

Azumamides A-E: Isolation, Synthesis, Biological Activity, and Structure–Activity Relationship


Abstract
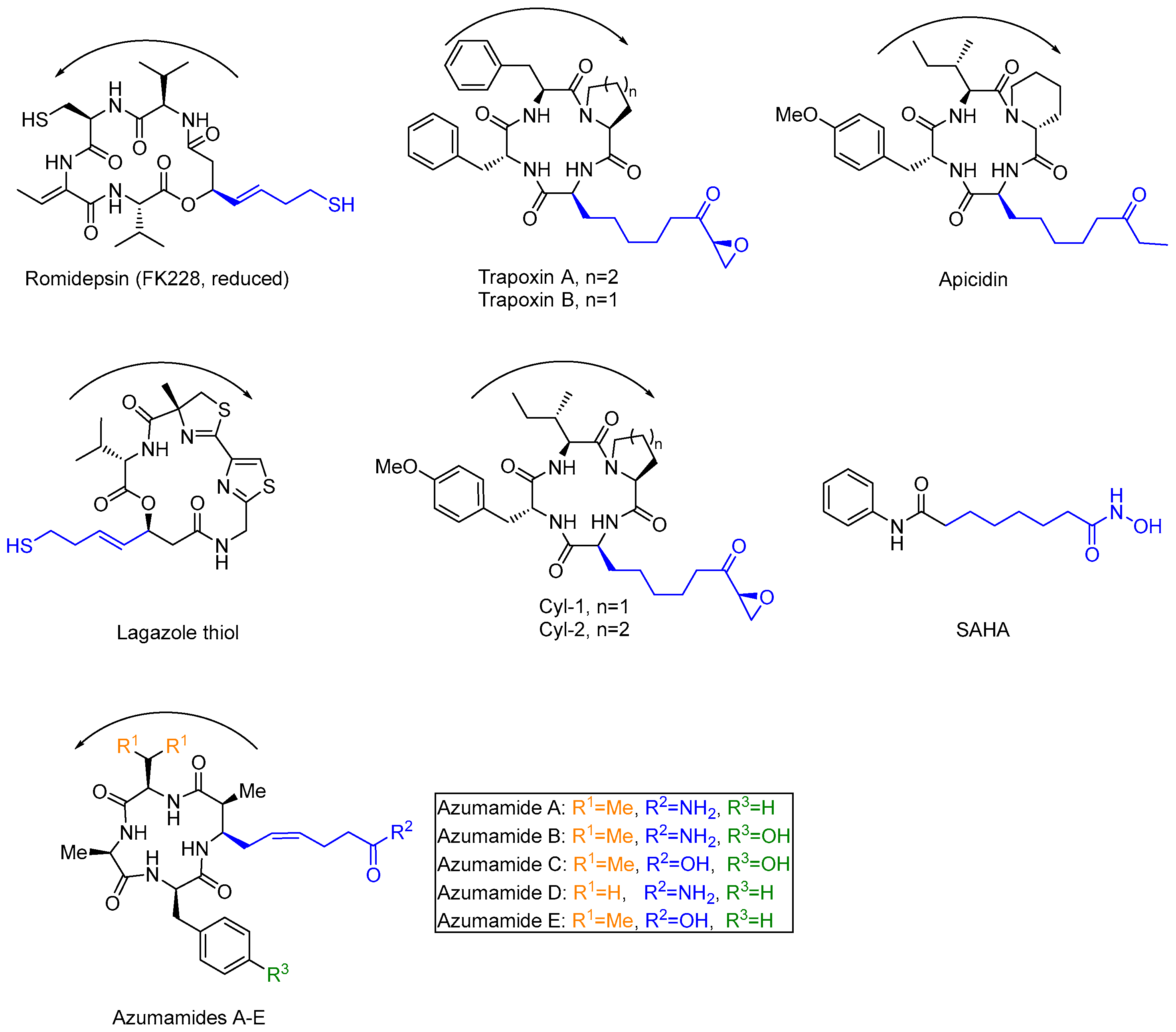
1. Introduction
2. Isolation and Structural Determination
3. Synthesis of Azumamides A-E
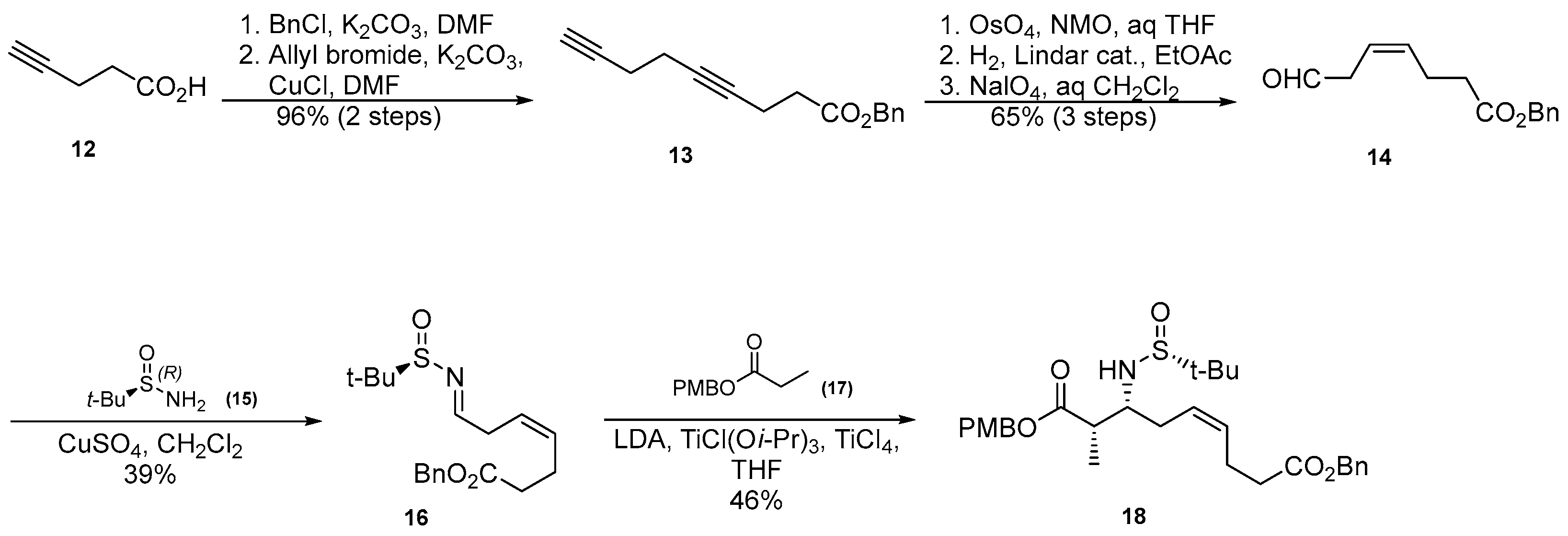
3.1. Synthesis of β-Amino Acids Amnna and Amnda
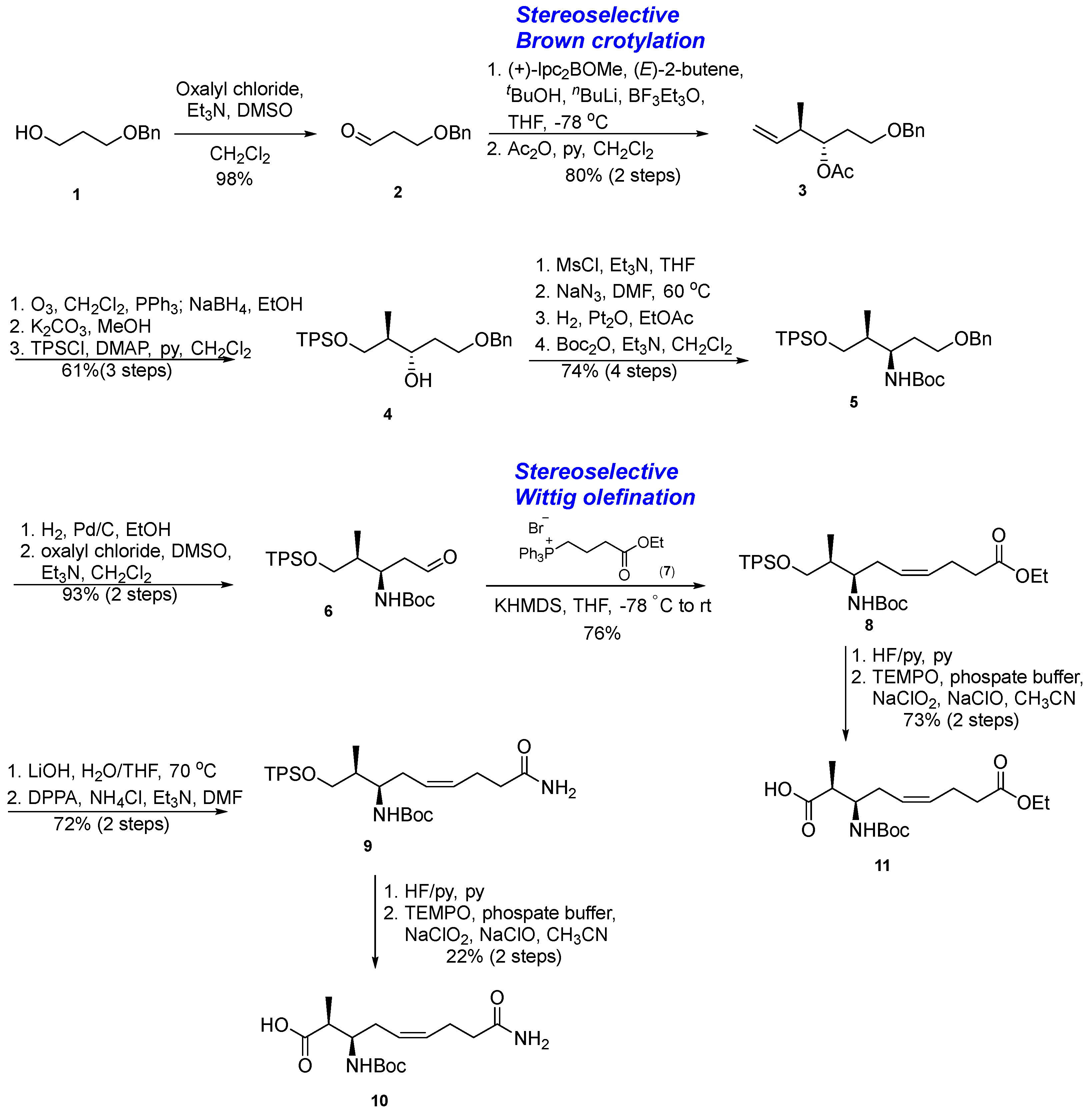
3.1.1. Synthesis of β-Amino Acid via a Stereoselective Brown’s Crotylboration and Wittig Olefination
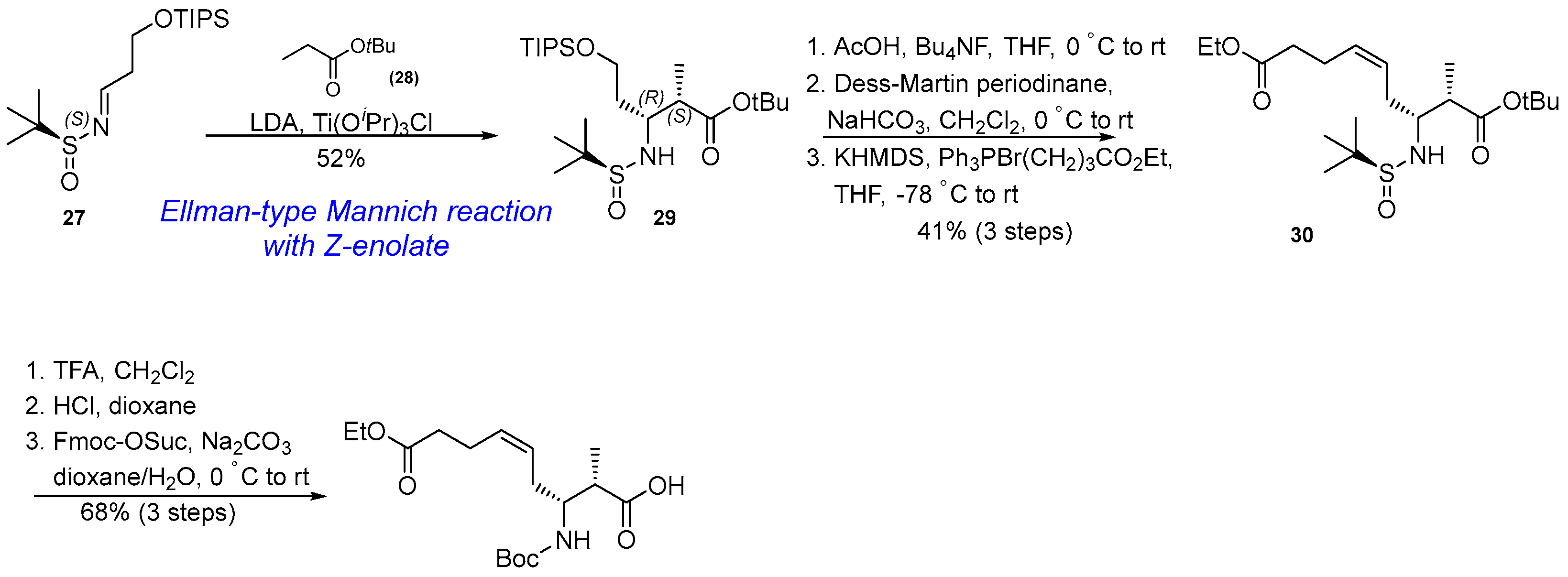
3.1.2. Synthesis of β-Amino Acid via Mannich Reaction with Ellman’s tert-Butylsulfinyl Auxiliary
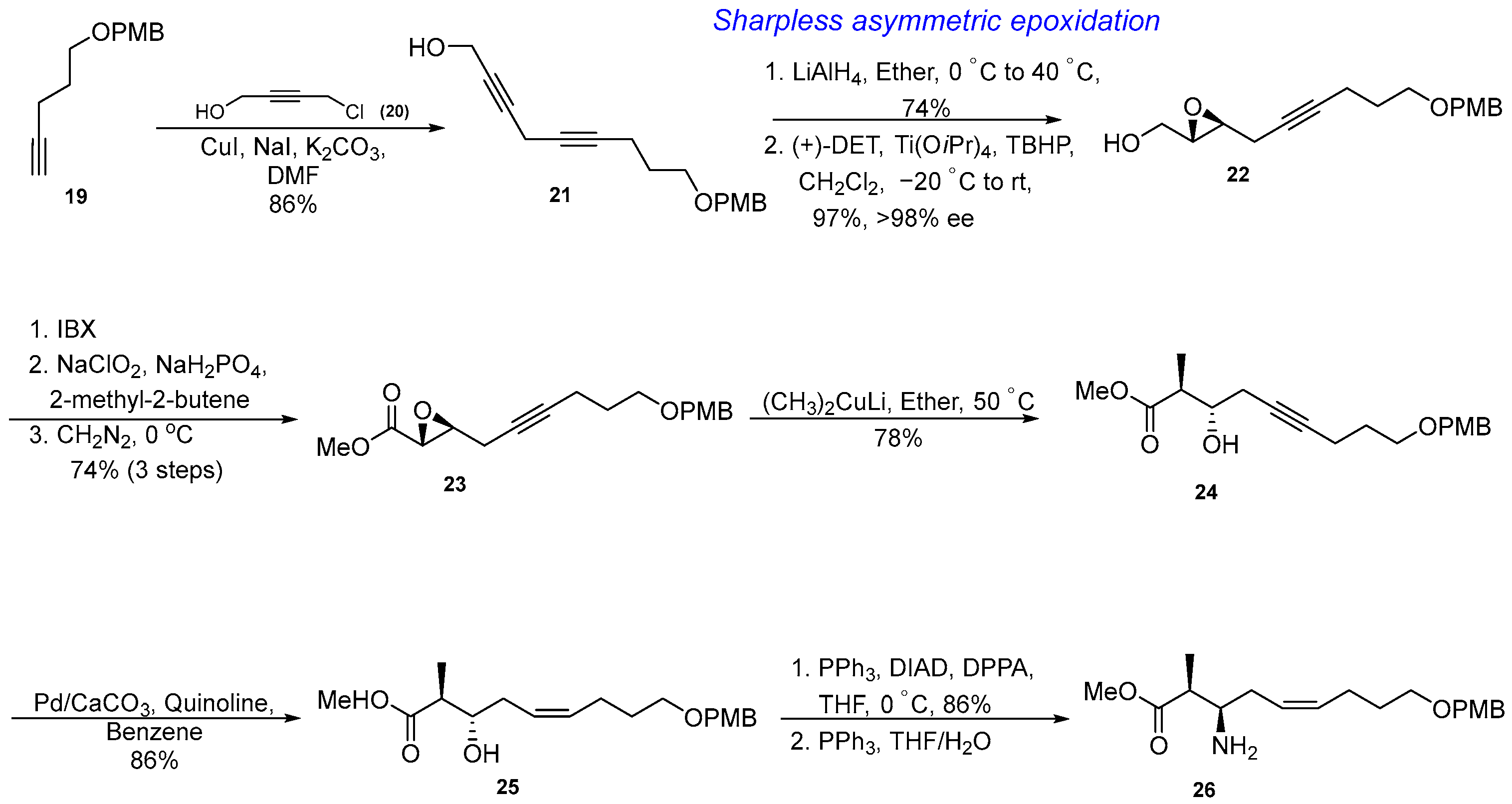
3.1.3. Synthesis of β-Amino Acid via an Asymmetric Epoxidation, Diastereo- and Regioselective Epoxide Opening, and Partial Reduction
3.1.4. Synthesis of β-Amino Acid via Ellman-Type Mannich Reaction and Wittig Olefination
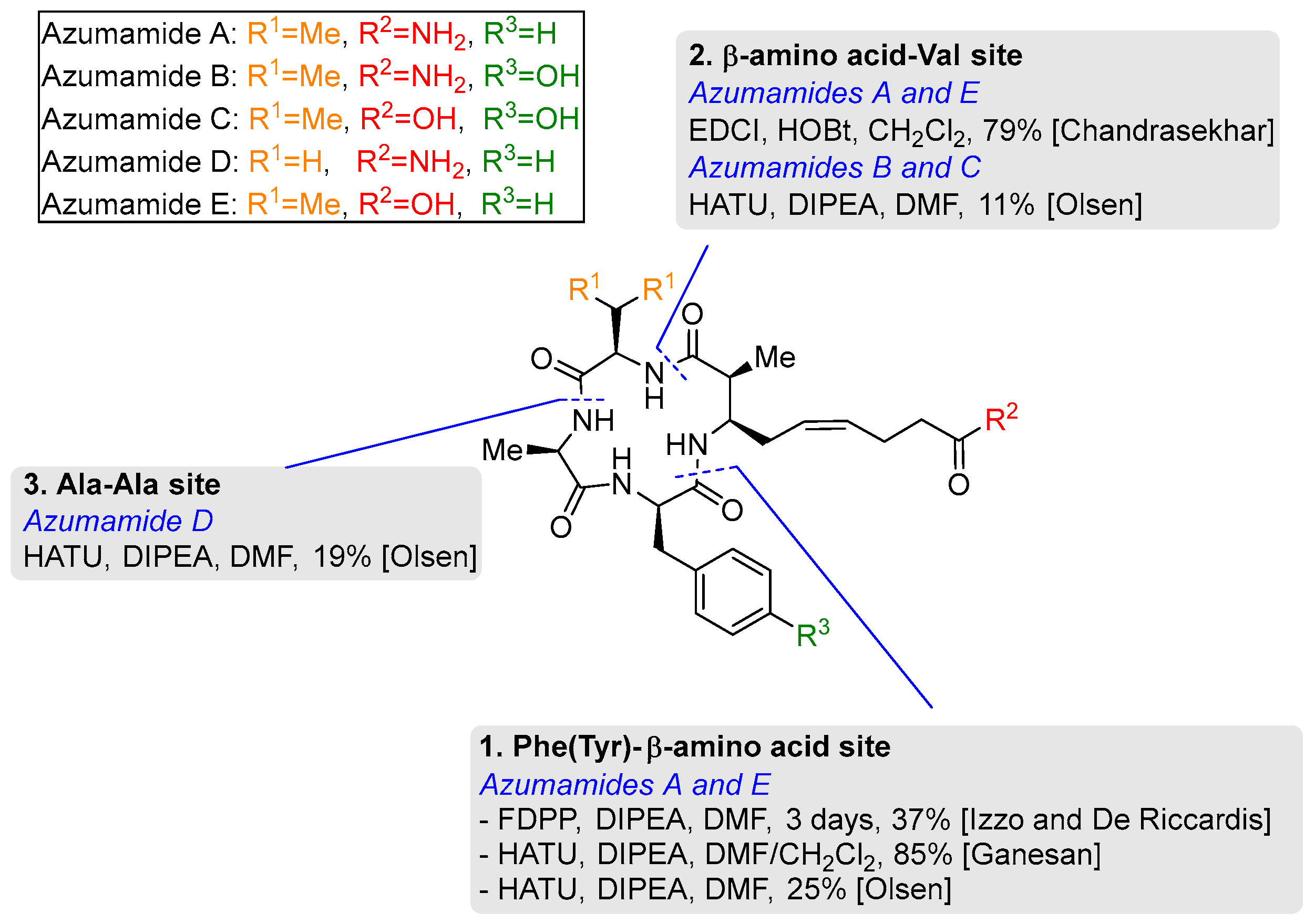
3.2. Macrocyclization of Azumamides A-E
3.2.1. Phe (Tyr)–β-Amino Acid Site
3.2.2. β-Amino Acid-Val Site
3.2.3. Ala-Ala Site
4. Biological Activity and Structure–Activity Relationship
4.1. Biological Role of Histone Deacetylase
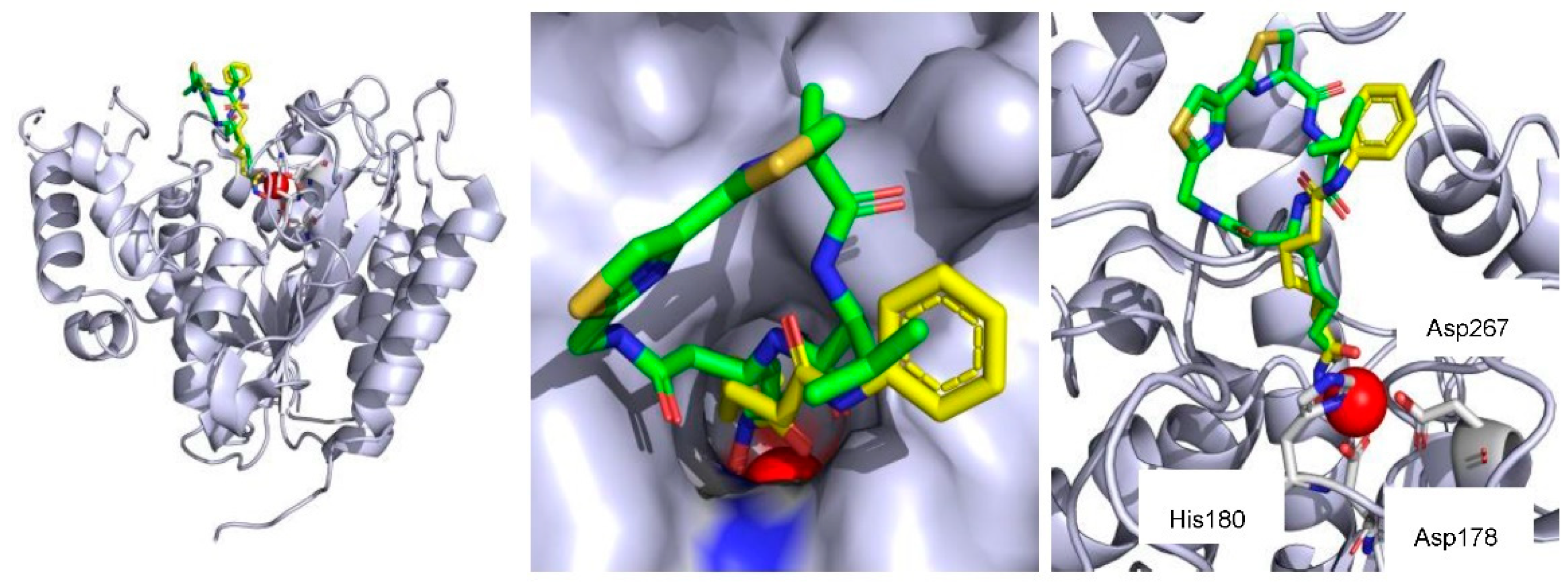
4.2. HDAC Inhibition of Azumamides and Structure–Activity Relationship
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Torres-Perez, J.V.; Irfan, J.; Febrianto, M.R.; Di Giovanni, S.; Nagy, I. Histone post-translational modifications as potential therapeutic targets for pain management. Trends Pharmacol. Sci. 2021, 42, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Fan, X.G.; Tang, D. Release and activity of histone in diseases. Cell Death Dis. 2014, 5, e1370. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y.W. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.T. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 1978, 17, 5524–5531. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef]
- Hotchkiss, R.D. The Quantitative Separation of Purines, Pyrimidines, and Nucleosides by Paper Chromatography. J. Biol. Chem. 1948, 175, 315–332. [Google Scholar] [CrossRef]
- Razin, A.; Riggs, A.D. DNA Methylation and Gene-Function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schioth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Brit. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a Novel Antitumor Bicyclic Depsipeptide Produced by Chromobacterium-Violaceum No-968.1. Taxonomy, Fermentation, Isolation, Physicochemical and Biological Properties, and Antitumor-Activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, N.; Nakajima, I.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Nakao, Y.; Yoshida, S.; Matsunaga, S.; Shindoh, N.; Terada, Y.; Nagai, K.; Yamashita, J.K.; Ganesan, A.; van Soest, R.W.M.; Fusetani, N. Azumamides A-E: Histone deacetylase inhibitory cyclic tetrapeptides from the marine sponge Mycale izuensis. Angew. Chem. Int. Ed. 2006, 45, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Izzo, I.; Maulucci, N.; Bifulco, G.; De Riccardis, F. Total synthesis of azumamides A and E. Angew. Chem. Int. Ed. 2006, 45, 7557–7560. [Google Scholar] [CrossRef]
- Wen, S.; Carey, K.L.; Nakao, Y.; Fusetani, N.; Packham, G.; Ganesan, A. Total synthesis of azumamide A and azumamide E, evaluation as histone deacetylase inhibitors, and design of a more potent analogue. Org. Lett. 2007, 9, 1105–1108. [Google Scholar] [CrossRef]
- Villadsen, J.S.; Stephansen, H.M.; Maolanon, A.R.; Harris, P.; Olsen, C.A. Total synthesis and full histone deacetylase inhibitory profiling of Azumamides A-E as well as beta(2)- epi-Azumamide E and beta(3)-epi-Azumamide E. J. Med. Chem. 2013, 56, 6512–6520. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, S.; Rao, C.L.; Seenaiah, M.; Naresh, P.; Jagadeesh, B.; Manjeera, D.; Sarkar, A.; Bhadra, M.P. Total synthesis of azumamide E and sugar amino acid-containing analogue. J. Org. Chem. 2009, 74, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Ellman, J.A. Applications of tert-butanesulfinamide in the asymmetric synthesis of amines. Pure Appl. Chem. 2003, 75, 39–46. [Google Scholar] [CrossRef]
- Diaz, D.D.; Betancort, J.M.; Crisostomo, F.R.P.; Martin, T.; Martin, V.S. Stereoselective synthesis of syn-2,7-disubstituted-4,5-oxepenes. Tetrahedron 2002, 58, 1913–1919. [Google Scholar] [CrossRef]
- Chong, J.M. Enantioselective Synthesis of Sitophilate, the Granary Weevil Aggregation Pheromone. Tetrahedron 1989, 45, 623–628. [Google Scholar] [CrossRef]
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Ellmeier, W.; Seiser, C. Histone deacetylase function in CD4(+) T cells. Nat. Rev. Immunol. 2018, 18, 617–634. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Evans, R.M. Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol. Cell 2000, 5, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Carbone, R.; Pearson, M.; Pruneri, G.; Viale, G.; Appella, E.; Pelicci, P.; Minucci, S. Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. Embo J. 2004, 23, 1144–1154. [Google Scholar] [CrossRef]
- San Jose-Eneriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef]
- Itazaki, H.; Nagashima, K.; Sugita, K.; Yoshida, H.; Kawamura, Y.; Yasuda, Y.; Matsumoto, K.; Ishii, K.; Uotani, N.; Nakai, H.; et al. Isolation and Structural Elucidation of New Cyclotetrapeptides, Trapoxin-a and Trapoxin-B, Having Detransformation Activities as Antitumor Agents. J. Antibiot. 1990, 43, 1524–1532. [Google Scholar] [CrossRef]
- Singh, S.B.; Zink, D.L.; Polishook, J.D.; Dombrowski, A.W.; DarkinRattray, S.J.; Schmatz, D.M.; Goetz, M.A. Apicidins: Novel cyclic tetrapeptides as coccidiostats and antimalarial agents from Fusarium pallidoroseum. Tetrahedron Lett. 1996, 37, 8077–8080. [Google Scholar] [CrossRef]
- Decroos, C.; Clausen, D.J.; Haines, B.E.; Wiest, O.; Williams, R.M.; Christianson, D.W. Variable Active Site Loop Conformations Accommodate the Binding of Macrocyclic Largazole Analogues to HDAC8. Biochemistry 2015, 54, 2126–2135. [Google Scholar] [CrossRef]
- Hirota, A.; Suzuki, A.; Suzuki, H.; Tamura, S. Isolation and Biological-Activity of Cyl-2, a Metabolite of Cylindrocladium-Scoparium. Agric. Biol. Chem. 1973, 37, 643–647. [Google Scholar] [CrossRef][Green Version]
- Takayama, S.; Isogai, A.; Nakata, M.; Suzuki, H.; Suzuki, A. Structure of Cyl-1, a Novel Cyclotetrapeptide from Cylindrocladium-Scoparium. Agric. Biol. Chem. 1984, 48, 839–842. [Google Scholar] [CrossRef]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, N.; Chini, M.G.; Di Micco, S.; Izzo, I.; Cafaro, E.; Russo, A.; Gallinari, P.; Paolini, C.; Nardi, M.C.; Casapullo, A.; et al. Molecular insights into azumamide E histone deacetylases inhibitory activity. J. Am. Chem. Soc. 2007, 129, 3007–3012. [Google Scholar] [CrossRef] [PubMed]
- Maolanon, A.R.; Villadsen, J.S.; Christensen, N.J.; Hoeck, C.; Friis, T.; Harris, P.; Gotfredsen, C.H.; Fristrup, P.; Olsen, C.A. Methyl Effect in Azumamides Provides Insight Into Histone Deacetylase Inhibition by Macrocycles. J. Med. Chem. 2014, 57, 9644–9657. [Google Scholar] [CrossRef] [PubMed]
- Villadsen, J.S.; Kitir, B.; Wich, K.; Friis, T.; Madsen, A.S.; Olsen, C.A. An azumamide C analogue without the zinc-binding functionality. MedChemComm 2014, 5, 1849–1855. [Google Scholar] [CrossRef]
- Fruhauf, A.; Meyer-Almes, F.J. Non-Hydroxamate Zinc-Binding Groups as Warheads for Histone Deacetylases. Molecules 2021, 26, 5151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | HDAC Inhibitors | Structure | HDAC Inhibition |
|---|---|---|---|
| Entry 1 | Azumamide A |  | Natural-HDACs b: 0.045 μM [17] |
| Synthetic HDAC1 c: >5 μM [21] HDAC2: >5 μM [21] HDAC3: 3.2 μM [21] HDACs: 5.8 μM [20] | |||
| Entry 2 | Azumamide B |  | Natural-HDACs: 0.11 μM [17] |
| Synthetic HDAC1: 5.0 μM [21] HDAC2: 3.0 μM [21] HDAC3: 3.0 μM [21] | |||
| Entry 3 | Azumamide C |  | Natural-HDACs: 0.11 μM [17] |
| Synthetic HDAC1: 0.032 μM [21] HDAC2: 0.040 μM [21] HDAC3: 0.014 μM [21] HDAC10: 0.010 μM [21] HDAC11: 0.035 μM [21] | |||
| Entry 4 | Azumamide D |  | Natural-HDACs: 1.3 μM [17] |
| Synthetic HDAC1: >5 μM [21] HDAC2: >5 μM [21] HDAC3: 3.7 μM [21] | |||
| Entry 5 | (+)-(2S,3R)-azumamide E [Natural] |  | Natural-HDACs: 0.033 μM [17] |
| Synthetic HDAC1: 0.067 μM [21] HDAC2: 0.050 μM [21] HDAC3: 0.025 μM [21] HDAC10: 0.020 μM [21] HDAC11: 0.060 μM [21] HDACs: 0.11 μM [20] | |||
| Entry 6 | Azumamide E hydroxamic acid |  | HDACs: 0.007 μM [20] |
| Entry 7 | Carboxylic acid- truncated azumamide C |  | HDAC1: 2.4 μM [44] HDAC2: 1.4 μM [44] HDAC3: 3.0 μM [44] |
| Entry 8 | β2-epi-azumamide E |  | HDAC1: N/A [21] HDAC2: N/A [21] HDAC3: N/A [21] |
| Entry 9 | β3-epi-azumamide E |  | HDAC1: N/A [21] HDAC2: N/A [21] HDAC3: N/A [21] |
| Entry 10 | (2R,3S)-azumamide E |  | HDACs: N/A at 50 μM [42] |
| Entry 11 | (-)-azumamide E |  | HDACs: 26.0 μM [42] |
| Entry 12 | Dimethyl azumamide E |  | HDAC1: 2–20%inhibition (10 μM) [43] HDAC2: <5%inhibition (10 μM) [43] HDAC3: <5%inhibition (10 μM) [43] |
| Entry 13 | Desmethyl azumamide E |  | HDAC1: 0.6 μM [43] HDAC2: 0.9 μM [43] HDAC3: 0.8 μM [43] HDAC10: 0.7 μM [43] HDAC11: 1.5 μM [43] |
| Entry 14 | Saturated desmethyl azumamide E |  | HDAC1: 0.9 μM [43] HDAC2: 0.8 μM [43] HDAC3: 0.7 μM [43] HDAC10: 1.0 μM [43] HDAC11: 2.0 μM [43] |
| Entry 15 | Azumamide E-SAA (sugar amino acid) analogue |  | HDACs: 48% inhibition (10 μM); 96% inhibition (20 μM) [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, S.; Kim, J.-H.; Lee, J.; Park, Y.; Jang, J. Azumamides A-E: Isolation, Synthesis, Biological Activity, and Structure–Activity Relationship. Molecules 2022, 27, 8438. https://doi.org/10.3390/molecules27238438
Jo S, Kim J-H, Lee J, Park Y, Jang J. Azumamides A-E: Isolation, Synthesis, Biological Activity, and Structure–Activity Relationship. Molecules. 2022; 27(23):8438. https://doi.org/10.3390/molecules27238438
Chicago/Turabian StyleJo, Sooheum, Jin-Hee Kim, Jiyeon Lee, Youngjun Park, and Jaebong Jang. 2022. "Azumamides A-E: Isolation, Synthesis, Biological Activity, and Structure–Activity Relationship" Molecules 27, no. 23: 8438. https://doi.org/10.3390/molecules27238438
APA StyleJo, S., Kim, J.-H., Lee, J., Park, Y., & Jang, J. (2022). Azumamides A-E: Isolation, Synthesis, Biological Activity, and Structure–Activity Relationship. Molecules, 27(23), 8438. https://doi.org/10.3390/molecules27238438