
Reactions of 3-Hydroxy-2-phenyl-1H-benzo[e]isoindol-1-one: A Route to 3-Hydroxy-/3-anilinobenzo[e]indan-1-ones and Benzo[f]phthalazin-1(2H)-ones


Abstract
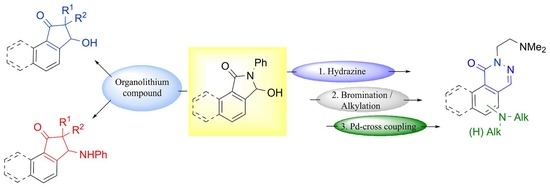
1. Introduction
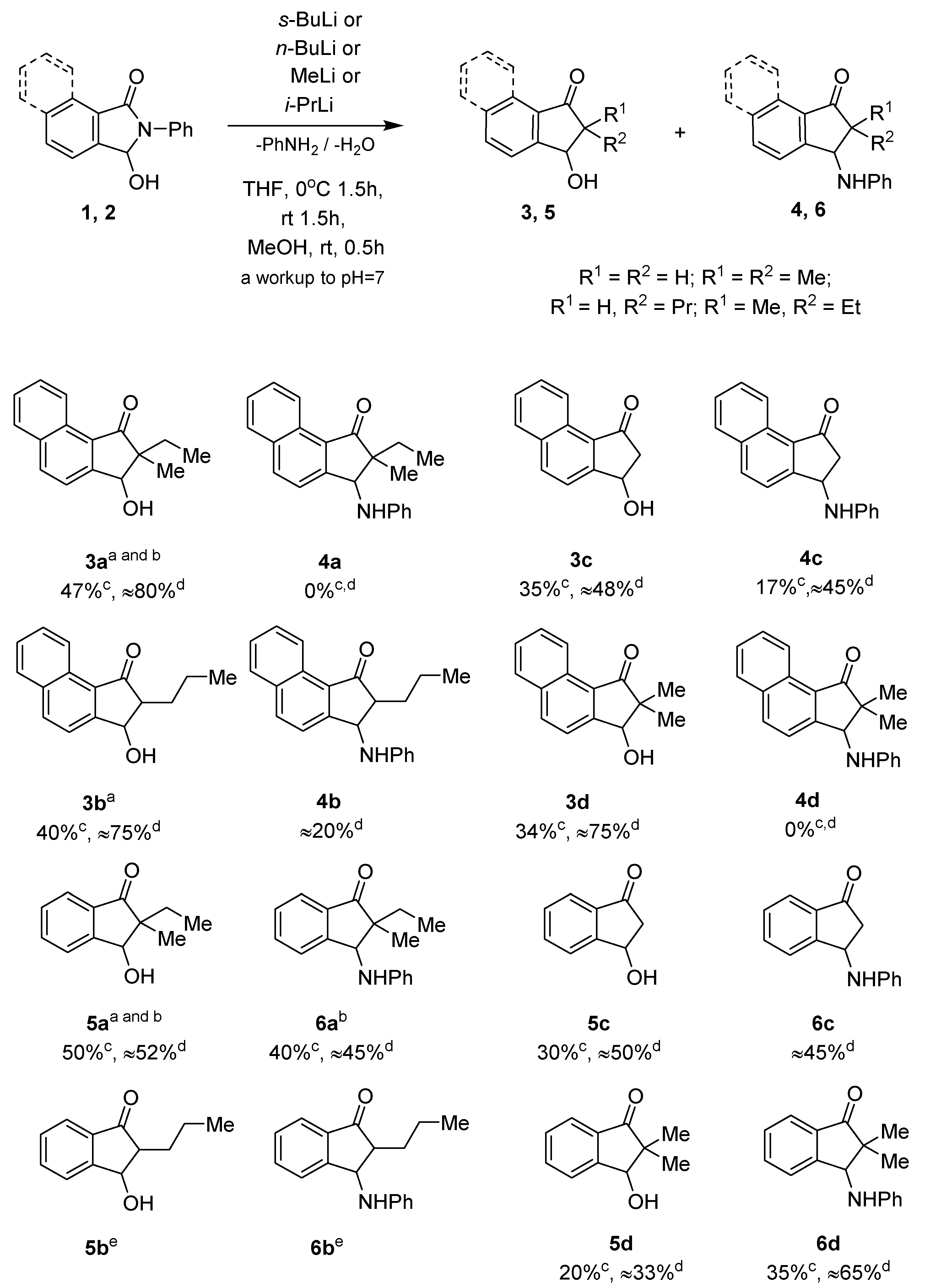
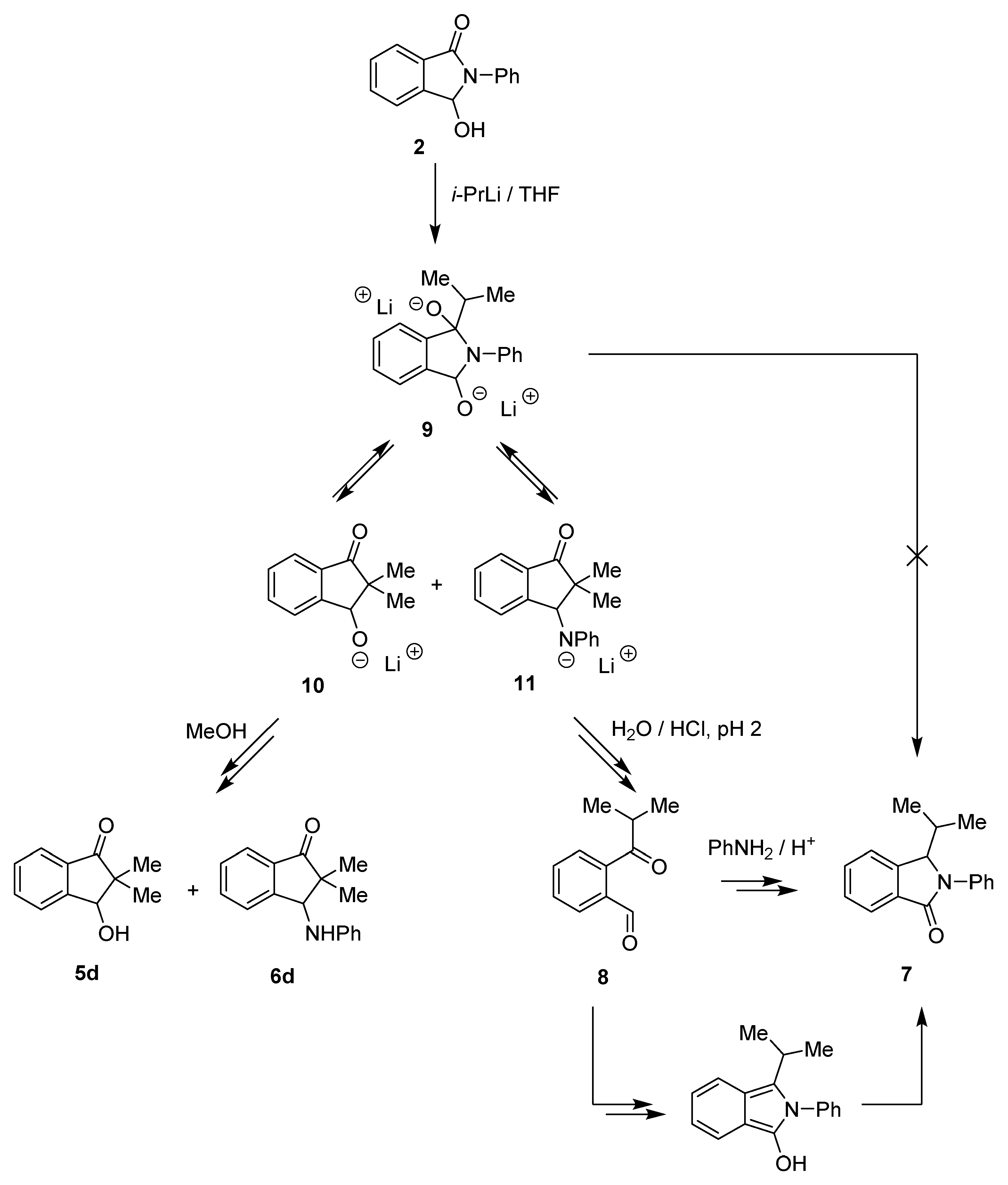
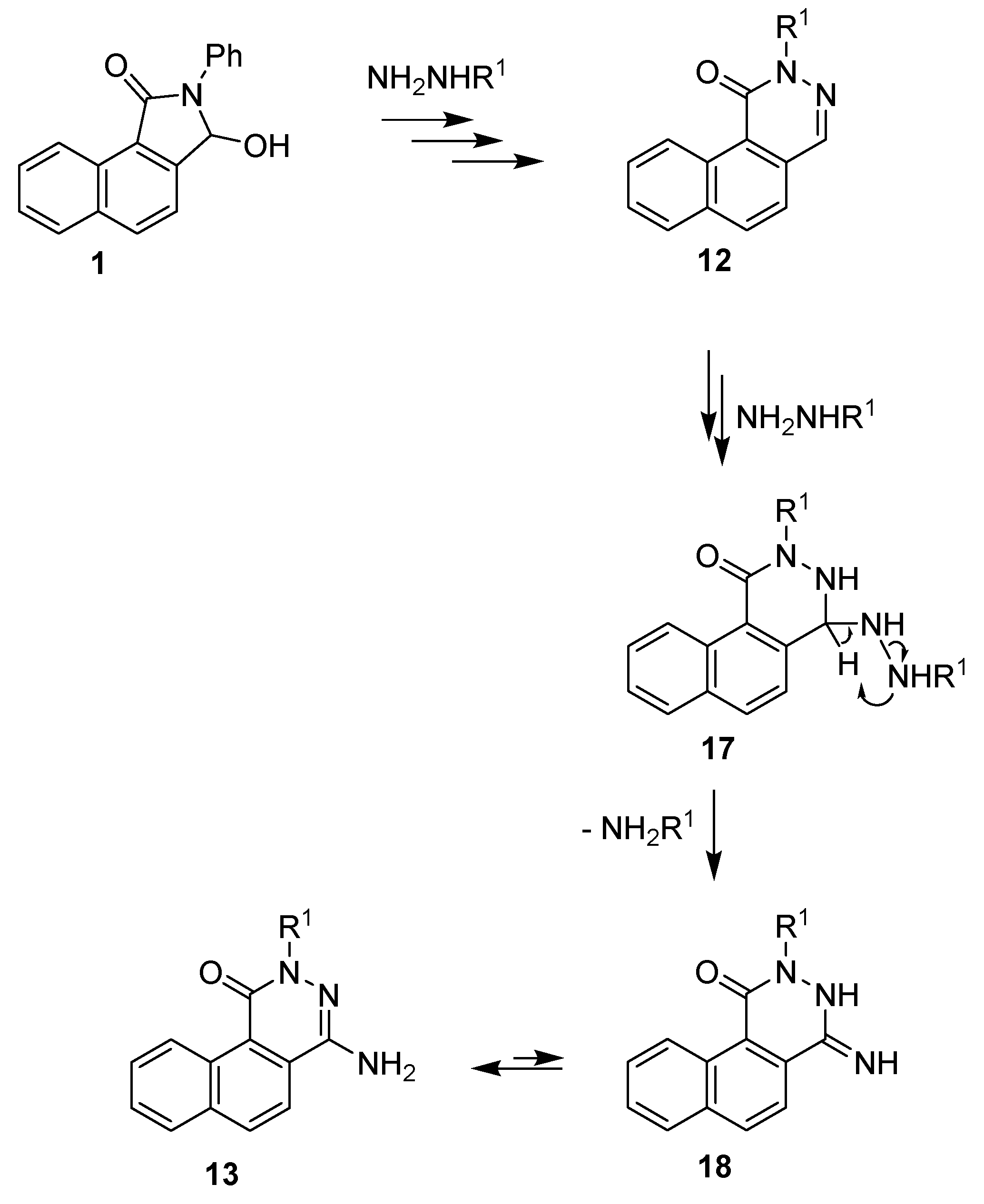
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 3-Hydroxy-2-phenyl-2,3-dihydro-1H-benzo[e]isoindol-1-one (1)
3.3. Synthesis of 3-Hydroxy-2,3-dihydro-1H-inden-1-one 3 and 5 and 3-Anilino-2,3-dihydro-1H-inden-1-one 4 and 6 Derivatives
3.4. Synthesis of benzo[f]phthalazin-1(2H)-ones 12 and 4-amino-benzo[f]phthalazin-1(2H)-ones 13
3.5. Synthesis of 7-bromo-2-[2-(dimethylamino)ethyl]benzo[f]phthalazin-1(2H)-one (15)
3.6. Synthesis of 2-[2-(dimethylamino)ethyl]-7-(morpholin-4-yl)benzo[f]phthalazin-1(2H)-one (16)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Speck, K.; Magauer, T. The chemistry of isoindole natural products. Beilstein J. Org. Chem. 2013, 9, 2048–2078. [Google Scholar] [CrossRef] [PubMed]
- Prakash, C.R.; Raja, S. Indolinones as promising scaffold as kinase inhibitors: A review. Mini Rev. Med. Chem. 2012, 12, 98–119. [Google Scholar] [CrossRef] [PubMed]
- Csende, F.; Porkolab, A. A review on antibacterial activity of some isoindole derivatives. Der Pharma. Chem. 2018, 10, 43–50. Available online: https://www.derpharmachemica.com/pharma-chemica/a-review-on-antibacterial-activity-of-some-isoindole-derivatives.pdf (accessed on 25 November 2022).
- Omura, S.; Sasaki, Y.; Iwai, Y.; Takeshima, H. Staurosporine, a potentially important gift from a microorganism. J. Antibiot. 1995, 48, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Nishiyama, T.; Yamada, Y. The antinociceptive effects and pharmacological properties of JM-1232(-): A novel isoindoline derivative. Anesth. Analg. 2009, 108, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, I.; Masayuki, S.; Yonamire, R.; Kazama, T. The effect of a new water-soluble sedative-hypnotic drug, JM-1232(−), on long-term potentiation in the CA1 region of the mouse hippocampus. Anesth. Analg. 2011, 113, 1043–1049. [Google Scholar] [CrossRef]
- de Wit, H.; Vieni, L.; Haig, G.M.; Hunt, T. Evaluation of the abuse potential of pagoclone, a partial GABAA agonist. J. Clin. Psychopharmacol. 2006, 26, 268–273. [Google Scholar] [CrossRef]
- Sorbera, L.A.; Leeson, P.A.; Silvestre, J.S.; Castaner, J. Pagoclone. Drugs Fut. 2001, 26, 651–657. [Google Scholar] [CrossRef]
- Alvarez-Mico, X.; Jensen, P.R.; Fenical, W.; Hughes, C.C. Chlorizidine, a cytotoxic 5H-pyrrolo[2,1-a]isoindol-5-one containing alkaloid from a Marine Streptomyces sp. Org. Lett. 2013, 15, 988–991. [Google Scholar] [CrossRef]
- Teryuya, T.; Nakagawa, S.; Koyama, T.; Arimoto, H.; Kita, M.; Uemura, D. Nakiterpiosin and nakiterpiosinone, novel cytotoxic C-nor-D-homosteroids from the Okinawan sponge Terpios hoshinota. Tetrahderon 2004, 60, 6989–6993. [Google Scholar] [CrossRef]
- Gao, S.; Wang, Q.; Chen, C. Synthesis and structure revision of nakiterpiosin. J. Am. Chem. Soc. 2009, 131, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Patil, R.; Patil, S.A. Recent developments in biological activities of indanones. Eur. J. Med. Chem. 2017, 138, 182–198. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. Available online: https://www.dovepress.com/getfile.php?fileID=939 (accessed on 25 November 2022). [PubMed]
- Ernest-Russell, M.A.; Chai, C.L.L.; Wardlaw, J.H.; Elix, J.A. Euplectin and Coneuplectin, new naphthopyrones from the lichen Flavoparmelia euplecta. J. Nat. Prod. 2000, 63, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Brzeziński, J.Z.; Epsztajn, J.; Bakalarz, A.D.; Łajszczak, A.; Malinowski, Z. Application of organolithium and related reagents in synthesis. Part 221. Synthetic strategies bsed on ortho-aromatic metallation. A concise regiospecific conversion of benzoic acids into the 4-pyridyl-2H-phthalazin-1-ones. Synth. Commun. 1999, 29, 457–473. [Google Scholar] [CrossRef]
- Epsztajn, J.; Malinowski, Z.; Urbaniak, P.; Andrijewski, G. A practical approach for preparation of 2-[(dialkylamino)-methyl]-4-aryl-2H-phthalazin-1-ones via Mannich reaction of 4-aryl-2H-phthalazin-1-ones. Synth. Commun. 2005, 35, 179–192. [Google Scholar] [CrossRef]
- Jóźwiak, A.; Ciechańska, M. The behaviour of 2-substituted-3-hydroxyisoindolinones in the reaction with sec-butyllithium. J. Het. Chem. 2014, 51, 357–362. [Google Scholar] [CrossRef]
- Achmatowicz, M.; Thiel, O.R.; Wheeler, P.; Bernard, C.; Huang, J.; Larsen, R.D.; Faul, M.M. Practical synthesis of a p38 MAP kinase inhibitor. J. Org. Chem. 2009, 74, 795–809. [Google Scholar] [CrossRef]
- Metallinos, C.; Nerdinger, S.; Snieckus, V. N-Cumyl benzamide, sulfonamide, and aryl O-carbamate directed metalation groups. Mild hydrolytic lability for facile manipulation of directed ortho metalation derived aromatics. Org. Lett. 1999, 1, 1183–1186. [Google Scholar] [CrossRef]
- Clayden, J.; Frampton, C.S.; McCarthy, C.; Westlund, N. Perilithiation and the synthesis of 8-substituted-1-naphthamides. Tetrahedron 1999, 55, 14161–14184. [Google Scholar] [CrossRef]
- Eppley, R.L.; Dixon, J.A. The addition to and alkylation of naphthalene by tert-butyllithium. Kinetics and mechanism. J. Am. Chem. Soc. 1968, 90, 1606–1612. [Google Scholar] [CrossRef]
- Pinto, D.J.P.; Quan, M.L.; Smith II, L.M.; Orwat, M.J.; Gilligan, P.J. Bristol-Myers Squibb Co. Dipeptide Analogs as Coagulation Factor Inhibitors. WO Patent 2008/157162, 24 December 2008. Available online: https://patentimages.storage.googleapis.com/8c/99/dd/b1181fa9b12372/US8367709.pdf (accessed on 25 November 2022).
- Heiss, C.; Schlosser, M. Organometallic control over the regiospecificity of functionalization reactions: 1,2,3-trifluorobenzene and bromo derivatives thereof as substrates. Eur. J. Org. Chem. 2003, 2003, 447–451. [Google Scholar] [CrossRef]
- Novák, J.; Salemink, C.A. Cannabis xxiv. A new convenient synthesis of cannabinol. Tetrahedron Lett. 1982, 23, 253–254. [Google Scholar] [CrossRef]
- Ciechańska, M.; Jóźwiak, A.; Nazarski, R.B.; Skorupska, E.A. Unexpected rearrangement of dilithiated isoindoline-1,3-diols into 3-aminoindan-1-ones via N-lithioaminoarylcarbenes: A combined synthetic and computational study. J. Org. Chem. 2019, 84, 11425–11440. [Google Scholar] [CrossRef] [PubMed]
- Phan, D.H.T.; Kim, B.; Dong, V.M. Phthalides by rhodium-catalyzed ketone hydroacylation. J. Am. Chem. Soc. 2009, 131, 15608–15609. [Google Scholar] [CrossRef] [PubMed]
- Augner, D.; Schmalz, H.-G. Biomimetic synthesis of isoindolinones related to the marilines. Synlett 2015, 26, 1395–1397. [Google Scholar] [CrossRef]
- Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Synthesis of 4-alkylsulfanylphthalazin-1(2H)-ones via palladium catalyzed sulfanylation of substituted 4-bromophthalazin-1(2H)-ones. Tetrahedron 2016, 72, 7942–7951. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Q.; Luo, M. Reduction of hydrazines to amines with aqueous solution of titanium(III) trichloride. Org. Biomol. Chem. 2011, 9, 4977–4982. [Google Scholar] [CrossRef]
- Jia, L.; Tang, Q.; Luo, M.; Zeng, X. Direct ortho-selective amination of 2-naphthol and its analogues with hydrazines. J. Org. Chem. 2018, 83, 5082–5091. [Google Scholar] [CrossRef]
- Malinowski, Z.; Fornal, E.; Sumara, A.; Kontek, R.; Bukowski, K.; Pasternak, B.; Sroczyński, D.; Kusz, J.; Małecka, M.; Nowak, M. Amino- and polyaminophthalazin-1(2H)-ones: Synthesis, coordination properties, and biological activity. Beilstein J. Org. Chem. 2021, 17, 558–568. [Google Scholar] [CrossRef]
- Nowak, M.; Malinowski, Z.; Fornal, E.; Jozwiak, A.; Parfieniuk, E.; Gajek, G.; Kontek, R. Substituted benzoquinazolinones. Part 2: Synthesis of amino-, and sulfanyl-derivatives of benzo[f]- and benzo[h]quinazolinones. Tetrahedron 2015, 71, 9463–9473. [Google Scholar] [CrossRef]
- Teipel, J.; Gottstein, V.; Hölzle, E.; Kaltenbach, K.; Lachenmeier, D.W.; Kuballa, T. An easy and reliable method for the mitigation of deuterated chloroform decomposition to stabilise susceptible NMR samples. Chemistry 2022, 4, 776–785. [Google Scholar] [CrossRef]
- Kawai, S. Discussion on decomposition of chloroform. Yakugaku Zasshi 1966, 86, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Badawi, H.M.; Förner, W.; Ali, S.A. The molecular structure and vibrational, 1H and 13C NMR spectra of lidocaine hydrochloride monohydrate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 152, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Holzer, W. Unambiguous assignment of the 1H- and 13C-NMR spectra of propafenone and a thiophene analogue. Molecules 2001, 6, 796–802. [Google Scholar] [CrossRef]
- Morishima, I.; Yoshikawa, K.; Okada, K.; Yonezawa, T.; Goto, K. Conformational dependence of the inductive effect in the.sigma.-electron system as studied by carbon-13 nuclear magnetic resonance. J. Am. Chem. Soc. 1973, 95, 165–171. [Google Scholar] [CrossRef]
- Sarneski, J.E.; Surprenant, H.L.; Molen, F.K.; Rellley, C.N. Chemical shifts and protonation shifts in carbon-13 nuclear magnetic resonance studies of aqueous amines. Anal. Chem. 1975, 47, 2116–2124. [Google Scholar] [CrossRef]
- Pakulska, W.; Malinowski, Z.; Szcześniak, A.K.; Czarnecka, E.; Epsztajn, J. Synthesis and pharmacological evaluation of N-(dimethylamino)ethyl derivatives of benzo- and pyridopyridazinones. Arch. Pharm. 2009, 342, 41–47. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Kofron, W.G.; Baclawski, L.M. A convenient method for estimation of alkyllithium concentrations. J. Org. Chem. 1976, 41, 1879–1880. [Google Scholar] [CrossRef]
- Wangngae, S.; Duangkamol, C.; Pattarawarapan, M.; Phakhodee, W. Significance of reagent addition sequence in the amidation of carboxylic acids mediated by PPh3 and I2. RSC Adv. 2015, 5, 25789–25793. [Google Scholar] [CrossRef]
- Pan, C.; Wang, L.; Han, J. Palladium-Catalyzed Annulation of Arylbenzamides with Diaryliodonium Salts. Adv. Synth. Catal. 2022, 364, 268–273. [Google Scholar] [CrossRef]
- He, Z.; Yan, C.; Zhang, M.; Irfan, M.; Wang, Z.; Zeng, Z. Palladium-Catalyzed Desulfurative Hiyama Coupling of Thioureas to Achieve Amides via Selective C–N Bond Cleavage. Synthesis 2022, 54, 705–710. [Google Scholar] [CrossRef]
- Epsztajn, J.; Jóźwiak, A.; Szcześniak, A.K. Application of organolithium and related reagents in synthesis. Part 13. Synthetic strategies based on aromatic metallation. A concise regiospecific conversion of benzoic acids into 4-hydroxy-1-arylnaphthalenes. Tetrahedron 1993, 49, 929–938. [Google Scholar] [CrossRef]
- He, G.; Wu, C.; Zhou, J.; Yang, Q.; Zhang, C.; Zhou, Y.; Zhang, H.; Liu, H. A Method for Synthesis of 3-Hydroxy-1-indanones via Cu-Catalyzed Intramolecular Annulation Reactions. J. Org. Chem. 2018, 83, 13356–13362. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Conditions | Molar Ratio of Products b | ||||
|---|---|---|---|---|---|---|
| Temperature/Time | 2 | 5d | 6d | 7 | 8 | |
| 1 | (1) i-PrLi 0 °C and 10 min at 0 °C; (2) MeOH 0 °C, 10 min at 0 °C. a | 0.73 | 0.82 | 1.00 | - c | - c |
| 2 | (1) i-PrLi 0 °C and 1.5 h at 0 °C; (2) MeOH 0 °C, 0.5 h at 0 °C. a | 0.31 | 0.48 | 1.00 | - c | - c |
| 3 | (1) i-PrLi 0 °C and 1.5 h at 0 °C; (2) 0 °C → rt, 1,5 h at rt; (3) MeOH rt, 0.5 h at rt. a | Trace | 0.47 | 1.00 | - c | - c |
| 4 | (1) i-PrLi 0 °C and 1.5 h 0 °C; (2) 0 °C → rt, 4 h at rt; (3) MeOH rt, 0.5 h at rt. a | 0.23 | 0.48 | 1.00 | - c | - c |
| 5 | (1) i-PrLi 0 °C and 1.5 h at 0 °C; (2) 0 °C → rt, 20 h at rt; (3) MeOH rt, 0.5 h at rt. a | 0.18 | 0.10 | 1.00 | - c | - c |
| 6 | (1) i-PrLi 0 °C and 1.5 h at 0 °C; (2) 0°C → rt, 36 h at rt; (3) MeOH rt, 0.5 h at rt. a | 0.21 | 0.10 | 1.00 | - c | - c |
| 7 | (1) i-PrLi rt and 1.5 h at rt; (2) MeOH rt, 0.5 h at rt. a | 0.04 | 0.52 | 1.00 | - c | - c |
| 8 | (1) i-PrLi −78 °C and 1.5 h at −78 °C; (2) MeOH, −78 °C, 0.5 h at −78 °C. a | 1.01 | 0.47 | 1.00 | - c | - c |
| 9 | (1) i-PrLi 0 °C and 1.5 h at 0 °C; (2) 0 °C → rt, 0.5 h at rt; (3) MeOH rt, 0.5 h at rt. a | 0.18 | 0.56 | 1.00 | - c | - c |
| 10 | (1) i-PrLi 0 °C and 1.5 h at 0 °C; (2) 0 °C → rt, 1,5 h at rt; (3) H2O rt, 0.5 h at rt, next to pH ≈ 7 (HClaq, 1:1, v/v). | 1.30 | 0.29 | 1.00 | - c | - c |
| 11 | (1) i-PrLi −78 °C and 1.5 h at −78 °C; (2) −78 °C → rt, 0.5 h at rt; (3) H2O rt, 0.5 h at rt, next to pH ≈ 2 (HClaq, 1:1, v/v). | 0.85 | Trace | Trace | 0.35 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malinowski, Z.; Fornal, E.; Stachniuk, A.; Nowak, M. Reactions of 3-Hydroxy-2-phenyl-1H-benzo[e]isoindol-1-one: A Route to 3-Hydroxy-/3-anilinobenzo[e]indan-1-ones and Benzo[f]phthalazin-1(2H)-ones. Molecules 2022, 27, 8319. https://doi.org/10.3390/molecules27238319
Malinowski Z, Fornal E, Stachniuk A, Nowak M. Reactions of 3-Hydroxy-2-phenyl-1H-benzo[e]isoindol-1-one: A Route to 3-Hydroxy-/3-anilinobenzo[e]indan-1-ones and Benzo[f]phthalazin-1(2H)-ones. Molecules. 2022; 27(23):8319. https://doi.org/10.3390/molecules27238319
Chicago/Turabian StyleMalinowski, Zbigniew, Emilia Fornal, Anna Stachniuk, and Monika Nowak. 2022. "Reactions of 3-Hydroxy-2-phenyl-1H-benzo[e]isoindol-1-one: A Route to 3-Hydroxy-/3-anilinobenzo[e]indan-1-ones and Benzo[f]phthalazin-1(2H)-ones" Molecules 27, no. 23: 8319. https://doi.org/10.3390/molecules27238319
APA StyleMalinowski, Z., Fornal, E., Stachniuk, A., & Nowak, M. (2022). Reactions of 3-Hydroxy-2-phenyl-1H-benzo[e]isoindol-1-one: A Route to 3-Hydroxy-/3-anilinobenzo[e]indan-1-ones and Benzo[f]phthalazin-1(2H)-ones. Molecules, 27(23), 8319. https://doi.org/10.3390/molecules27238319