


Purification and Characterization of Novel Anti-MRSA Peptides Produced by Brevibacillus sp. SPR-20

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
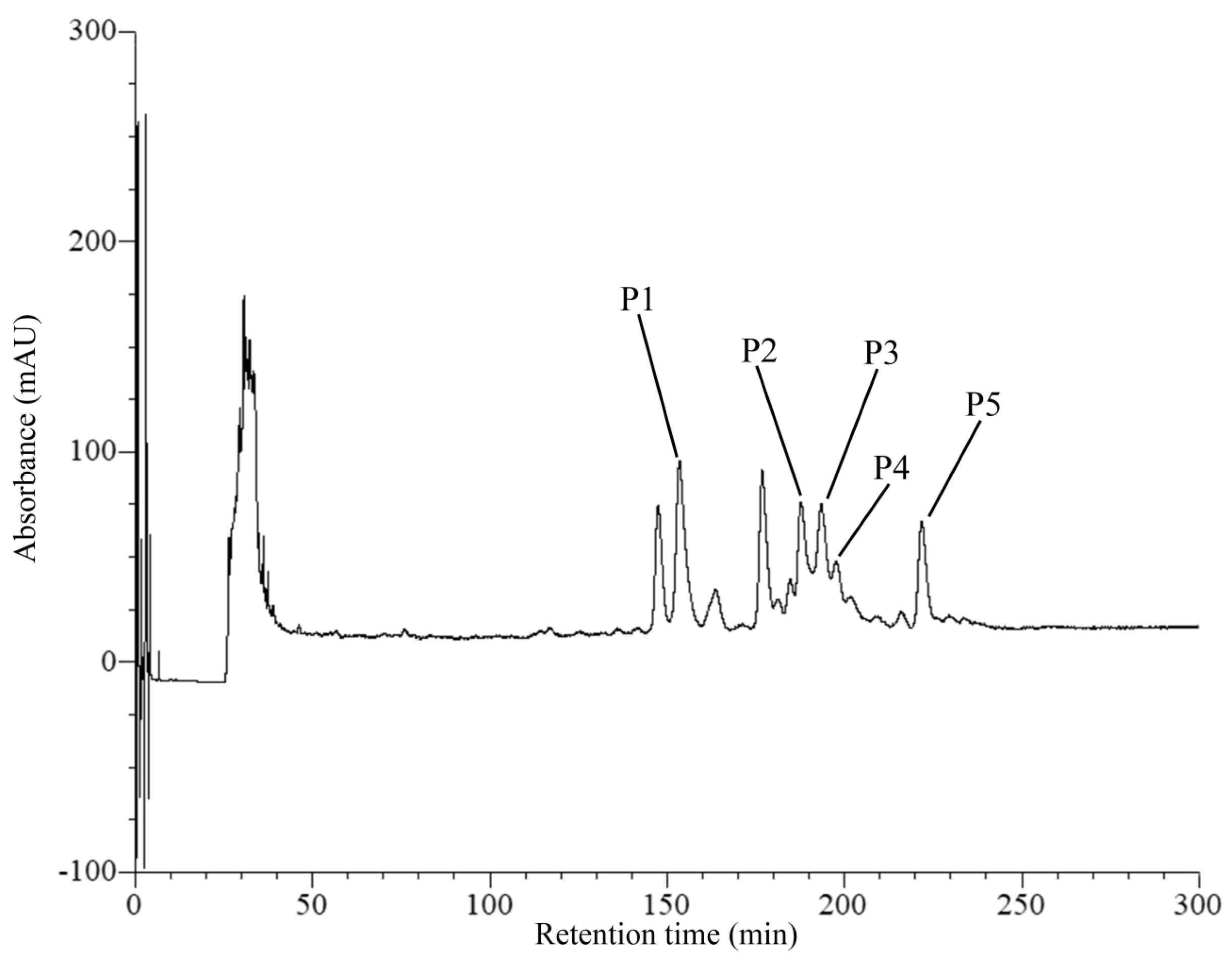
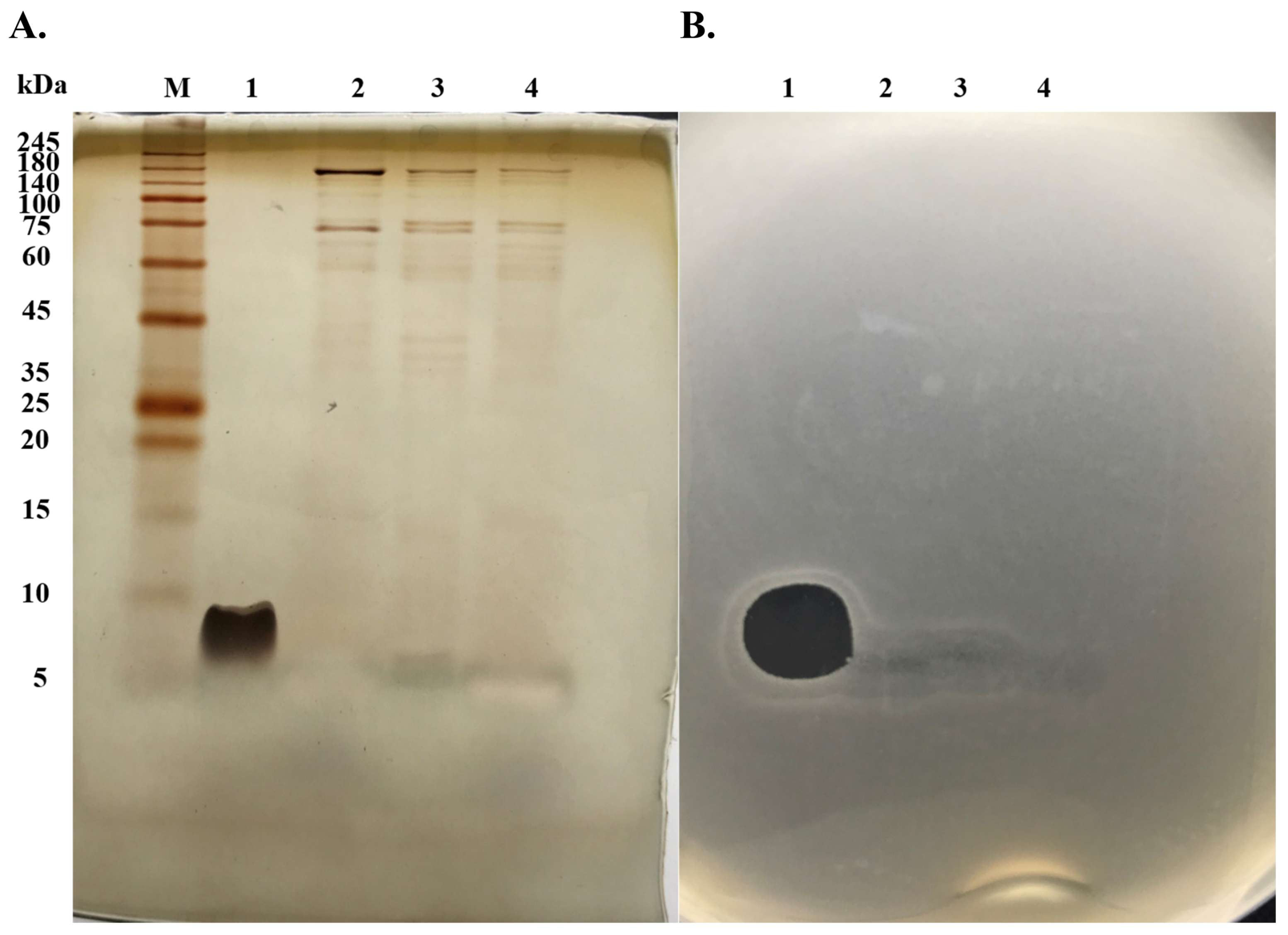
2.1. Purification of Anti-MRSA Substances
2.2. Determination of Antimicrobial Activity by Microdilution Method
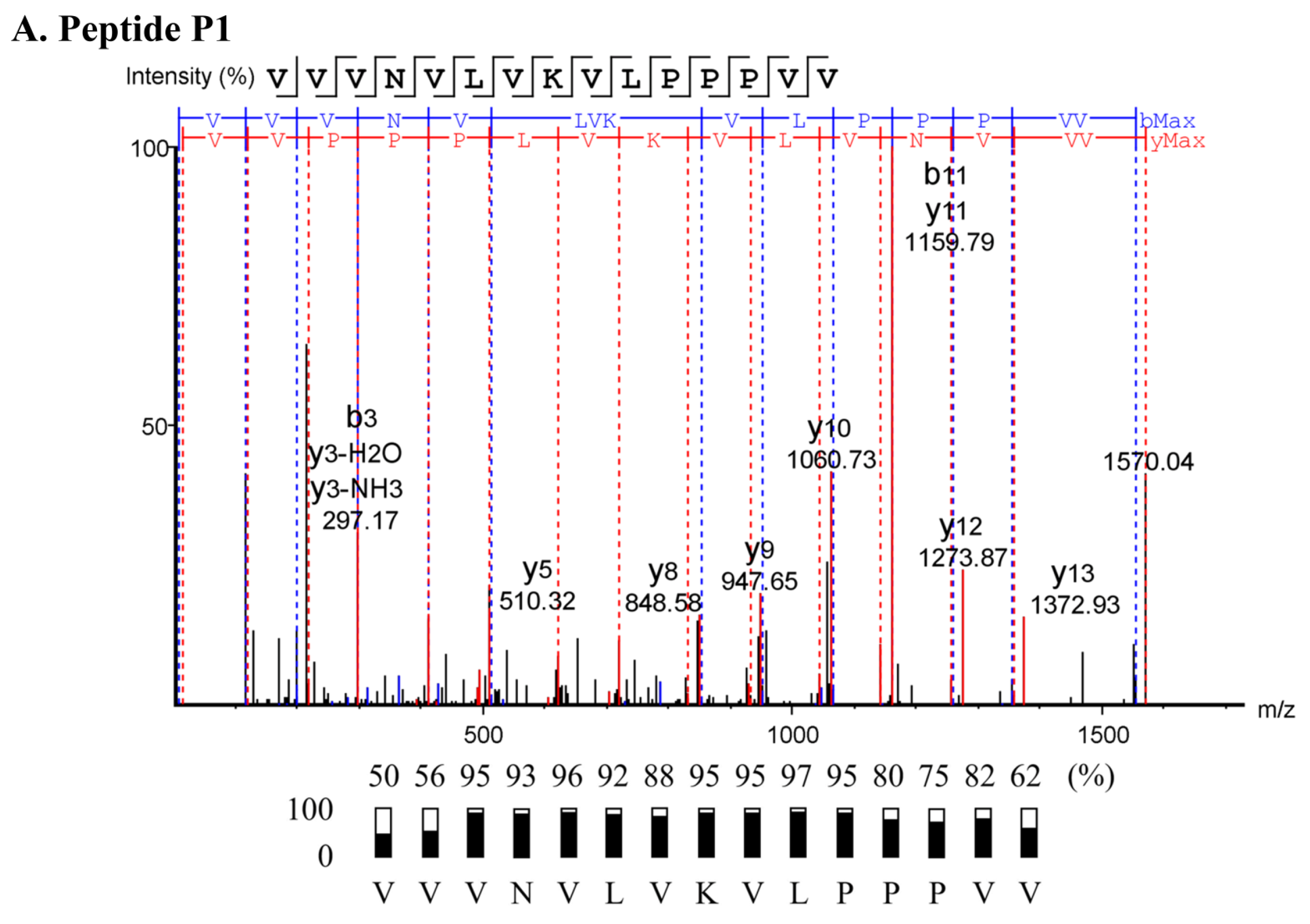
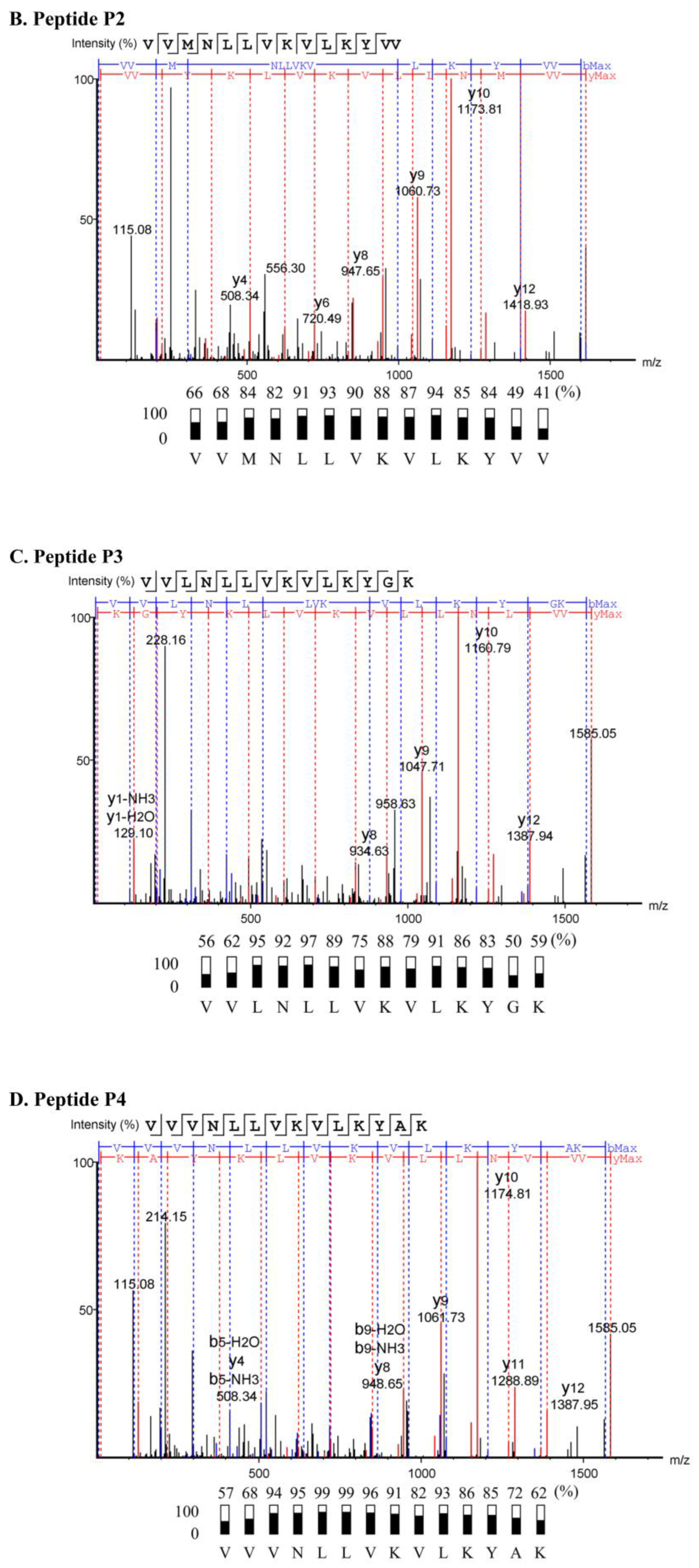
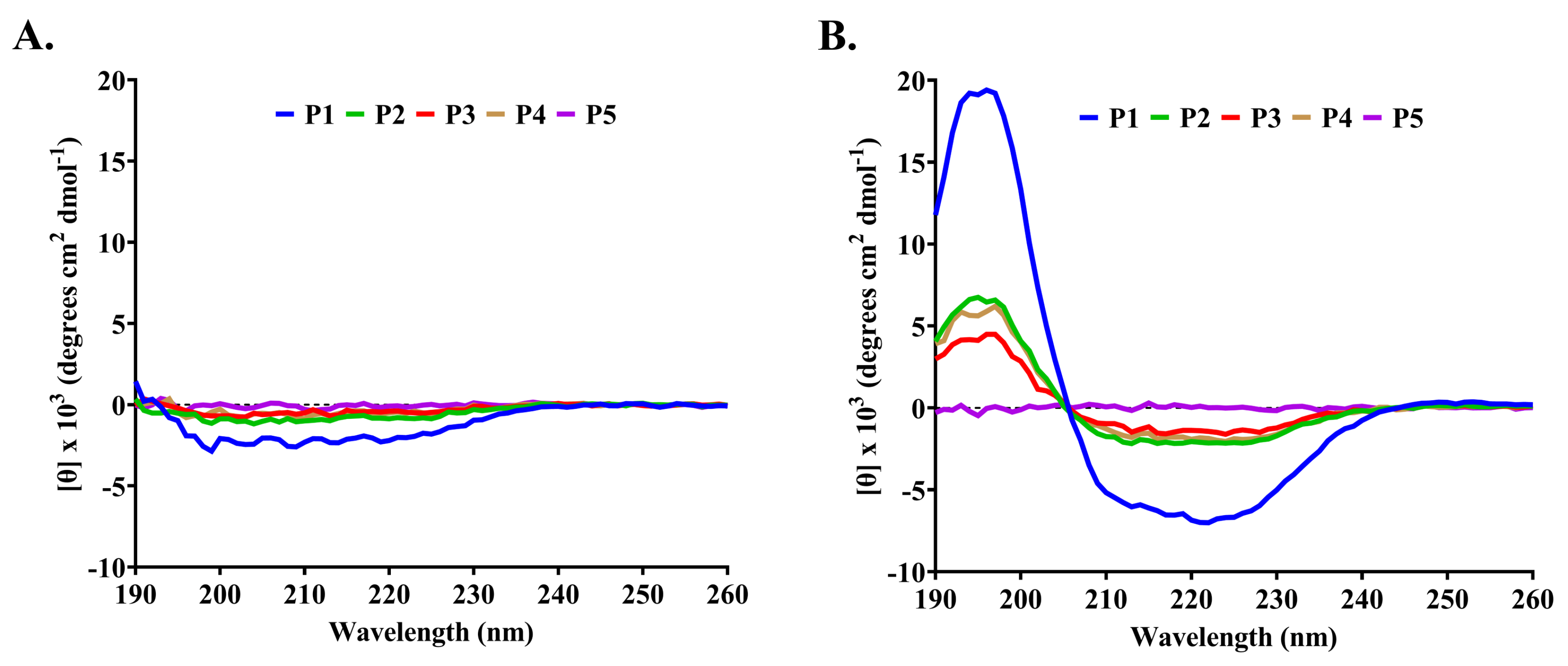
2.3. Characterization of Anti-MRSA Substances
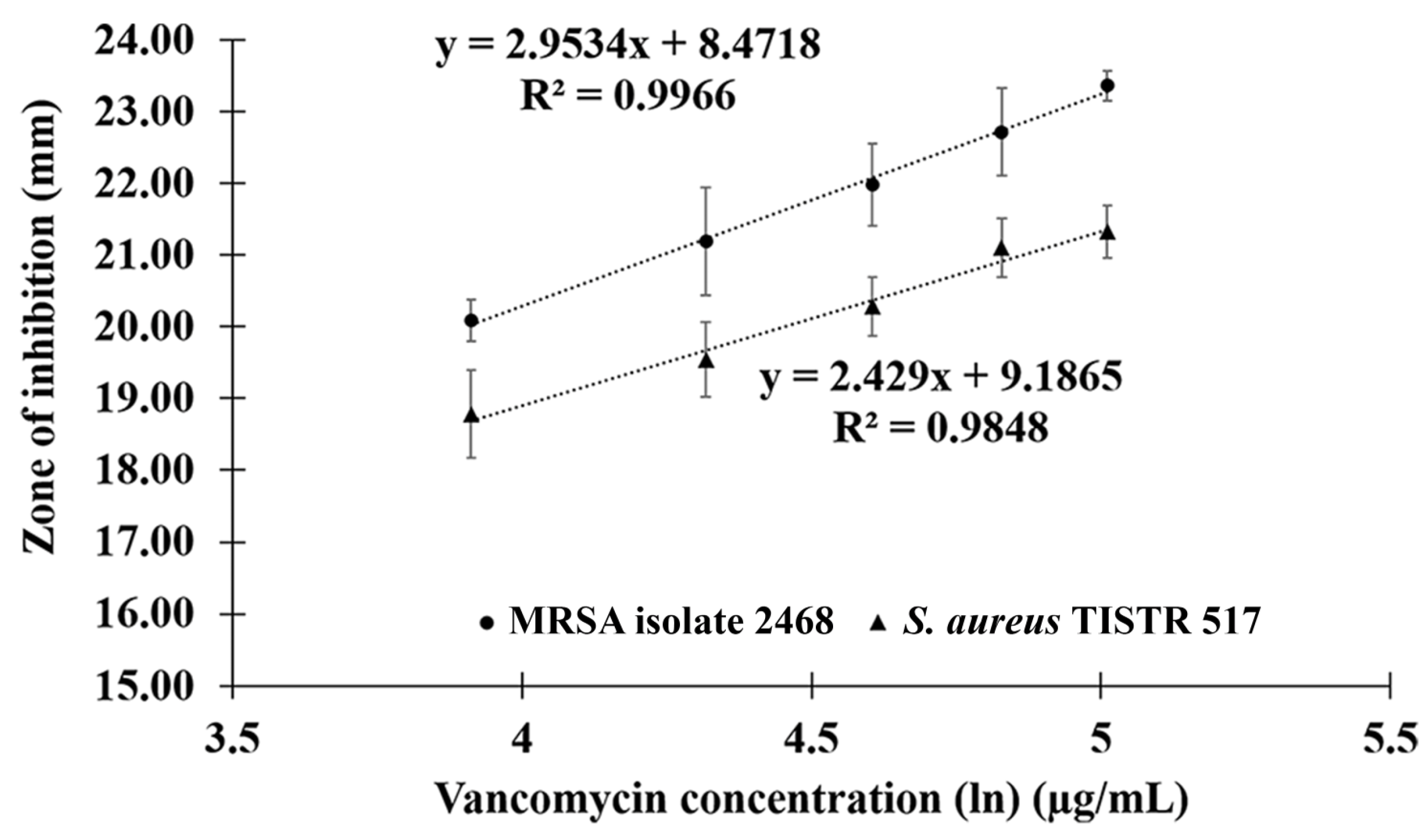
2.4. Comparison of Potency between Anti-MRSA Peptide and Vancomycin
2.5. Scanning Electron Microscopy (SEM) of Anti-MRSA Peptide-Treated Pathogens
2.6. Stability Profiles of Anti-MRSA Peptide
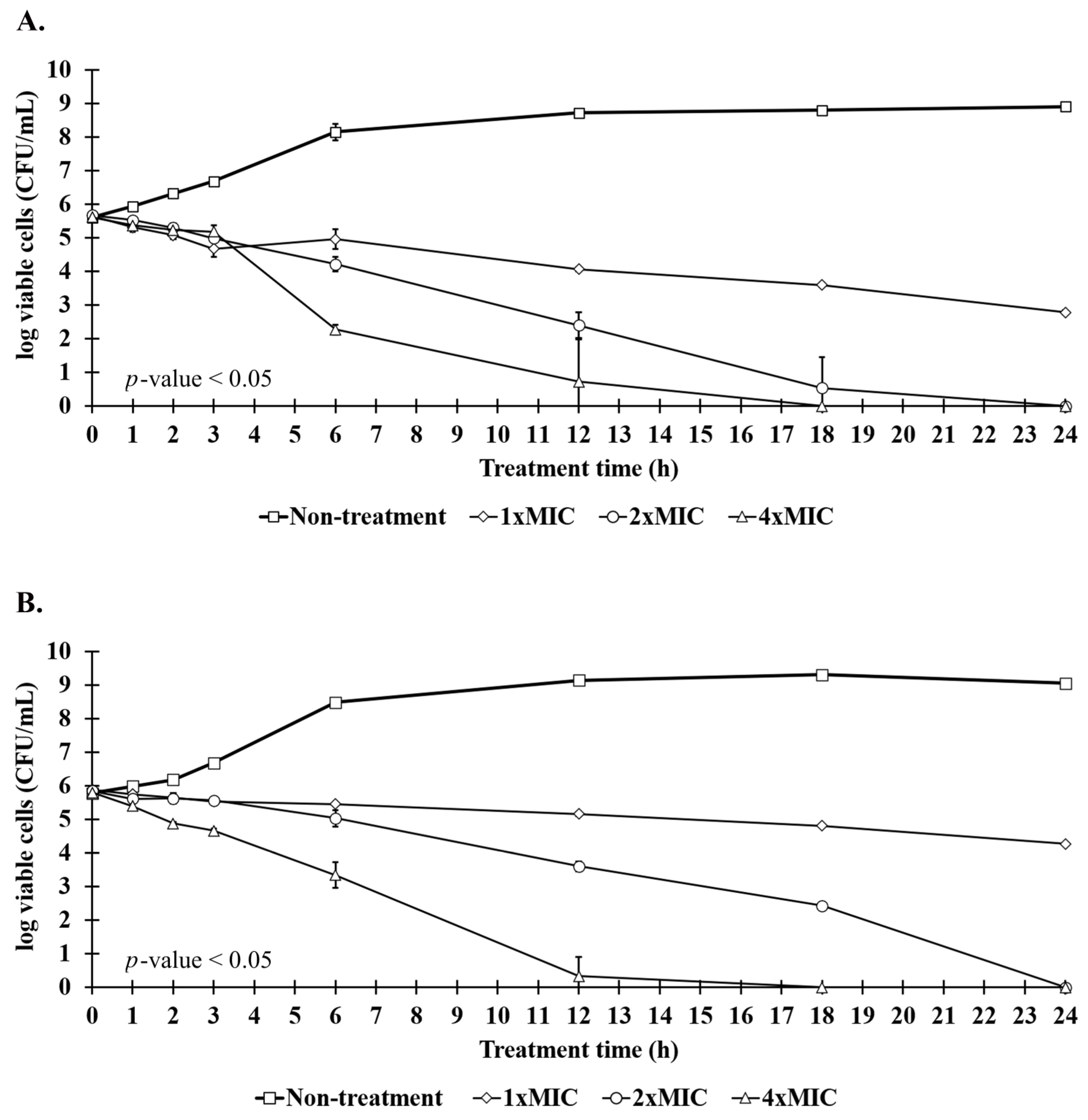
2.7. Killing Kinetics of Anti-MRSA Peptide
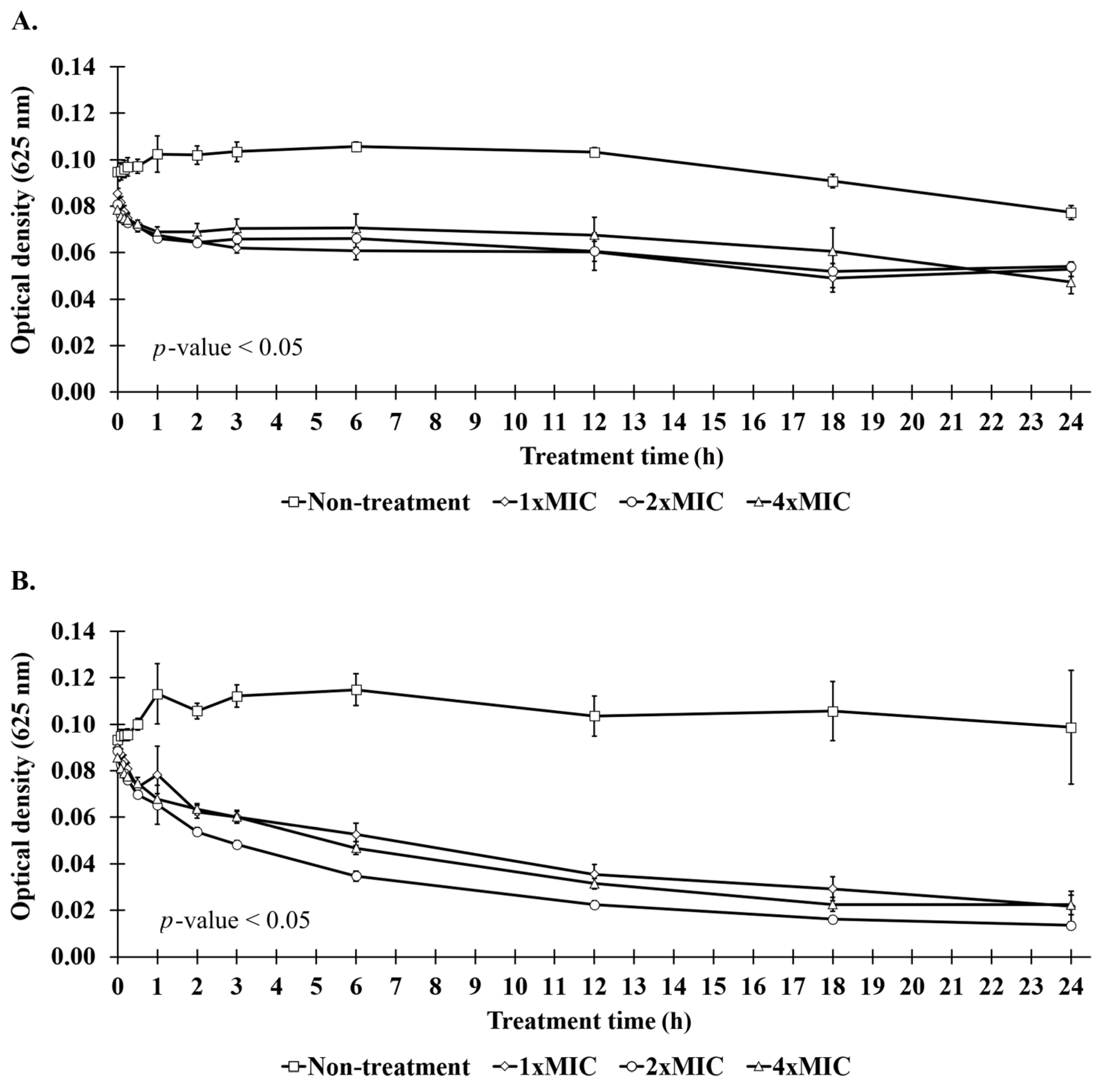
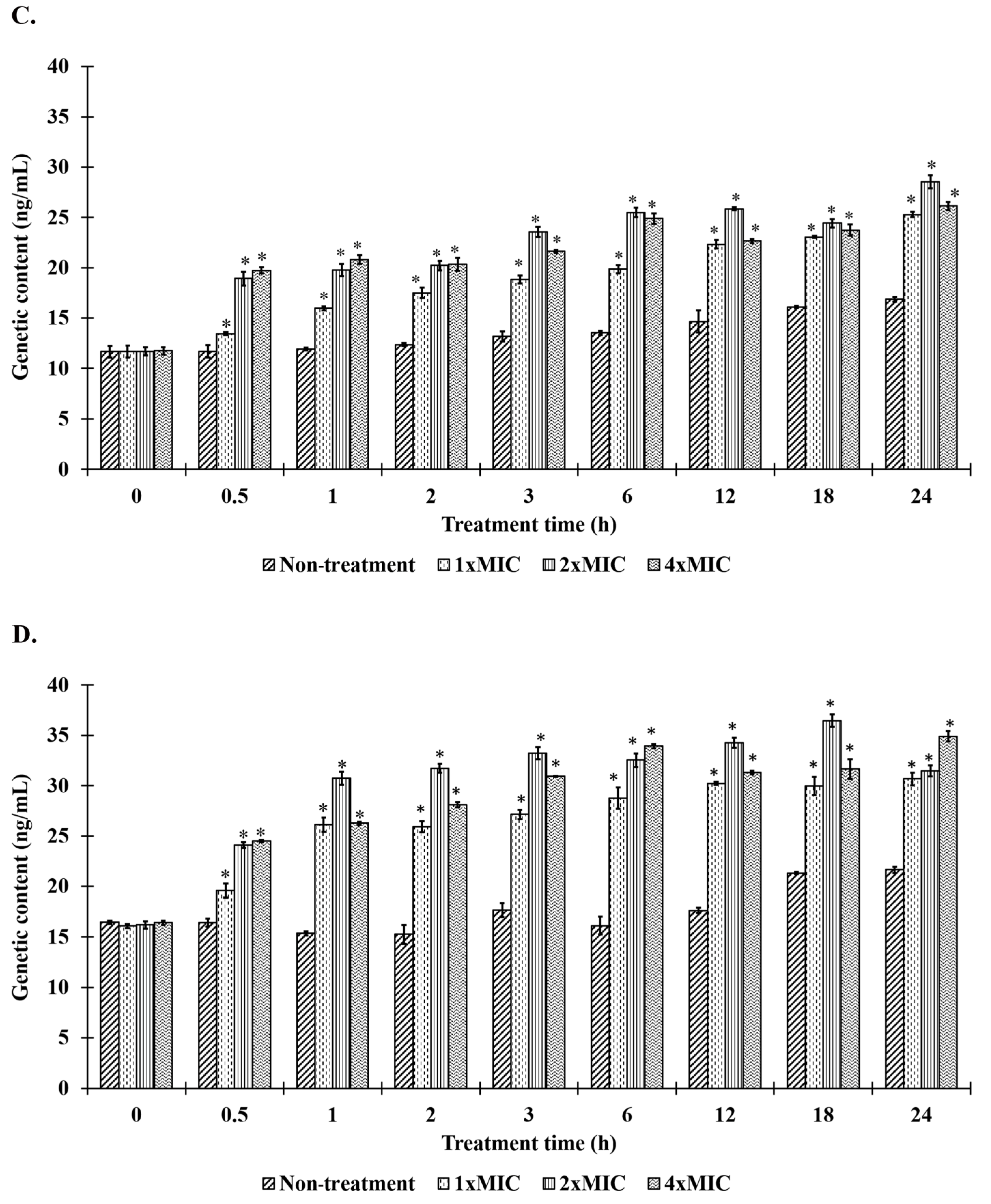
2.8. Bacteriolysis and Leakage of Genetic Materials
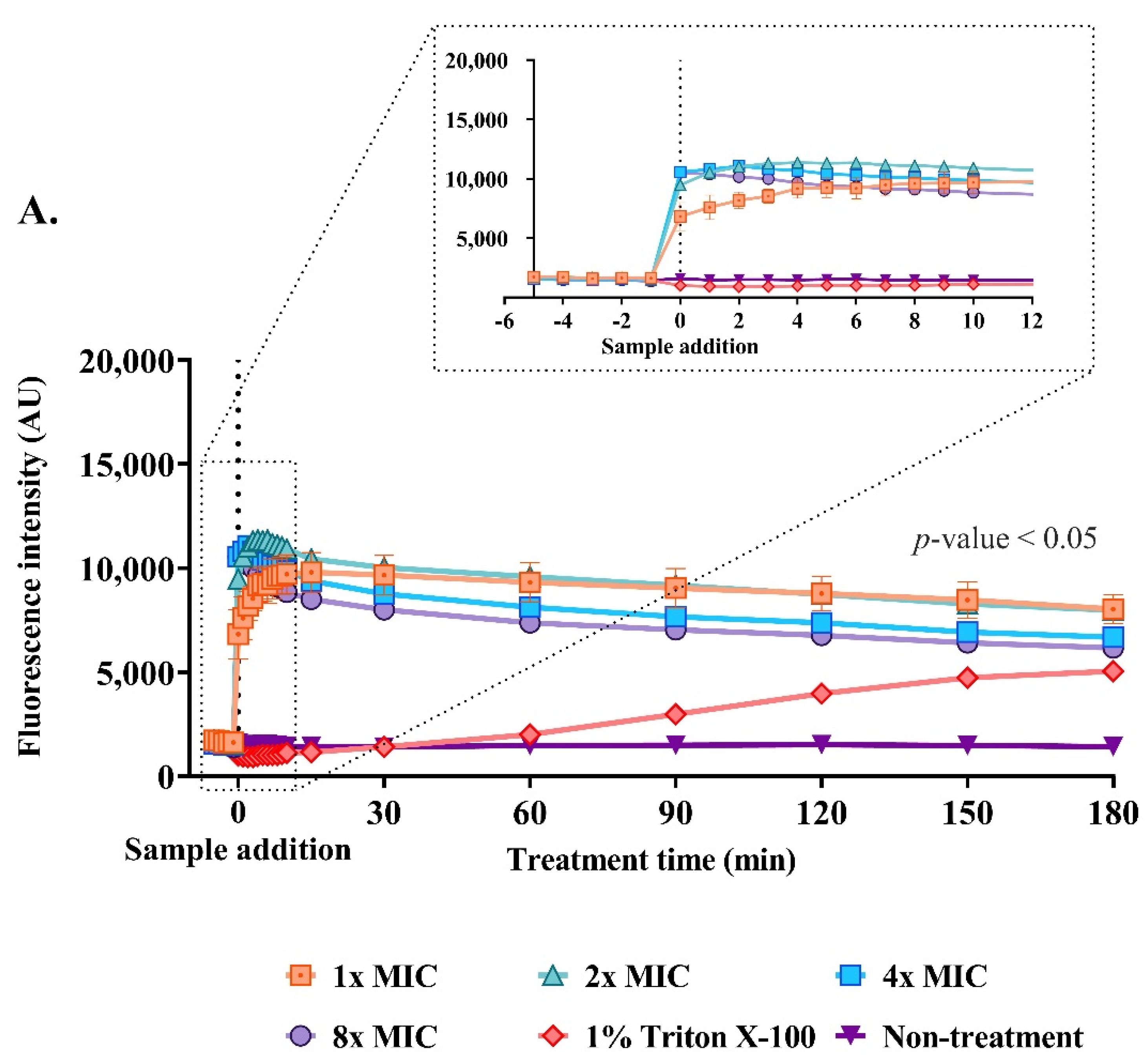
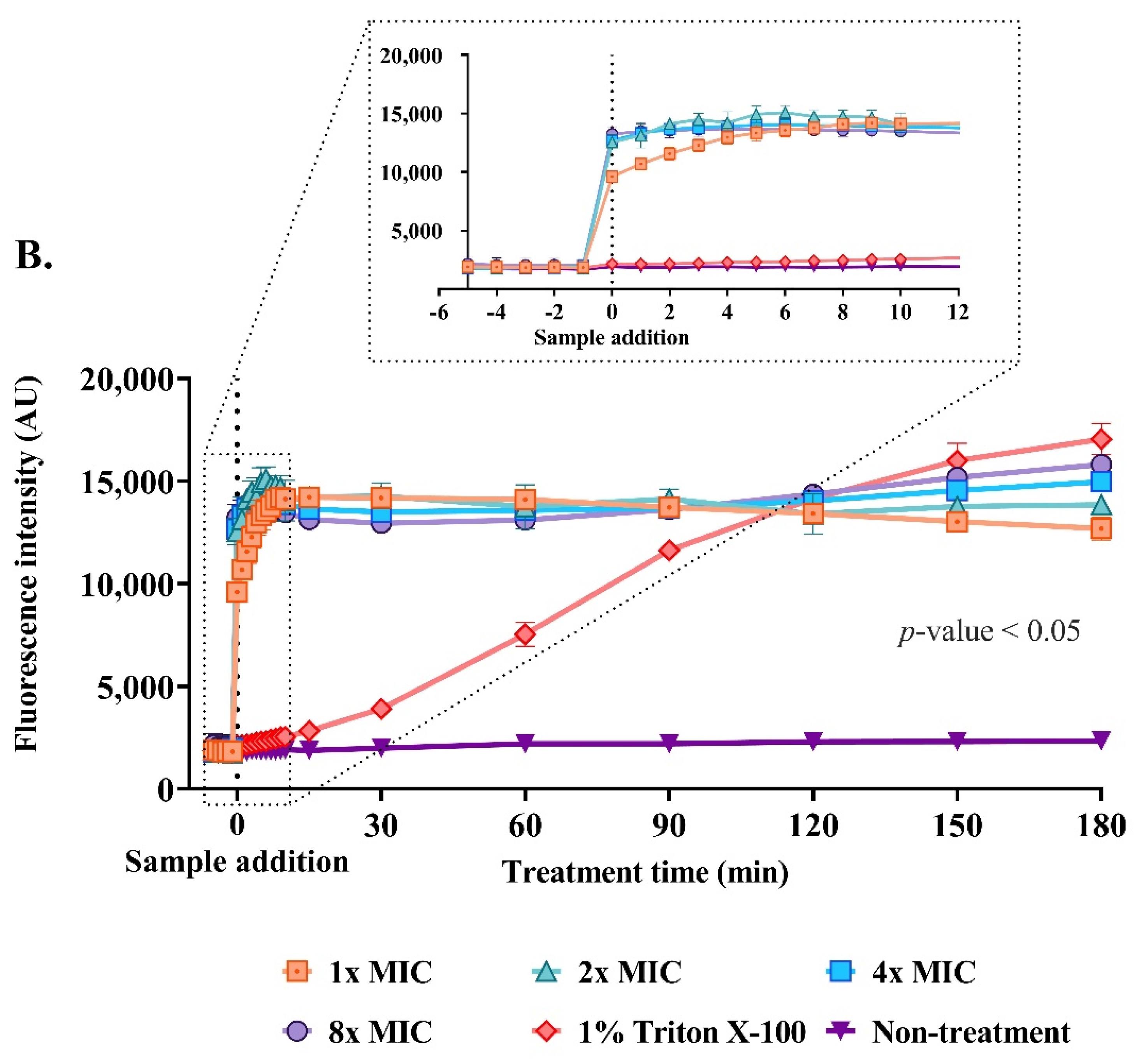
2.9. Effect of the Anti-MRSA Peptide on Cell Permeability
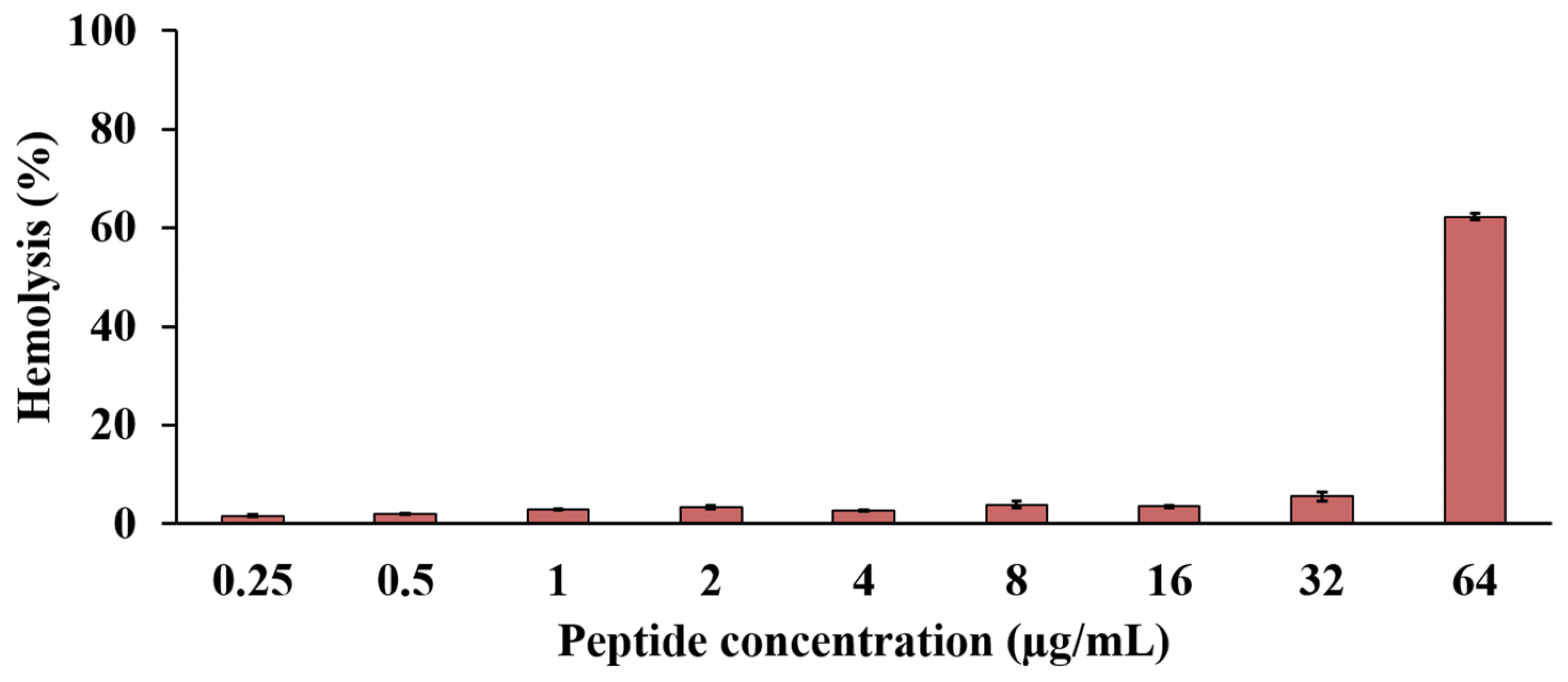
2.10. Effect of the Anti-MRSA Peptide on Hemolysis
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Condition
4.2. Purification of Anti-MRSA Substances
4.3. Microdilution Antimicrobial Assay
4.4. Agar Well Diffusion Assay
4.5. Peptide Sequencing
4.6. Determination of Peptide Secondary Structure
4.7. Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Gel Overlay Assay
4.8. Potency Comparison between Anti-MRSA Peptide and Antibiotic
4.9. Scanning Electron Microscopy (SEM) of Antimicrobial Peptide-Treated Cells
4.10. Stability Study of Anti-MRSA Peptide
4.11. Killing Kinetics of Anti-MRSA Peptide
4.12. Cell Lysis and Release of the Genetic Materials
4.13. Effect of the Anti-MRSA Peptide on Membrane Permeability
4.14. Effect of the Anti-MRSA Peptide on Hemolysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Harkins, C.P.; Pichon, B.; Doumith, M.; Parkhill, J.; Westh, H.; Tomasz, A.; de Lencastre, H.; Bentley, S.D.; Kearns, A.M.; Holden, M.T.G. Methicillin-resistant staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome Biol. 2017, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Junnila, J.; Hirvioja, T.; Rintala, E.; Auranen, K.; Rantakokko-Jalava, K.; Silvola, J.; Lindholm, L.; Gröndahl-Yli-Hannuksela, K.; Marttila, H.; Vuopio, J. Changing epidemiology of methicillin-resistant staphylococcus aureus in a low endemicity area-new challenges for MRSA control. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- De la Calle, C.; Morata, L.; Cobos-Trigueros, N.; Martinez, J.A.; Cardozo, C.; Mensa, J.; Soriano, A. Staphylococcus aureus bacteremic pneumonia. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 3, 318–327. [Google Scholar] [CrossRef]
- Penesyan, A.; Gillings, M.; Paulsen, I.T. Antibiotic discovery: Combatting bacterial resistance in cells and in biofilm communities. Molecules 2015, 20, 5286–5298. [Google Scholar] [CrossRef]
- Stoica, R.; Moscovici, M.; Tomulescu, C.; Cășărică, A.; Băbeanu, N.; Popa, O.; Kahraman, H. Antimicrobial compounds of the genus Bacillus: A review. Rom. Biotechnol. Lett. 2019, 24, 1111–1119. [Google Scholar] [CrossRef]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Songnaka, N.; Lertcanawanichakul, M.; Atipairin, A. Promising anti-MRSA activity of Brevibacillus sp. isolated from soil and strain improvement by UV mutagenesis. Sci. Pharm. 2020, 89, 1. [Google Scholar] [CrossRef]
- Erckes, V.; Steuer, C.A. Story of peptides, lipophilicity and chromatography—Back and forth in time. RSC Med. Chem. 2022, 13, 676–687. [Google Scholar] [CrossRef]
- Shibue, M.; Mant, C.T.; Hodges, R.S. Effect of anionic ion-pairing reagent hydrophobicity on selectivity of peptide separations by reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1080, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, B. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Campanella, J.J.; Bitincka, L.; Smalley, J. MatGAT: An application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinform. 2003, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Toda, H.; Ikeguchi, M. Dependence of α-helical and β-sheet amino acid propensities on the overall protein fold type. BMC Struct. Biol. 2012, 12, 18. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M. Antimicrobial peptides: From design to clinical application. Antibiotics 2022, 11, 349. [Google Scholar] [CrossRef]
- North, S.H.; Taitt, C.R. Application of circular dichroism for structural analysis of surface-immobilized cecropin A interacting with lipoteichoic acid. Langmuir 2015, 31, 10791–10798. [Google Scholar] [CrossRef]
- Prasad, A.K.; Tiwari, C.; Ray, S.; Holden, S.; Armstrong, D.A.; Rosengren, K.J.; Rodger, A.; Panwar, A.S.; Martin, L.L. Secondary structure transitions for a family of amyloidogenic, antimicrobial uperin 3 peptides in contact with sodium dodecyl sulfate. ChemPlusChem 2022, 87, e202100408. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Selsted, M.E.; White, S.H. CD spectra of indolicidin antimicrobial peptides suggest turns, not polyproline helix. Biochemistry 1999, 38, 12313–12319. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, M.; Muhammad, I.; Cui, Q.; Zhang, H.; Jia, Y.; Xu, Q.; Kong, L.; Ma, H. An antibacterial peptide with high resistance to trypsin obtained by substituting D-amino acids for trypsin cleavage sites. Antibiotics 2021, 10, 1465. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Vishnepolsky, B.; Grigolava, M.; Managadze, G. Physicochemical features and peculiarities of interaction of AMP with the membrane. Pharmaceuticals 2021, 14, 471. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein. Cell. 2010, 1, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Ohgita, T.; Takechi-Haraya, Y.; Okada, K.; Matsui, S.; Takeuchi, M.; Saito, C.; Nishitsuji, K.; Uchimura, K.; Kawano, R.; Hasegawa, K.; et al. Enhancement of direct membrane penetration of arginine-rich peptides by polyproline II helix structure. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183403. [Google Scholar] [CrossRef]
- Hong, Z.; Damodaran, K.; Asher, S.A. Sodium dodecyl sulfate monomers induce XAO peptide polyproline II to α-helix transition. J. Phys. Chem. B 2014, 118, 10565–10575. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef]
- Mkrtchyan, H.; Gibbons, S.; Heidelberger, S.; Zloh, M.; Limaki, H.K. Purification, characterisation and identification of acidocin LCHV, an antimicrobial peptide produced by Lactobacillus acidophilus n.v. Er 317/402 strain Narine. Int. J. Antimicrob. Agents 2010, 35, 255–260. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Han, P.; Liu, Y.; Hong, D.; Li, S.; Ma, A.; Jia, Y. Discovery of novel antimicrobial peptides, Brevilaterin V, from Brevibacillus laterosporus S62-9 after regulated by exogenously-added L-valine. LWT 2022, 155, 112962. [Google Scholar] [CrossRef]
- Keil, B. Specificity of Proteolysis, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 43–227. [Google Scholar] [CrossRef]
- Arsène, M.M.J.; Davares, A.K.L.; Viktorovna, P.I.; Andreevna, S.L.; Sarra, S.; Khelifi, I.; Sergueïevna, D.M. The public health issue of antibiotic residues in food and feed: Causes, consequences, and potential solutions. Vet. World 2022, 15, 662–671. [Google Scholar] [CrossRef]
- Liu, K.; Yang, L.; Peng, X.; Gong, H.; Wang, J.; Lu, J.R.; Xu, H. Effects of conventional surfactants on the activity of designed antimicrobial peptide. Langmuir 2020, 36, 3531–3539. [Google Scholar] [CrossRef]
- Ravensdale, J.; Wong, Z.; O’Brien, F.; Gregg, K. Efficacy of antibacterial peptides against peptide-resistant MRSA is restored by permeabilization of bacteria membranes. Front. Microbiol. 2016, 7, 1745. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Chen, Z.; Onishi, H.R.; Kohler, J.; Silver, L.L.; Kerns, R.; Fukuzawa, S.; Thompson, C.; Kahne, D. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science 1999, 284, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 1997, 63, 2421–2431. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Dutta, D.; Willcox, M.D.P. Mode of action of the antimicrobial peptide Mel4 is independent of Staphylococcus aureus cell membrane permeability. PLoS ONE 2019, 14, e0215703. [Google Scholar] [CrossRef] [PubMed]
- Odah, K.A.; Dong, W.L.; Lei, L.; Atiah, L.A.; Wang, Y.; Kong, L.C.; Ma, H.X. Isolation, identification, and characterization of a novel bacteriocin produced by Brevibacillus laterosporus DS-3 against methicillin-resistant Staphylococcus aureus (MRSA). Int. J. Pept. Res. Ther. 2020, 26, 709–715. [Google Scholar] [CrossRef]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide-membrane interactions of three related antimicrobial peptides. Colloids Surf. B Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef]
- Panteleev, P.V.; Bolosov, I.A.; Balandin, S.V.; Ovchinnikova, T.V. Design of antimicrobial peptide arenicin analogs with improved therapeutic indices. J. Pept. Sci. 2015, 21, 105–113. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef]
- Atipairin, A.; Songnaka, N.; Krobthong, S.; Yingchutrakul, Y.; Chinnawong, T.; Wanganuttara, T. Identification and characterization of a potential antimicrobial peptide isolated from soil Brevibacillus sp. WUL10 and its activity against MRSA pathogens. Trop. Med. Infect. Dis. 2022, 7, 93. [Google Scholar] [CrossRef]
- Xi, Q.; Wang, J.; Du, R.; Zhao, F.; Han, Y.; Zhou, Z. Purification and characterization of bacteriocin produced by a strain of Enterococcus faecalis TG2. Appl. Biochem. Biotechnol. 2018, 184, 1106–1119. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 11th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018; pp. 15–52. [Google Scholar]
- Goh, H.F.; Philip, K. Purification and characterization of bacteriocin produced by Weissella confusa A3 of dairy origin. PLoS ONE 2015, 10, e0140434. [Google Scholar] [CrossRef]
- Kirby-Bauer Disk Diffusion Susceptibility Test Protocol. Available online: https://asm.org/getattachment/2594ce26-bd44-47f6-8287-0657aa9185ad/Kirby-Bauer-Disk-Diffusion-Susceptibility-Test-Protocol-pdf.pdf (accessed on 22 August 2022).
- Krobthong, S.; Yingchutrakul, Y. Identification and enhancement of antioxidant P1-peptide isolated from Ganoderma lucidum hydrolysate. Food Biotechnol. 2020, 34, 338–351. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteom. 2012, 11, 10587. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Shi, J.; Chen, C.; Wang, D.; Tong, Z.; Wang, Z.; Liu, Y. Amphipathic peptide antibiotics with potent activity against multidrug-resistant pathogens. Pharmaceutics 2021, 13, 438. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Chalasani, A.G.; Dhanarajan, G.; Nema, S.; Sen, R.; Roy, U. An antimicrobial metabolite from Bacillus sp.: Significant activity against pathogenic bacteria including multidrug-resistant clinical strains. Front. Microbiol. 2015, 6, 1335. [Google Scholar] [CrossRef]
- Dafale, N.A.; Semwal, U.P.; Rajput, R.K.; Singh, G.N. Selection of appropriate analytical tools to determine the potency and bioactivity of antibiotics and antibiotic resistance. J. Pharm. Anal. 2016, 6, 207–213. [Google Scholar] [CrossRef]
- United States Pharmacopeia. United States Pharmacopeia and National Formulary (USP 41–NF 36); United States Pharmacopeial Convention: Rockville, MD, USA, 2018; pp. 5991–6005. [Google Scholar]
- Xu, L.; Shao, C.; Li, G.; Shan, A.; Chou, S.; Wang, J.; Ma, Q.; Dong, N. Conversion of broad-spectrum antimicrobial peptides into species-specific antimicrobials capable of precisely targeting pathogenic bacteria. Sci. Rep. 2020, 10, 944. [Google Scholar] [CrossRef]
- Baindara, P.; Chaudhry, V.; Mittal, G.; Liao, L.M.; Matos, C.O.; Khatri, N.; Franco, O.L.; Patil, P.B.; Korpole, S. Characterization of the antimicrobial peptide Penisin, a class Ia novel lantibiotic from Paenibacillus sp. strain A3. Antimicrob. Agents. Chemother. 2016, 60, 580–591. [Google Scholar] [CrossRef]
- Meng, F.; Alayash, A.I. Determination of extinction coefficients of human hemoglobin in various redox states. Anal. Biochem. 2017, 521, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Oddo, A.; Hansen, P.R. Hemolytic activity of antimicrobial peptides. In Antimicrobial Peptides. Methods in Molecular Biology, 1st ed.; Hansen, P., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1548, pp. 427–435. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification Step | Volume (mL) | Total Protein (mg) | Total Activity (AU) | Specific Activity (AU/mg) | Purification Factor | %Yield |
|---|---|---|---|---|---|---|
| Cell-free supernatant | 1740 | 446 | 69,600 | 156 | 1 | 100 |
| Ammonium sulfate precipitation | 205 | 231 | 65,600 | 284 | 2 | 94 |
| CIEX | 277 | 49 | 22,160 | 452 | 3 | 32 |
| RPC (P1) | 23 | 9 | 14,720 | 1694 | 11 | 21 |
| Active Substance | Tested Strains | MIC (µg/mL) | MBC (µg/mL) |
|---|---|---|---|
| P1 | S. aureus TISTR 517 | 2 | 4 |
| MRSA isolate 142 | 2 | 4 | |
| MRSA isolate 1096 | 2 | 4 | |
| MRSA isolate 2468 | 2 | 4 | |
| P2 | S. aureus TISTR 517 | 8 | 16 |
| MRSA isolate 142 | 8 | 64 | |
| MRSA isolate 1096 | 8 | 64 | |
| MRSA isolate 2468 | 8 | 64 | |
| P3 | S. aureus TISTR 517 | 4 | 4 |
| MRSA isolate 142 | 4 | 8 | |
| MRSA isolate 1096 | 4 | 8 | |
| MRSA isolate 2468 | 4 | 8 | |
| P4 | S. aureus TISTR 517 | 8 | 8 |
| MRSA isolate 142 | 8 | 16 | |
| MRSA isolate 1096 | 8 | 16 | |
| MRSA isolate 2468 | 8 | 16 | |
| P5 | S. aureus TISTR 517 | 32 | 32 |
| MRSA isolate 142 | 32 | 32 | |
| MRSA isolate 1096 | 32 | 32 | |
| MRSA isolate 2468 | 32 | 32 | |
| Vancomycin | S. aureus TISTR 517 | 2 | 2 |
| MRSA isolate 142 | 2 | 2 | |
| MRSA isolate 1096 | 2 | 2 | |
| MRSA isolate 2468 | 2 | 2 | |
| Cefoxitin | S. aureus TISTR 517 | 2 | 2 |
| MRSA isolate 142 | >64 | ND | |
| MRSA isolate 1096 | >64 | ND | |
| MRSA isolate 2468 | >64 | ND |
| Sample | Sequence (N→C) | Length (Residues) | Mass (Da) | pI | Net Charge at pH 7.4 | Hydrophobicity (H) |
|---|---|---|---|---|---|---|
| P1 | VVVNVLVKVLPPPVV | 15 | 1570.02 | 8.62 | +1 | 0.915 |
| P2 | VVMNLLVKVLKYVV | 14 | 1616.01 | 9.51 | +2 | 0.859 |
| P3 | VVLNLLVKVLKYGK | 14 | 1585.03 | 9.93 | +3 | 0.648 |
| P4 | VVVNLLVKVLKYAK | 14 | 1585.03 | 9.93 | +3 | 0.636 |
| P5 | VVLNLVVKLLKYAK | 14 | 1599.05 | 9.93 | +3 | 0.670 |
| Peptide | Solvents | α-Helix (%) | β-Strand (%) | Turn (%) | Random (%) | Total (%) |
|---|---|---|---|---|---|---|
| P1 | H2O | 4.1 | 44.0 | 19.5 | 32.4 | 100.0 |
| 50 mM SDS | 25.5 | 29.3 | 10.6 | 34.6 | 100.0 | |
| P2 | H2O | 3.4 | 39.8 | 20.5 | 36.3 | 100.0 |
| 50 mM SDS | 3.7 | 48.8 | 20.8 | 26.7 | 100.0 | |
| P3 | H2O | 3.9 | 40.3 | 22.6 | 33.2 | 100.0 |
| 50 mM SDS | 12.4 | 29.0 | 10.6 | 48.0 | 100.0 | |
| P4 | H2O | 3.5 | 42.0 | 20.8 | 33.7 | 100.0 |
| 50 mM SDS | 4.9 | 38.7 | 22.7 | 33.7 | 100.0 | |
| P5 | H2O | 3.1 | 43.1 | 21.2 | 32.6 | 100.0 |
| 50 mM SDS | 2.6 | 40.7 | 19.3 | 37.4 | 100.0 |
| Concentration | Corrected Zone of Inhibition (Mean ± SD; mm) | |
|---|---|---|
| S. aureus TISTR 517 | MRSA Isolate 2468 | |
| 50 µg/mL of vancomycin | 18.78 ± 0.61 * | 20.09 ± 0.29 * |
| 75 µg/mL of vancomycin | 19.54 ± 0.52 | 21.19 ± 0.76 * |
| 100 µg/mL of vancomycin | 20.27 ± 0.41 | 21.98 ± 0.57 |
| 125 µg/mL of vancomycin | 21.09 ± 0.41 * | 22.71 ± 0.61 * |
| 150 µg/mL of vancomycin | 21.32 ± 0.36 * | 23.36 ± 0.21 * |
| 15 µg/mL of P1 | 19.88 ± 0.16 | 21.98 ± 0.35 |
| Conditions | % Remaining Activity | |||
|---|---|---|---|---|
| S. aureus TISTR 517 | MRSA Isolate 2468 | |||
| P1 | Vancomycin | P1 | Vancomycin | |
| Untreated Sample | 100.00 ± 3.48 | 100.00 ± 4.74 | 100.00 ± 3.25 | 100.00 ± 3.24 |
| Thermal Stability | ||||
| Sample 60 °C, 1 h | 100.49 ± 0.84 | 99.25 ± 0.65 | 100.00 ± 0.79 | 98.30 ± 1.18 |
| Sample 80 °C, 1 h | 100.00 ± 0.84 | 97.39 ± 1.12 * | 99.08 ± 0.00 | 98.30 ± 1.56 |
| Sample 100 °C, 1 h | 100.00 ± 0.84 | 94.40 ± 1.29 * | 98.62 ± 0.79 | 96.94 ± 2.04 |
| Sample 121 °C, 15 psi, 15 min (autoclave) | 98.54 ± 1.69 | 91.79 ± 1.12 * | 98.62 ± 0.79 | 93.54 ± 1.56 * |
| Enzyme Stability | ||||
| Sample + Proteinase K | 94.20 ± 1.45 * | 100.99 ± 2.50 | 91.20 ± 0.80 * | 99.69 ± 1.95 |
| Sample + Trypsin | 94.20 ± 1.45 * | 101.32 ± 2.63 | 91.20 ± 0.80 * | 100.94 ± 2.17 |
| Sample + α-Chymotrypsin | 94.69 ± 1.67 * | 100.33 ± 0.99 | 94.44 ± 0.00 * | 99.06 ± 1.95 |
| Surfactant Stability | ||||
| Sample + 1% SDS | 114.49 ± 3.53 * | 97.82 ± 1.67 | 117.13 ± 1.60 * | 98.99 ± 0.59 |
| Sample + 1% Triton X-100 | 125.23 ± 2.92 * | 99.27 ± 2.18 | 115.74 ± 0.80 * | 98.99 ± 1.17 |
| 1% SDS | 114.95 ± 2.80 * | 89.45 ± 2.18 * | 112.96 ± 0.80 * | 82.43 ± 0.59 * |
| 1% Triton X-100 | 0.00 ± 0.00 * | 0.00 ± 0.00 * | 0.00 ± 0.00 * | 0.00 ± 0.00 * |
| pH Stability | ||||
| pH 2 | 98.48 ± 3.17 | 101.74 ± 1.60 | 97.57 ± 1.46 | 100.66 ± 1.15 |
| pH 4 | 97.97 ± 5.77 | 98.61 ± 1.21 | 99.03 ± 1.46 | 99.01 ± 0.57 |
| pH 5 | 97.46 ± 2.64 | 97.91 ± 1.21 | 97.09 ± 1.68 | 101.32 ± 1.72 |
| pH 6 | 97.97 ± 3.17 | 98.26 ± 2.09 | 97.57 ± 3.85 | 99.34 ± 1.99 |
| pH 7 | 97.97 ± 1.76 | 99.65 ± 0.60 | 97.57 ± 5.25 | 100.00 ± 2.48 |
| pH 8 | 102.54 ± 3.17 | 98.90 ± 1.90 | 97.09 ± 2.22 | 99.03 ± 0.56 |
| pH 9 | 100.00 ± 1.49 | 99.27 ± 1.68 | 98.03 ± 1.71 | 98.05 ± 1.12 |
| pH 11 | 98.01 ± 2.28 | 97.57 ± 3.66 | 98.03 ± 2.26 | 101.32 ± 2.05 |
| pH 12 | 96.52 ± 2.28 | 97.09 ± 2.18 | 97.54 ± 0.00 | 96.41 ± 1.50 * |
| pH 13 | 98.51 ± 1.49 | 93.82 ± 2.18 * | 99.51 ± 0.85 | 92.16 ± 0.98 * |
| pH 14 | 101.00 ± 1.72 | 88.00 ± 0.63 * | 100.00 ± 3.08 | 85.29 ± 1.70 * |
| Peptide | APD3 Database | Sequence | Sequence Similarity (%) | DBAASP Database | Sequence | Sequence Similarity (%) |
|---|---|---|---|---|---|---|
| P1 | Acidocin LCHV (Lactobacillus acidophilus n.v. Er 317/402 Narine) | NVGVLNPPPLV | 60.00 | Brevilaterin V (Brevibacillus laterosporus S62-9) | XVXVVVKVLKYLX | 53.30 |
| P2 | Bogorol L (Brevibacillus laterosporus MG64) | TVKIIVKVVKYLV | 78.60 | Brevilaterin B (Brevibacillus laterosporus S62-9) | XMXIVVKVLKYLX | 71.40 |
| P3 | Brevibacillin (Brevibacillus laterosporus OSY-I1) | TLKIIVKVVKYLV | 64.30 | Bogorol B (Brevibacillus laterosporus PNG-276) | XVXIVVKVLKYLX | 64.30 |
| P4 | Bogorol L (Brevibacillus laterosporus MG64) | TVKIIVKVVKYLV | 64.30 | Bogorol B (Brevibacillus laterosporus PNG-276) | XVXIVVKVLKYLX | 64.30 |
| P5 | Brevibacillin V (Brevibacillus laterosporus fmb70) | TLKIVVKVVKYLV | 64.30 | Bogorol B (Brevibacillus laterosporus PNG-276) | XVXIVVKVLKYLX | 64.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Songnaka, N.; Lertcanawanichakul, M.; Hutapea, A.M.; Krobthong, S.; Yingchutrakul, Y.; Atipairin, A. Purification and Characterization of Novel Anti-MRSA Peptides Produced by Brevibacillus sp. SPR-20. Molecules 2022, 27, 8452. https://doi.org/10.3390/molecules27238452
Songnaka N, Lertcanawanichakul M, Hutapea AM, Krobthong S, Yingchutrakul Y, Atipairin A. Purification and Characterization of Novel Anti-MRSA Peptides Produced by Brevibacillus sp. SPR-20. Molecules. 2022; 27(23):8452. https://doi.org/10.3390/molecules27238452
Chicago/Turabian StyleSongnaka, Nuttapon, Monthon Lertcanawanichakul, Albert M. Hutapea, Sucheewin Krobthong, Yodying Yingchutrakul, and Apichart Atipairin. 2022. "Purification and Characterization of Novel Anti-MRSA Peptides Produced by Brevibacillus sp. SPR-20" Molecules 27, no. 23: 8452. https://doi.org/10.3390/molecules27238452
APA StyleSongnaka, N., Lertcanawanichakul, M., Hutapea, A. M., Krobthong, S., Yingchutrakul, Y., & Atipairin, A. (2022). Purification and Characterization of Novel Anti-MRSA Peptides Produced by Brevibacillus sp. SPR-20. Molecules, 27(23), 8452. https://doi.org/10.3390/molecules27238452