I.V.T. and O.A.I.; supervision, O.A.I. and I.V.T.; project administration, O.A.I.; funding acquisition, O.A.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (grant 21-73-20095).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The NMR measurements were carried out in the Laboratory of Magnetic Tomography and Spectroscopy, Faculty of Fundamental Medicine of Moscow State University. X-ray study was performed using a STOE STADI VARI PILATUS-100K diffractometer purchased through the MSU Development Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Baxendale, I.R.; Ley, S.V.; Nikbin, N. An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. Beilstein J. Org. Chem. 2011, 7, 442–495. [Google Scholar] [CrossRef] [PubMed]
- Walji, A.; Berger, R.; Stump, C.A.; Schlegel, K.A.S.; Mulhearn, J.J.; Greshock, T.J.; Wang, D.; Fraley, M.E.; Jones, K.G. 3-Aryl and Heteroaryl Substituted 5-Trifluoromethyl Oxadiazoles as Histone Deacetylase 6 (HDAC6) Inhibitors. WO Pat. 2017222951A1, 28 December 2017. [Google Scholar]
- Walji, A.; Berger, R.; Stump, C.A.; Schlegel, K.A.S.; Mulhearn, J.J.; Greshock, T.J.; Ginnetti, A.T.; Wang, D.; Stachel, S.J.; Fraley, M.E. 3-Heterocyclyl Substituted 5-Trifluoromethyl Oxadiazoles as Histone Deacetylase 6 (HDAC6) Inhibitors. WO Pat. 2017222950A1, 28 December 2017. [Google Scholar]
- Mandegar, M.A.; Patel, S.; Ding, P.; Bhatt, U.; Holan, M.; Lee, J.; Li, Y.; Medina, J.; Nerurkar, A.; Seidl, F.; et al. Fluoroalkyl-Oxadiazoles and Uses Thereof. WO Pat. 2021127643A1, 24 June 2021. [Google Scholar]
- Carpino, P.A.; Sanner, M.A. Cannabinoid Receptors and Uses Thereof. WO Pat. 2007020502A2, 19 April 2007. [Google Scholar]
- Liu, H.; He, X.; Phillips, D.; Zhu, X.; Yang, K.; Lau, T.; Wu, B.; Xie, Y.; Nguyen, T.N.; Wang, X. Compounds and Compositions as Inhibitors of Cannabinoid Receptor 1 Activity. WO Pat. 2008076754A2, 22 December 2008. [Google Scholar]
- Pevarello, P.; Brasca, M.G.; Orsini, P.; Traquandi, G.; Longo, A.; Nesi, M.; Orzi, F.; Piutti, C.; Sansonna, P.; Varasi, M.; et al. 3-Aminopyrazole inhibitors of CDK2/cyclin A as antitumor agents. 2. Lead optimization. J. Med. Chem. 2005, 48, 2944–2956. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Chakka, N.; Guzman-Perez, A.; Gunaydin, H.; Gu, Y.; Huang, X.; Berry, V.; Liu, J.; Teffera, Y.; Huang, L.; et al. Discovery of novel, induced-pocket binding oxazolidinones as potent, selective, and orally bioavailable tankyrase inhibitors. J. Med. Chem. 2013, 56, 4320–4342. [Google Scholar] [CrossRef]
- Heiser, U.; Ramsbeck, D.; Sommer, R.; Meyer, A.; Hoffmann, T.; Boehme, L.; Demuth, H.U. Novel Inhibitors. US Pat. 20110092501A1, 21 April 2011. [Google Scholar]
- Lee, E.C.; Tu, M.; Stevens, B.D.; Bian, J.; Aspnes, G.; Perreault, C.; Sammons, M.F.; Wright, S.W.; Litchfield, J.; Kalgutkar, A.S.; et al. Identification of a novel conformationally constrained glucagon receptor antagonist. Bioorg. Med. Chem. Lett. 2014, 24, 839–844. [Google Scholar] [CrossRef]
- Sifferlen, T.; Boller, A.; Chardonneau, A.; Cottreel, E.; Gatfield, J.; Treiber, A.; Roch, C.; Jenck, F.; Aissaoui, H.; Williams, J.T.; et al. Substituted pyrrolidin-2-ones: Centrally acting orexin receptor antagonists promoting sleep. Part 2. Bioorg. Med. Chem. Lett. 2015, 25, 1884–1891. [Google Scholar] [CrossRef]
- Sifferlen, T.; Boller, A.; Chardonneau, A.; Cottreel, E.; Hoecker, J.; Aissaoui, H.; Williams, J.T.; Brotschi, C.; Heidmann, B.; Siegrist, R.; et al. Discovery of substituted lactams as novel dual orexin receptor antagonists. Synthesis, preliminary structure-activity relationship studies and efforts towards improved metabolic stability and pharmacokinetic properties. Part 1. Bioorg. Med. Chem. Lett. 2014, 24, 1201–1208. [Google Scholar] [CrossRef]
- Crowley, B.; Fraley, M.; Potteiger, C.; Gilfillan, R.; Patel, M.; Arrington, K.; Mitchell, H.; Shirripa, K.; McWerther, M.; Biftu, T.; et al. Benzamide CGPR Receptor Antagonists. WO Pat. 2015161011A1, 22 October 2015. [Google Scholar]
- Kise, N.; Hamada, Y.; Sakurai, T. Electroreductive coupling of aromatic ketones, aldehydes, and aldimines with α,β-unsaturated esters: Synthesis of 5-aryl substituted γ-butyrolactones and lactams. Tetrahedron 2017, 73, 1143–1156. [Google Scholar] [CrossRef]
- Ogiwara, Y.; Uchiyama, T.; Sakai, N. Reductive amination/cyclization of keto acids using a hydrosilane for selective production of lactams versus cyclic amines by switching of the indium catalyst. Angew. Chem. Int. Ed. 2016, 55, 1864–1867. [Google Scholar] [CrossRef]
- Yeh, C.H.; Korivi, R.P.; Cheng, C.H. Regioselective synthesis of γ-amino esters, nitriles, sulfones, and pyrrolidinones by nickel-catalyzed reductive coupling of aldimines and activated alkenes. Angew. Chem. Int. Ed. 2008, 47, 4892–4895. [Google Scholar] [CrossRef]
- Dugar, S.; Crouse, J.R.; Das, P.R. Isolation and characterization of a unique hydrated gamma-lactam. J. Org. Chem. 1992, 57, 5766–5768. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, X.; Feng, X. Asymmetric catalytic reactions of donor–acceptor cyclopropanes. Angew. Chem. Int. Ed. 2021, 60, 9192–9204. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Dey, A.; Banerjee, P. Relieving the stress together: Annulation of two different strained rings towards the formation of biologically significant heterocyclic scaffolds. Chem. Commun. 2021, 57, 5359–5373. [Google Scholar] [CrossRef] [PubMed]
- Augustin, A.U.; Werz, D.B. Exploiting heavier organochalcogen compounds in donor–acceptor cyclopropane chemistry. Acc. Chem. Res. 2021, 54, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Das, S. Recent advances in ring-opening of donor acceptor cyclopropanes using C-nucleophiles. Org. Biomol. Chem. 2021, 19, 965–982. [Google Scholar] [CrossRef]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic enantioselective ring-opening reactions of cyclopropanes. Chem. Rev. 2021, 121, 227–263. [Google Scholar] [CrossRef]
- Sarkar, T.; Das, B.K.; Talukdar, K.; Shah, T.A.; Punniyamurthy, T. Recent advances in stereoselective ring expansion of spirocyclopropanes: Access to the spirocyclic compounds. ACS Omega 2020, 5, 26316–26328. [Google Scholar] [CrossRef]
- Werz, D.B.; Biju, A.T. Uncovering the neglected similarities of arynes and donor-acceptor cyclopropanes. Angew. Chem. Int. Ed. 2020, 59, 3385–3398. [Google Scholar] [CrossRef]
- Singh, P.; Varshnaya, R.K.; Dey, R.; Banerjee, P. Donor-acceptor cyclopropanes as an expedient building block towards the construction of nitrogen-containing molecules: An update. Adv. Synth. Catal. 2020, 362, 1447–1484. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Trushkov, I.V. Donor-acceptor cyclopropanes in the synthesis of carbocycles. Chem. Rec. 2019, 19, 2189–2208. [Google Scholar] [CrossRef]
- Tomilov, Y.V.; Menchikov, L.G.; Novikov, R.A.; Ivanova, O.A.; Trushkov, I.V. Methods for the synthesis of donor-acceptor cyclopropanes. Russ. Chem. Rev. 2018, 87, 201–250. [Google Scholar] [CrossRef]
- Budynina, E.M.; Ivanov, K.L.; Sorokin, I.D.; Melnikov, M.Ya. Ring opening of donor-acceptor cyclopropanes with N-nucleophiles. Synthesis 2017, 49, 3035–3068. [Google Scholar] [CrossRef]
- Pagenkopf, B.L.; Vemula, N. Cycloadditions of Donor-Acceptor Cyclopropanes and Nitriles. Eur. J. Org. Chem. 2017, 2017, 2561–2567. [Google Scholar] [CrossRef]
- Grover, H.K.; Emmett, M.; Kerr, M.A. Carbocycles from donor-acceptor cyclopropanes. Org. Biomol. Chem. 2015, 13, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Novikov, R.A.; Tomilov, Y.V. Dimerization of donor-acceptor cyclopropanes. Mendeleev Commun. 2015, 25, 1–10. [Google Scholar] [CrossRef]
- Schneider, T.F.; Kaschel, J.; Werz, D.B. A new golden age for donor-acceptor cyclopropanes. Angew. Chem. Int. Ed. 2014, 53, 5504–5523. [Google Scholar] [CrossRef] [PubMed]
- Vartanova, A.E.; Levina, I.I.; Ratmanova, N.K.; Andreev, I.A.; Ivanova, O.A.; Trushkov, I.V. Ambident reactivity of 5-aminopyrazoles towards donor-acceptor cyclopropanes. Org. Biomol. Chem. 2022, 20, 7795–7802. [Google Scholar] [CrossRef] [PubMed]
- Vartanova, A.E.; Plodukhin, A.Y.; Ratmanova, N.K.; Andreev, I.A.; Anisimov, M.N.; Gudimchuk, N.B.; Rybakov, V.B.; Levina, I.I.; Ivanova, O.A.; Trushkov, I.V.; et al. Expanding Stereoelectronic Limits of endo-tet Cyclizations: Synthesis of Benz[b]azepines from Donor-Acceptor Cyclopropanes. J. Am. Chem. Soc. 2021, 143, 13952–13961. [Google Scholar] [CrossRef]
- Vartanova, A.E.; Levina, I.I.; Rybakov, V.B.; Ivanova, O.A.; Trushkov, I.V. Donor-Acceptor Cyclopropane Ring Opening with 6-Amino-1,3-dimethyluracil and Its Use in Pyrimido[4,5-b]azepines Synthesis. J. Org. Chem. 2021, 86, 12300–12308. [Google Scholar] [CrossRef]
- Boichenko, M.A.; Ivanova, O.V.; Andreev, I.A.; Chagarovskiy, A.O.; Levina, I.I.; Rybakov, V.B.; Skvortsov, D.A.; Trushkov, I.V. Convenient approach to polyoxygenated dibenzo[c,e]pyrrolo[1,2-a]azepines from donor-acceptor cyclopropanes. Org. Chem. Front. 2018, 5, 2829–2834. [Google Scholar] [CrossRef]
- Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Skvortsov, D.A.; Trushkov, I.V.; Melnikov, M.Ya. Concise approach to pyrrolizino[1,2-b]indoles from indole-derived donor-acceptor cyclopropanes. RSC Adv. 2016, 6, 62014–62018. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V.; Melnikov, M.Ya. Ring opening of donor-acceptor cyclopropanes with the azide ion: A tool for construction of N-heterocycles. Chem. Eur. J. 2015, 21, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.F.G.; O’Connor, N.R.; Craig, R.A.; Stoltz, B.M. Lewis Acid Mediated (3 + 2) Cycloadditions of Donor-Acceptor Cyclopropanes with Heterocumulenes. Org. Lett. 2012, 14, 5314–5317. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ishida, T.; Tsuji, J. Palladium(0)-catalyzed Cycloaddition of Activated Vinylcyclopropanes with Aryl Isocyanates. Chem. Lett. 1987, 16, 1157–1158. [Google Scholar] [CrossRef]
- Sahu, A.K.; Biswas, S.; Bora, S.K.; Saikia, A.K. Synthesis of 3C-alkylated active methylene substituted 2H-indazole derivatives via sequential ring opening of donor-acceptor cyclopropanes and reductive cyclization reaction. New J. Chem. 2022, 46, 12456–12460. [Google Scholar] [CrossRef]
- Unnava, R.; Chahal, K.; Reddy, K.R. Synthesis of substituted 1,2-dihydroisoquinolines via Ni(II) and Cu(I)/Ag(I) catalyzed double nucleophilic addition of arylamines to ortho-alkynyl donor-acceptor cyclopropanes (o-ADACs). Org. Biomol. Chem. 2021, 19, 6025–6029. [Google Scholar] [CrossRef]
- Chang, F.; Shen, B.; Wang, S.; Lin, L.; Feng, X. Lewis acid catalysed asymmetric cascade reaction of cyclopropyl ketones: Concise synthesis of pyrrolobenzothiazoles. Chem. Commun. 2020, 56, 13429–13432. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, N.; Banerjee, P. Regioselective Bronsted acid-catalyzed annulation of cyclopropane aldehydes with N′-aryl anthranil hydrazides: Domino construction of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)ones. J. Org. Chem. 2020, 85, 3393–3406. [Google Scholar] [CrossRef]
- Augustin, A.U.; Jones, P.G.; Werz, D.B. Ring-Opening 1,3-Aminochalcogenation of Donor–Acceptor Cyclopropanes: A Three-Component Approach. Chem. Eur. J. 2019, 25, 11620–11624. [Google Scholar] [CrossRef]
- Li, S.K.; Huang, L.L.; Lv, Y.D.; Feng, H.D. Synthesis of γ-(Arylamino)butyric Acid Derivatives via Ring-Opening Addition of Arylamines to Cyclopropane-1,1-Dicarboxylates. Russ. J. Org. Chem. 2019, 55, 1432–1438. [Google Scholar] [CrossRef]
- Nambu, H.; Hirota, W.; Fukumoto, M.; Tamura, T.; Yakura, T. An Efficient Route to Highly Substituted Indoles via Tetrahydroindol-4(5H)-one Intermediates Produced by Ring-Opening Cyclization of Spirocyclopropanes with Amines. Chem. Eur. J. 2017, 23, 16799–16805. [Google Scholar] [CrossRef] [PubMed]
- Garve, L.K.B.; Jones, P.G.; Werz, D.B. Ring-Opening 1-Amino-3-aminomethylation of Donor–Acceptor Cyclopropanes via 1,3-Diazepanes. Angew. Chem. Int. Ed. 2017, 56, 9226–9230. [Google Scholar] [CrossRef]
- Xia, Y.; Lin, L.; Chang, F.; Liao, Y.; Liu, X.; Feng, X. Asymmetric ring opening/cyclization/retro-Mannich reaction of cyclopropyl ketones with aryl 1,2-diamines for the synthesis of benzimidazole derivatives. Angew. Chem. Int. Ed. 2016, 55, 12228–12232. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Q.; Zhang, H.H.; Xu, P.F.; Luo, Y.C. Lewis acid and (hypo)iodite relay catalysis allows a strategy for the synthesis of polysubstituted azetidines and tetrahydroquinolines. Org. Lett. 2016, 18, 5212–5215. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, X.; Zheng, H.; Lin, L.; Feng, X. Asymmetric synthesis of 2,3-dihydropyrroles by ring-opening/cyclization of cyclopropyl ketones using primary amines. Angew. Chem. Int. Ed. 2015, 54, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Nambu, H.; Fukumoto, M.; Hirota, W.; Yakura, T. Ring-opening cyclization of cyclohexane-1,3-dione-2-spirocyclopropanes with amines: Rapid access to 2-substituted 4-hydroxyindole. Org. Lett. 2014, 16, 4012–4015. [Google Scholar] [CrossRef]
- Lebold, T.P.; Leduc, A.B.; Kerr, M.A. Zn(II)-Catalyzed Synthesis of Piperidines from Propargyl Amines and Cyclopropanes. Org. Lett. 2009, 11, 3770–3772. [Google Scholar] [CrossRef]
- Lifchits, O.; Charette, A.B. A Mild Procedure for the Lewis Acid-Catalyzed Ring-Opening of Activated Cyclopropanes with Amine Nucleophiles. Org. Lett. 2008, 10, 2809–2812. [Google Scholar] [CrossRef]
- Schobert, R.; Gordon, G.J.; Bieser, A.; Milius, W. 3-Functionalized Tetronic Acids from Domino Rearrangement/Cyclization/Ring-Opening Reactions of Allyl Tetronates. Eur. J. Org. Chem. 2003, 2003, 3637–3647. [Google Scholar] [CrossRef]
- Jacoby, D.; Celerier, J.P.; Haviari, G.; Petit, H.; Lhommet, G. Regiospecific synthesis of dihydropyrroles. Synthesis 1992, 1992, 884–887. [Google Scholar] [CrossRef]
- Blanchard, L.A.; Schneider, J.A. Diethylaluminum Chloride–Amine Complex Mediated Aminolysis of Activated Cyclopropanes. J. Org. Chem. 1986, 51, 1372–1374. [Google Scholar] [CrossRef]
- Akaev, A.A.; Melnikov, M.Y.; Budynina, E.M. Chameleon-like Activating Nature of the Spirooxindole Group in Donor-Acceptor Cyclopropanes. Org. Lett. 2019, 21, 9795–9799. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Sun, Z.; Fernando, E.H.N.; Nesterov, V.N.; Cundari, T.R.; Wang, H. Asymmetric ring-opening of donor-acceptor cyclopropanes with primary arylamines catalyzed by a chiral heterobimetallic catalyst. ACS Catal. 2019, 9, 8285–8293. [Google Scholar] [CrossRef]
- Das, S.; Daniliuc, C.G.; Studer, A. Stereospecific 1,3-aminobromination of donor-acceptor cyclopropanes. Angew. Chem. Int. Ed. 2017, 56, 11554–11558. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.C.; Patil, D.V.; France, S. Functionalized 4-carboxy- and 4-keto-2,3-dihydropyrroles via Ni(II)-catalyzed nucleophilic amine ring-opening cyclizations of cyclopropanes. J. Org. Chem. 2014, 79, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- So, S.S.; Auvil, T.J.; Garza, V.J.; Mattson, A.E. Boronate urea activation of nitrocyclopropane carboxylates. Org. Lett. 2012, 14, 444–447. [Google Scholar] [CrossRef]
- Stewart, J.M.; Pagenkopf, G.K. Transmission of Conjugation by the Cyclopropane Ring. J. Org. Chem. 1969, 34, 7–11. [Google Scholar] [CrossRef]
- Badarinarayana, V.; Mahmud, H.; Lovely, C.J. An asymmetric total synthesis of martinellic acid. Heterocycles 2017, 95, 1082–1105. [Google Scholar]
- Gratia, S.; Mosesohn, K.; Diver, S.T. Highly Selective Ring Expansion of Bicyclo[3.1.0]hexenes. Org. Lett. 2016, 18, 5320–5323. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, W.; Yuan, M.; Wang, H.; Ding, W.; Shao, M.; Xu, X. The reaction of electron-deficient cyclopropane derivatives with aromatic amines. Synth. Commun. 2008, 38, 3346–3353. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; O’Hare, S.M. Total Synthesis of (±)-Martinellic Acid. Org. Lett. 2001, 3, 4217–4220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ding, W.; Cao, W.; Yu, C. The stereoselective synthesis of N-aryl-trans,trans-α-carboxyl-β-methoxy carbonyl-γ-aryl-γ-butyrolactams. Synth. Commun. 2001, 31, 3107–3112. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; Foxman, B.M. Synthesis of the Tricyclic Triamine Core of Martinelline and Martinellic Acid. Tetrahedron Lett. 1999, 40, 3339–3342. [Google Scholar] [CrossRef]
- Abaev, V.T.; Trushkov, I.V.; Uchuskin, M.G. The Butin reaction. Chem. Heterocycl. Compd. 2016, 52, 973–995. [Google Scholar] [CrossRef]
- Trushkov, I.V.; Uchuskin, M.G.; Butin, A.V. Furan’s Gambit: Electrophile-Attack-Triggered Sacrifice of Furan Rings for the Intramolecular Construction of Azaheterocycles. Eur. J. Org. Chem. 2015, 2015, 2999–3016. [Google Scholar] [CrossRef]
- The Structures of 2c was Proved by Single-Crystal X-ray Crystallography. CCDC 2180495 Contains the Supplementary Crystallographic Data for this Paper; The Cambridge Crystallographic Data Centre: Cambridge, UK.
- Zhang, W.; Huang, L.; Wang, J. A Concise Synthesis of Pyrrolo- and Pyrrolidino[1,2-a]quinolin-1-ones via Diels-Alder Reactions of Acyliminium Cations with Olefins. Synthesis 2006, 2006, 2053–2063. [Google Scholar] [CrossRef]
- Fraser, W.; Suckling, C.J.; Wood, H.C.S. Latent inhibitors. Part 7. Inhibition of dihydro-orotate dehydrogenase by apirocyclopropanobarbiturates. J. Chem. Soc. Perkin Trans. 1 1990, 3137–3144. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium methylide((CH3)2SOCH2) and dimethylsulfonium methylide ((CH3)2SCH2). Formation and application to organic synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Maity, A.K.; Roy, S. A Multimetallic Piano-Stool Ir–Sn3 Catalyst for Nucleophilic Substitution Reaction of γ-Hydroxy Lactams through N-Acyliminium Ions. J. Org. Chem. 2012, 77, 2935–2941. [Google Scholar] [CrossRef]
- Meyer, W.L.; Vaughan, W.R. 1,5-Diaryl-2,3-pyrrolidinediones. VIII. Synthesis and Structure Proof. J. Org. Chem. 1957, 22, 1554–1560. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanov, K.L.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V. Reaction of dimethyl (S)-2-(p-tolyl)cyclopropane-1,1-dicarboxylate with acetonitrile. Chem. Heterocycl. Compd. 2012, 48, 825–827. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).