
Synthesis of Novel Cavitand Host Molecules via Palladium-Catalyzed Aryloxy- and Azidocarbonylation

Abstract
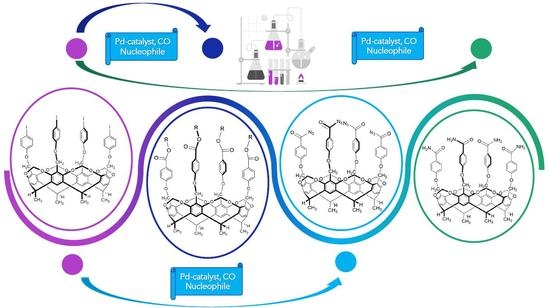
1. Introduction
2. Results and Discussion
2.1. Synthesis of Starting Material
2.2. Aryloxycarbonylation on Cavitand Scaffold
2.2.1. Effect of Temperature and Pressure on the Conversion
2.2.2. Aryloxycarbonylation with Ten Different Phenols
2.3. Azidocarbonylation on Cavitand Scaffold
3. Conclusions
4. Experimental
4.1. General Information
4.2. Synthesis and Characterization of Cavitand 2–13
Aryloxycarbonylation
Azidocarbonylation
- 2: Dark brown powder (49 mg, 54%), mp 281 °C. IR [cm−1] (KBr): 974, 1008, 1094, 1168, 1198, 1251, 1298, 1476, 1493, 1509, 1606, 1734, 2970 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.88 (12H, d, J 6.2 Hz, CH3CH ), 4.7 (4H, d, J 8.1 Hz, inner OCH2O), 5.06 (8H, s, ArCH2O), 5.14 (4H, q, J 6.3 Hz, CH3CH), 5.82 (4H, d, J 8.1 Hz, outer O2O), 7.03 (8H, d, J 6.0 Hz, Ar), 7.05–7.47 (24H, m, Ar), 8.19 (8H, d, J 8.3 Hz, Ar). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 46.2, 60.9 (OCH2O), 100.3 (ArCH2O), 114.8, 120.9, 121.7, 125.7, 128.3, 129.4, 132.4, 139.1, 151.0, 154.0, 162,8, 164.6 (ArC=O).
- 3: Light brown powder (34 mg, 32%), mp 287 °C. IR [cm−1] (KBr): 974, 1008, 1068, 1250, 1475, 1511, 1605, 1734, 2971 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.87 (12H, br s, CH3CH), 3.41 (8H, br s, ArCH2), 3.76 (12H, br s, ArOCH3), 4.68 (4 H, d, J 6.6 Hz, inner O2O), 5.03 (8H, s, Ar2O), 5.12 (4H, br s, CH3CH), 5.82 (4H, m, C=CH), 5.99 (4H, d J 6.9 Hz, outer OCH2O), 6.00 (4H, m, C=CH), 6.81 (4H, m, CH2CH=CH2), 7.03–8.2 (20H, m, Ar). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 40.1, 55.9, 60.6 (OCH2O), 100.1 (ArCH2O), 112.8, 114.2, 116.0, 120.9, 122.1, 122.6, 122.8, 132.5, 137.2, 138.3, 139.1, 151.2, 154.1, 162.7, 164.3 (ArC=O).
- 4: Beige powder (54 mg, 50%), mp 240 °C. IR [cm−1] (KBr): 974, 1008, 1068, 1250, 1475, 1510, 1604, 1701, 1735, 2942 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.87 (12H, br s, CH3CH), 3.85 (12H, s, AROCH3), 4.68 (4H, br s, inner OCH2O), 5.05 (8H, s, ArCH2), 5.12 (4H, br s, CH3CH), 5.81 (4H, br s, outer OCH2O), 7.03–8.17 (28H, m, Ar), 9.98 (4H, s, ArCHO). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.2 (CH3CH), 56.5, 60.9 (OCH2O, 100.0 (ArCH2O), 110.9, 114.2, 122.0, 124.6, 128.3, 129.2, 132.7, 139.1, 145.2, 12.3, 159.2, 165.6 (ArC=O), 190.9 (CHO).
- 5: Light brown powder (60.2 mg, 54%), mp 225 °C. IR [cm−1] (KBr): 976, 1008, 1057, 1240, 1255, 1511, 1605, 1701, 1733, 2972 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.89 (12H, d, J 7.3 Hz, CH3CH), 4.7 (4H, d J 7.0 Hz, inner OCH2O), 5.09 (8H, s, ArCH2), 5.16 (4H, q, J 7.1 Hz, CH3CH), 5.85 (4H, d J 7.1 Hz, outer OCH2O), 7.06 (8H, d, J 8.1 Hz, Ar), 7.46–7.81 (28H, m, Ar), 8.23 (8H, d, J 8.7 Hz, Ar). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 60.9 (OCH2O, 100.1 (ArCH2O), 114.5, 118.7, 120.9, 121.2, 121.3, 122.6, 125.5, 126.4, 127.9, 129.31, 131.4, 132.5, 133.8, 139.1, 148.6, 162.8, 164.8 (ArC=O).
- 6: Beige powder (70 mg, 50%), mp 235 °C. IR [cm−1] (KBr): 976, 1008, 1167, 1260, 1509, 1605, 1736, 2933 cm−1; 1H-NMR (500.15 MHz, CDCl3): 0.94 (12H, s, ArCH3), 1.86 (12H, d, J 6.7 Hz, CH3CH), 1.17–3.19 (48H, m, steroid skeleton protons), 4.65 (4H, d J 5.8 Hz, inner OCH2O), 5.1 (8H, s, ArCH2), 5.13 (4H, q, J 6.6 Hz, CH3CH), 5.81 (4H, d J 6.6 Hz, outer OCH2O), 6.92–7.35 (16H, m, Ar), 7.45 (4H, s Ar), 8.16 (8H, d, J 6.5 Hz, Ar). 13C-NMR (125.78 MHz, CDCl3): 8.5, 13.8, 16.2 (CH3CH), 21.6, 25.8, 26.39, 29.4, 32.3, 31.6 (CH3CH), 35.8, 38.0, 44.2, 47.9, 50.5, 60.8 (OCH2O, 100.0 (ArCH2O), 112.9, 114.3, 115.3, 118.9, 121.8, 128.3, 132.4, 137.9, 139.1, 148.9, 154.0, 432.8, 164.9 (ArC=O), 220.8 (C=O).
- 7: Beige powder (81.5 mg, 76%), mp 226 °C. IR [cm−1] (KBr): 973, 1068, 1168, 1161, 1267, 1460, 1604, 1734, 2949 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.87 (12H, d, J 7.1 Hz, CH3CH), 3.94 (12H, s, CH3COO), 4.69 (4H, d, J 6.8 Hz, inner OCH2O), 5.05 (8H, s, ArCH2O), 5.13 (4H, q, J 7.6 Hz, CH3CH), 5.81 (4H, d, J 7.6 Hz, outer O2O), 7.04 (8H, d, J 8.8 Hz, Ar), 7.25 (8H, d, J 9.6 Hz, Ar), 7.47 (4H, s, Ar), 8.07 (8H, d, J 8.1 Hz, Ar), 8.17 (8H, d, J 8.17 Hz, Ar). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 52.2 (OCH3), 60.9 (OCH2O), 100.4 (ArCH2O), 114.3, 121.03, 122.5 (overlapping signals), 122.6, 127.6, 131.1, 132.5, 139.1, 154.0, 154.9, 162.9, 164.0 (ArC=O), 164.3 (O=CCH3).
- 8: Beige powder (55 mg, 49%), mp 235 °C. IR [cm−1] (KBr): 978, 1008, 1094, 1255, 1509, 1736, 2971 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.89 (12H, d, J 7.5 Hz, CH3CH), 4.7 (4H, d, J 6.6 Hz, inner OCH2O), 5.04 (8H, s, ArCH2O), 5.15 (4H, q, J 6.6 Hz, CH3CH), 5.88 (4H, d, J 6.6 Hz, outer O2O), 7.12 (8H, d, J 9.8 Hz), 7.25–7.5 (28H, m, Ar), 7.72 (4H, d, J 8.9 Hz), 7.85 (8H, d, J 8.9 Hz), 8.31 (8H, d, J 8.9 Hz). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 60.9 (OCH2O), 100.0 (ArCH2O), 108.6, 114.1, 114.5, 118.2, 118.3, 121.0, 121.2, 122.0, 122.4, 125.4, 125.9, 126.4, 127.0, 132.6, 134.6, 139.2, 146.8, 154.0, 164.7 (ArC=O).
- 9: Light brown powder (75 mg, 70%), mp > 260 °C. IR [cm−1] (KBr): 974, 1007, 1165, 1251, 1272, 1300, 1478, 1604, 1701, 1740, 2971 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.87 (12H, d, J 7.7 Hz, CH3CH), 3.8 (12H, s, ArOCH3), 4.69 (4H, d, J 7.5 Hz, inner OCH2O), 5.05 (8H, s, ArCH2O), 5.14 (4H, q, J 7.3 Hz, CH3CH), 5.84 (4H, d, J 6.9 Hz, outer O2O), 7.02 (8H, d, Ar), 7.07 (8H, d, J 8.5 Hz), 7.22–7.47 (16H, m, Ar), 8.21 (8H, d, J 8.3 Hz, Ar), 10.15 (4H, s, ArCHO). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 52.2 (OCH3), 60.9 (OCH2O), 100.4 (ArCH2O), 114.1, 120.1, 121.0, 127.7, 131.2, 132.6, 138.3, 139.1, 154.0, 155.0, 162.9, 164.7 (ArC=O), 166.3 (ArCHO).
- 10: Beige powder (54 mg, 57%), mp > 260 °C. IR [cm−1] (KBr): 976, 1008, 1166, 1200, 1250, 1264, 1300, 1511, 1605, 1732, 2972 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.87 (12H, d, J 7.3 Hz, CH3CH), 2.37 (12H, s, ArCH3), 4.7 (4H, d, J 6.6 Hz, inner OCH2O), 5.04 (8H, s, ArCH2O), 5.13 (4H, q, J 7.1 Hz, CH3CH), 5.81 (4H, d, J 7.2 Hz, outer O2O), 7.01–7.04 (12H, m, Ar), 7.15 (8H, d, J 8.4 Hz, Ar), 7.46 (4H, s, Ar), 8.15 (8H, d, J 8.4 Hz, Ar). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 20.9, 31.3 (CH3CH), 60.8 (OCH2O), 100.0 (ArCH2O), 114.2, 120.8, 121.4, 122.6, 129.3, 132.4, 135.0, 139.1, 148.7, 154.0, 162.7, 164.8 (ArC=O).
- 11: Brown powder (102 mg), mp > 260 °C. IR [cm−1] (KBr): 978, 1008, 1064, 1266, 1475, 1606, 1744, 29720 cm−1; 1H-NMR (500.15 MHz, CDCl3): 1.85 (12H, br s, CH3CH), 2.09 (12H, s CH3), 2.10 (12H, s, CH3), 4.64 (4H, br s, inner OCH2O), 5.01 (8H, s, ArCH2O), 5.1 (4H, br s, CH3CH), 5.8 (4H, br s, outer O2O), 6.81–7.03 (20H, m, Ar), 7.45 (4H br s, Ar, 8.04 (4H, br s, Ar), 8.19 (8H, br s, Ar). 13C-NMR (125.78 MHz, CDCl3): 16.2 (CH3CH), 31.3 (CH3CH), 46.2, 60.9 (OCH2O), 100.3 (ArCH2O), 114.8, 120.9, 121.7, 125.7, 128.3, 129.4, 132.4, 139.1, 151.0, 154.1, 162,8, 164.6 (ArC=O).
- 12: Grey powder (60-80 mg, isolated from product mixture). IR [cm−1] (KBr): 973, 1090, 1160, 1245, 1482, 1597, 1685, 2134, 2876, 2968 cm−1; 1H-NMR (500.15 MHz, DMSO−d6): 1.89 (12H, d, J 6.9 Hz, CH), 4.44 (4H, q, J 7.6 Hz, CH3), 4.8–4.94 (12H, m, OO + ArO), 5.78 (4H, d, J 7.0 Hz, outer OO), 7.05 (8H, d, J 8.8 Hz, Ar), 7.9 (12H, m, Ar). 13C-NMR (125.78 MHz, DMSO−d6): 16.1 (CH3CH), 31.2 (CH3CH), 60.7 (ArO), 100.0 (OCH2O), 114.3, 116.9, 120.7, 131.9, 138.4, 154, 158.4, 163.5, 171.5 (C=O). MS: 1293.3 [M]+.
- 13: Light grey powder (59 mg, 75%), mp 240–250 °C. IR [cm−1] (KBr): 970, 1248, 1603, 1657, 2879, 2970 cm−1; 1H-NMR (500.15 MHz, DMSO−d6): 1.90 (12H, d, J 6.6 Hz, CH), 4.47 (4H, d, J 7.1 Hz, inner OO), 4.88 (2H, m, CH3 + ArO), 5.8 (4H, d, J 7.6 Hz, outer OO), 6.96 (8H, d, J 8.3 Hz, Ph), 7.1 (4H, br s), 7.8 (8H, d, J 8.3 Hz, Ph), 7.82 (4H, br s), 7.9 (4H, s). 13C-NMR (125.78 MHz, DMSO−d6): 16.5 (CH3CH), 31.8 (CH3CH), 60.9 (ArO), 99.8 (OCH2O), 114.4, 122.9, 127.1, 129.9 (overlapping signals), 139.5, 153.6, 161.2, 168.1 (C=O). MS: 1189.3 [M]+.
Author Contributions
Funding
Conflicts of Interest
References
- Cram, D.J.; Cram, J.M. Host-Guest Chemistry: Complexes between organic compounds simulate the substrate selectivity of enzymes. Science 1974, 183, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.R.; Karbach, S.; Cram, D.J. Cavitands: Synthetic molecular vessels. J. Am. Chem. Soc. 1982, 104, 5826–5828. [Google Scholar] [CrossRef]
- Cram, D.J. Molecular container compounds. Nature 1992, 356, 29. [Google Scholar] [CrossRef]
- Cram, D.J.; Cram, J.M. Container Molecules and Their Guests; Royal Society of Chemistry: Cambridge, UK, 1997. [Google Scholar]
- Timmerman, P.; Verboom, W.; Reinhoudt, D.N. Resorcinarenes. Tetrahedron 1996, 52, 2663–2704. [Google Scholar] [CrossRef]
- Ma, S.; Rudkevich, D.M.; Rebek, J. “Deep-Cavity” resorcinarenes dimerize through hydrogen bonding and self-inclusion. J. Am. Chem. Soc. 1998, 120, 4977–4981. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Schultheiss, N.; Desper, J. C-Pentyltetra (3-pyridyl) cavitand: A versatile building block for the directed assembly of hydrogen-bonded heterodimeric capsules. Org. Lett. 2006, 8, 2607–2610. [Google Scholar] [CrossRef]
- Hass, O.; Schierholt, A.; Jordan, M.; Lützen, A. Improved synthesis of monohalogenated cavitands and their use in the synthesis of further functionalized cavitands. Synthesis 2006, 2006, 519–527. [Google Scholar]
- Lin, Z.; Emge, T.J.; Warmuth, R. Multicomponent Assembly of Cavitand-Based Polyacylhydrazone Nanocapsules. Chem.-Eur. J. 2011, 17, 9395–9405. [Google Scholar] [CrossRef]
- Yu, J.T.; Huang, Z.T.; Zheng, Q.Y. Synthesis, structure, fullerene-binding and resolution of C 3-symmetric cavitands with rigid and deep cavities. Org. Biomol. Chem. 2012, 10, 1359–1364. [Google Scholar] [CrossRef]
- Ogoshi, T. Synthesis of novel pillar-shaped cavitands “Pillar [5] arenes” and their application for supramolecular materials. J. Incl. Phenom. Macrocycl. Chem. 2012, 72, 247–262. [Google Scholar] [CrossRef]
- Sémeril, D.; Matt, D.; Ramesh, R. Synthesis of the first resorcin [4] arene-functionalized triazolium salts and their use in Suzuki–Miyaura cross-coupling reactions. Catalysts 2019, 9, 388. [Google Scholar] [CrossRef]
- Kégl, T.R.; Kégl, T. Palladium-catalyzed carbonylative synthesis and theoretical study of elongated tubular cavitands. J. Organomet. Chem. 2020, 923, 121387. [Google Scholar] [CrossRef]
- Jánosi, T.Z.; Makkai, G.; Kégl, T.; Mátyus, P.; Kollár, L.; Erostyák, J. Light-Enhanced Fluorescence of Multi-Level Cavitands Possessing Pyridazine Upper rim. J. Fluoresc. 2016, 26, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Kégl, T.; Csekő, G.; Mikle, G.; Takátsy, A.; Kollár, L.; Kégl, T. The Role of Weak Interactions in Supramolecular Compounds: A Synthetic and Theoretical Study of Novel Elongated Cavitands. ChemistrySelect 2017, 2, 8337–8345. [Google Scholar] [CrossRef]
- Csók, Z.; Takátsy, A.; Kollár, L. Highly selective palladium-catalyzed aminocarbonylation and cross-coupling reactions on a cavitand scaffold. Tetrahedron 2012, 68, 2657–2661. [Google Scholar] [CrossRef]
- Wan, Y.; Song, F.; Ye, T.; Li, G.; Liu, D.; Lei, Y. Carbonylative Suzuki coupling and alkoxycarbonylation of aryl halides using palladium supported on phosphorus-doped porous organic polymer as an active and robust catalyst. Appl. Organomet. Chem. 2019, 33, e4714. [Google Scholar] [CrossRef]
- Geng, H.Q.; Wu, X.F. Copper-Catalyzed Alkoxycarbonylation of Alkyl Iodides for the Synthesis of Aliphatic Esters: Hydrogen Makes the Difference. Org. Lett. 2021, 23, 8062–8066. [Google Scholar] [CrossRef]
- Mikle, G.; Noveczky, P.; Mahó, S.; Kollár, L. Palladium-catalysed amino-vs. Alkoxycarbonylation of iodoalkenes using bifunctional N, O-nucleophiles. Tetrahedron 2021, 85, 132050. [Google Scholar] [CrossRef]
- Liu, N.; Wu, X.; Wang, C.; Qu, J.; Chen, Y. Nickel-catalyzed alkoxycarbonylation of aryl iodides with 1 atm CO. Chem. Commun. 2022, 58, 4643–4646. [Google Scholar] [CrossRef]
- Kégl, T.R.; Mika, L.T.; Kégl, T. 27 Years of Catalytic Carbonylative Coupling Reactions in Hungary (1994–2021). Molecules 2022, 27, 460. [Google Scholar] [CrossRef]
- Brennführer, A.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylation reactions of aryl halides and related compounds. Angew. Chem. Int. Ed. Eng. 2009, 48, 4114–4133. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylative coupling reactions between Ar–X and carbon nucleophiles. Chem. Soc. Rev. 2011, 40, 4986–5009. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Ikeda, M.; Miura, M.; Nomura, M. Palladium-catalyzed phenoxycarbonylation of aryl iodides: Electronic effect of the substituents on phenol. J. Mol. Catal. A-Chem. 1996, 111, 25–31. [Google Scholar] [CrossRef]
- Watson, D.A.; Fan, X.; Buchwald, S.L. Carbonylation of aryl chlorides with oxygen nucleophiles at atmospheric pressure. Preparation of phenyl esters as acyl transfer agents and the direct preparation of alkyl esters and carboxylic acids. J. Org. Chem. 2008, 73, 7096–7101. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Marton, B.; Albert, E.; Tóth, I.; Mika, L.T. Palladium-catalyzed aryloxy-and alkoxycarbonylation of aromatic iodides in γ-valerolactone as bio-based solvent. J. Organomet. Chem. 2020, 923, 121407. [Google Scholar] [CrossRef]
- Szuroczki, P.; Molnár, L.; Dörnyei, Á.; Kollár, L. Facile, High-Yielding Synthesis of 4-Functionalised 1, 2, 3-Triazoles via Amino-and Aryloxycarbonylation. ChemistrySelect 2020, 5, 448–451. [Google Scholar] [CrossRef]
- Seni, A.A.; Kollar, L.; Mika, L.T.; Pongracz, P. Rhodium-catalysed aryloxycarbonylation of iodo-aromatics by 4-substituted phenols with carbon monoxide or paraformaldehyde. Mol. Catal. 2018, 457, 67–73. [Google Scholar] [CrossRef]
- Miloserdov, F.M.; Grushin, V.V. Palladium-Catalyzed Aromatic Azidocarbonylation. Angew. Chem. Int. Ed. Eng. 2012, 51, 3668–3672. [Google Scholar] [CrossRef]
- Miloserdov, F.M.; McMullin, C.L.; Belmonte, M.M.; Benet-Buchholz, J.; Bakhmutov, V.I.; Macgregor, S.A.; Grushin, V.V. The challenge of palladium-catalyzed aromatic azidocarbonylation: From mechanistic and catalyst deactivation studies to a highly efficient process. Organometallics 2014, 33, 736–752. [Google Scholar] [CrossRef]
- Yadav, V.K.; Srivastava, V.P.; Yadav, L.D.S. One-Pot Synthesis of Carbamoyl Azides via Palladium-Catalysed Azidocarbonylation of Haloarenes Using N-Formylsaccharin as a CO Surrogate. Synlett 2016, 27, 2826–2830. [Google Scholar] [CrossRef]
- Li, M.; Yu, F.; Chen, P.; Liu, G. Palladium-catalyzed intermolecular azidocarbonylation of alkenes via a cooperative strategy. J. Org. Chem. 2017, 82, 11682–11690. [Google Scholar] [CrossRef] [PubMed]
- Mikle, G.; Skoda-Földes, R.; Kollar, L. Amino-and azidocarbonylation of iodoalkenes. Tetrahedron 2021, 100, 132495. [Google Scholar] [CrossRef]
- Sharma, A.K.; Bhattacherjee, D.; Sharma, N.; Giri, K.; Das, P. Supported-Pd catalyzed tandem approach for N-arylbenzamides synthesis. Mol. Catal. 2021, 516, 111948. [Google Scholar]
- Csók, Z.; Kégl, T.; Li, Y.; Skoda-Földes, R.; Kiss, L.; Kunsági-Máté, S.; Todd, M.H.; Kollár, L. Synthesis of elongated cavitands via click reactions and their use as chemosensors. Tetrahedron 2013, 69, 8186–8190. [Google Scholar] [CrossRef]
- Tunstad, L.M.; Tucker, J.A.; Dalcanale, E.; Weiser, J.; Bryant, J.A.; Sherman, J.C.; Helgeson, R.C.; Knobler, C.B.; Cram, D.J. Host-guest complexation. 48. Octol building blocks for cavitands and carcerands. J. Org. Chem. 1989, 54, 1305–1312. [Google Scholar] [CrossRef]
- Román, E.; Peinador, C.; Mendoza, S.; Kaifer, A.E. Improved Synthesis of Cavitands. J. Org. Chem. 1999, 64, 2577–2578. [Google Scholar] [CrossRef]
- Sorrell, T.N.; Pigge, F.C. A convenient synthesis of functionalized cavitands via free-radical bromination. J. Org. Chem. 1993, 58, 784–785. [Google Scholar] [CrossRef]
- Csók, Z.; Kégl, T.; Párkányi, L.; Varga, Á.; Kunsági-Máté, S.; Kollár, L. Facile, high-yielding synthesis of deepened cavitands: A synthetic and theoretical study. Supramol. Chem. 2011, 23, 710–719. [Google Scholar] [CrossRef]
- Körner, S.K.; Tucci, F.C.; Rudkevich, D.M.; Heinz, T.; Rebek Jr, J. A Self-Assembled Cylindrical Capsule: New Supramolecular Phenomena through Encapsulation. Chem. Eur. J. 2000, 6, 187–195. [Google Scholar] [CrossRef]
- Gangemi, C.M.A.; Pappalardo, A.; Trusso Sfrazzetto, G. Assembling of supramolecular capsules with resorcin [4] arene and calix [n] arene building blocks. Curr. Org. Chem. 2015, 19, 2281–2308. [Google Scholar] [CrossRef]
- Galan, A.; Ballester, P. Stabilization of reactive species by supramolecular encapsulation. Chem. Soc. Rev. 2016, 45, 1720–1737. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Temperature (℃) | Phosphine | Conversion (%) |
|---|---|---|---|
| 1 | 50 | PPh3 | <5 |
| 2 | 75 | PPh3 | 32 |
| 3 | 100 | PPh3 | 100 |
| 4 | 50 | Xantphos | <5 |
| 5 | 75 | Xantphos | 38 |
| 6 | 100 | Xantphos | 100 |
| Run | Cavitand | Phosphine | Chemoselectivity (%) |
|---|---|---|---|
| 1 | 2 | PPh3 | 72 |
| 2 | 2 | Xantphos | 85 |
| 3 | 3 | PPh3 | 65 |
| 4 | 3 | Xantphos | 77 |
| 5 | 4 | PPh3 | 83 |
| 6 | 4 | Xantphos | 77 |
| 7 | 5 | PPh3 | 68 |
| 8 | 5 | Xantphos | 78 |
| 9 | 6 | PPh3 | 75 |
| 10 | 6 | Xantphos | 72 |
| 11 | 7 | PPh3 | 82 |
| 12 | 7 | Xantphos | 100 |
| 13 | 8 | PPh3 | 58 |
| 14 | 8 | Xantphos | 74 |
| 15 | 9 | PPh3 | 70 |
| 16 | 9 | Xantphos | 90 |
| 17 | 10 | PPh3 | 90 |
| 18 | 10 | Xantphos | 75 |
| Run | Temp. (℃) | Cat. Ratio (mol%) | Reac. Time | Conv. (%) | Chemosel. (%) | Product |
|---|---|---|---|---|---|---|
| 1 | 50 | 2 | 16 h | 100 | 100 | 13 |
| 2 | rt | 4 | 16 h | 51 | 100 | 12 |
| 3 | rt | 8 | 16 h | 100 | 66 | 12 + 13 |
| 4 | rt | 2 | 2 days | 32 | 100 | 12 |
| 5 | rt | 8 | 2 days | 100 | 63 | 12 + 13 |
| 6 | rt | 8 | 3 days | 100 | 59 | 12 + 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akash; Kollár, L.; Kégl, T.R. Synthesis of Novel Cavitand Host Molecules via Palladium-Catalyzed Aryloxy- and Azidocarbonylation. Molecules 2022, 27, 8404. https://doi.org/10.3390/molecules27238404
Akash, Kollár L, Kégl TR. Synthesis of Novel Cavitand Host Molecules via Palladium-Catalyzed Aryloxy- and Azidocarbonylation. Molecules. 2022; 27(23):8404. https://doi.org/10.3390/molecules27238404
Chicago/Turabian StyleAkash, László Kollár, and Tímea R. Kégl. 2022. "Synthesis of Novel Cavitand Host Molecules via Palladium-Catalyzed Aryloxy- and Azidocarbonylation" Molecules 27, no. 23: 8404. https://doi.org/10.3390/molecules27238404
APA StyleAkash, Kollár, L., & Kégl, T. R. (2022). Synthesis of Novel Cavitand Host Molecules via Palladium-Catalyzed Aryloxy- and Azidocarbonylation. Molecules, 27(23), 8404. https://doi.org/10.3390/molecules27238404