

Computational Analysis of Triazole-Based Kojic Acid Analogs as Tyrosinase Inhibitors by Molecular Dynamics and Free Energy Calculations
 , and
, and 
Abstract
1. Introduction
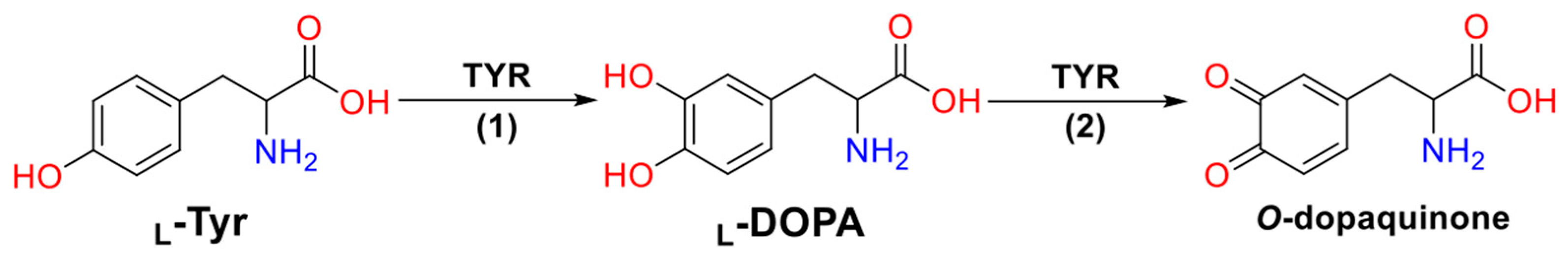
2. Results and Discussion
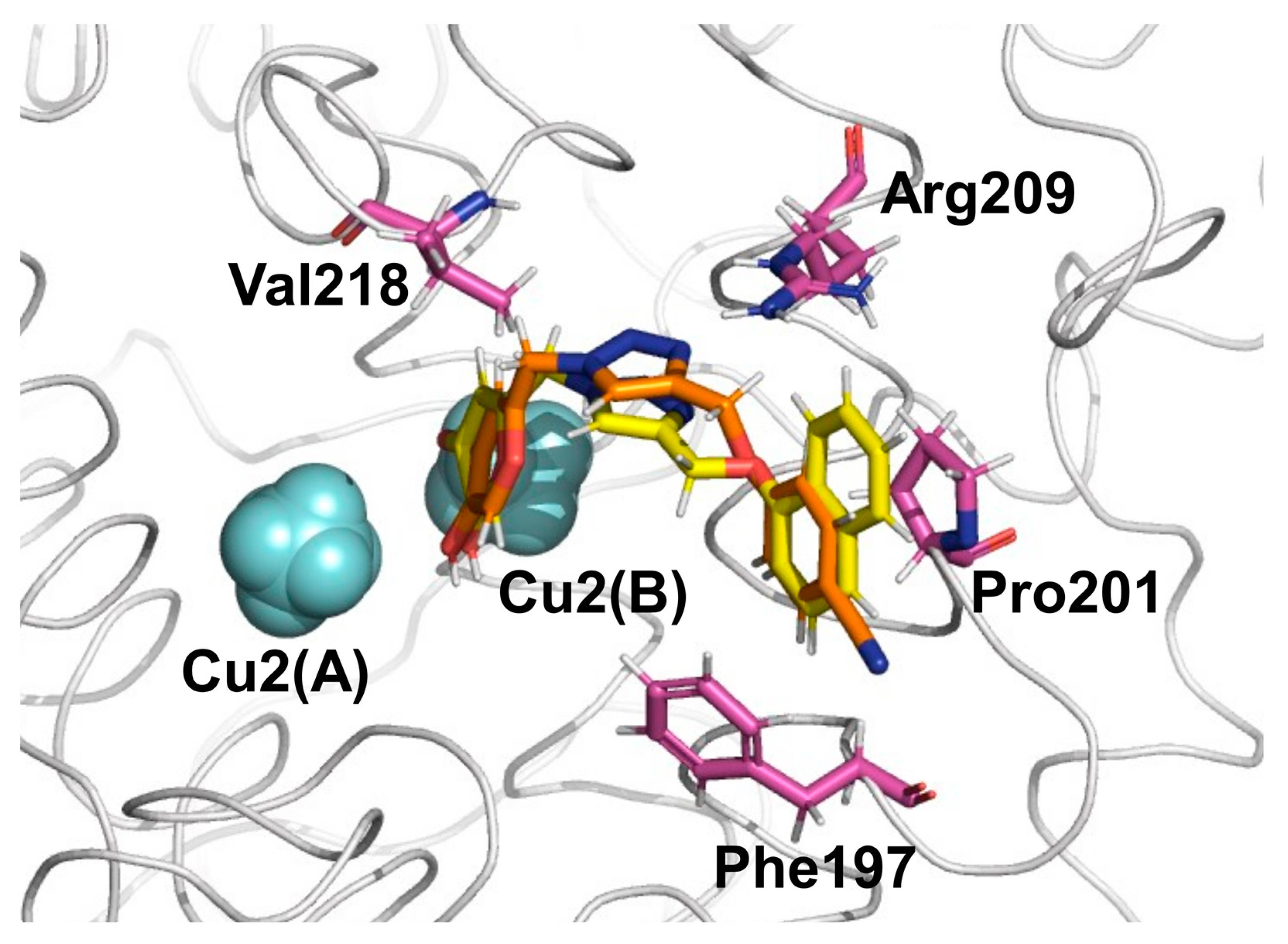
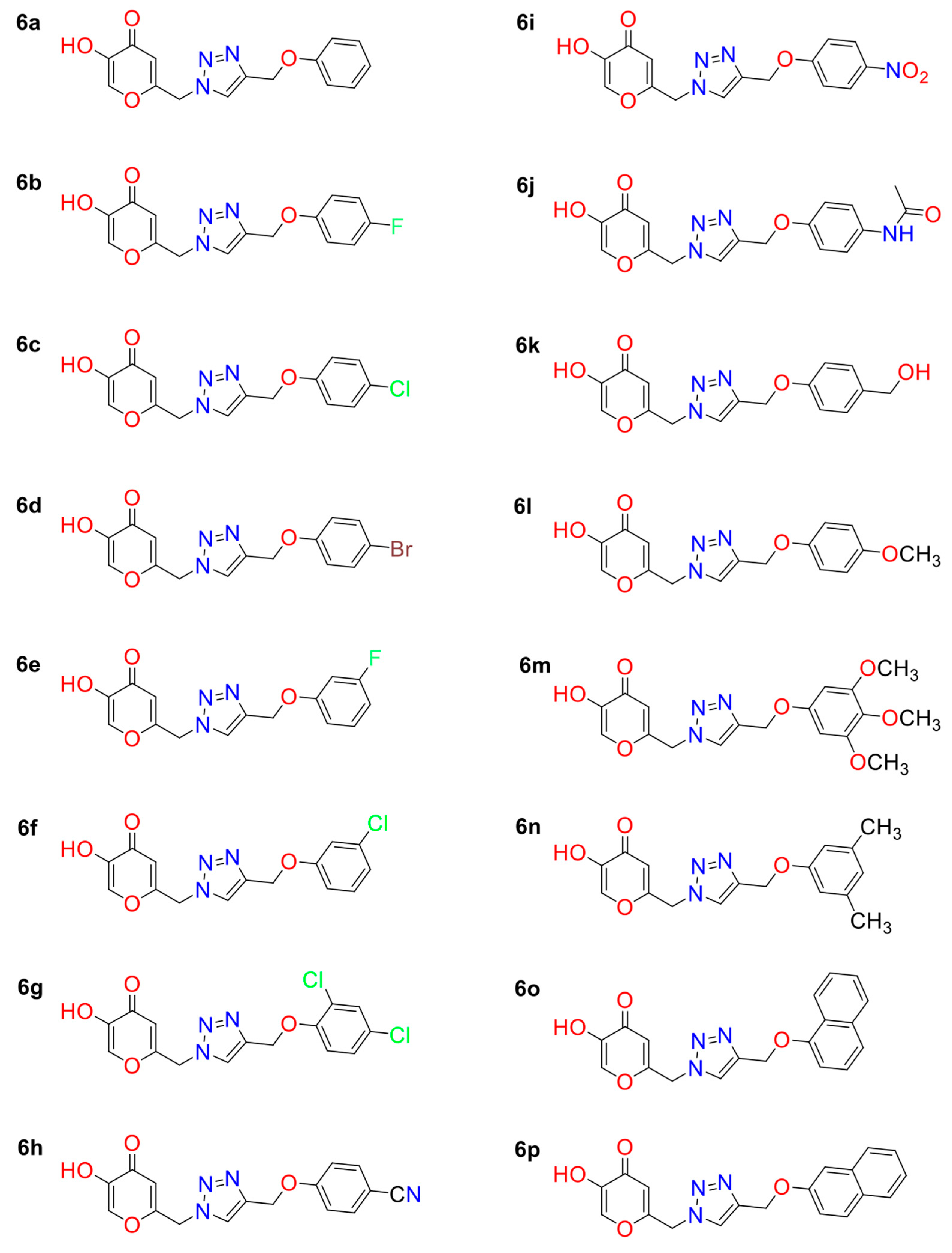
2.1. Molecular Docking and MD Simulations
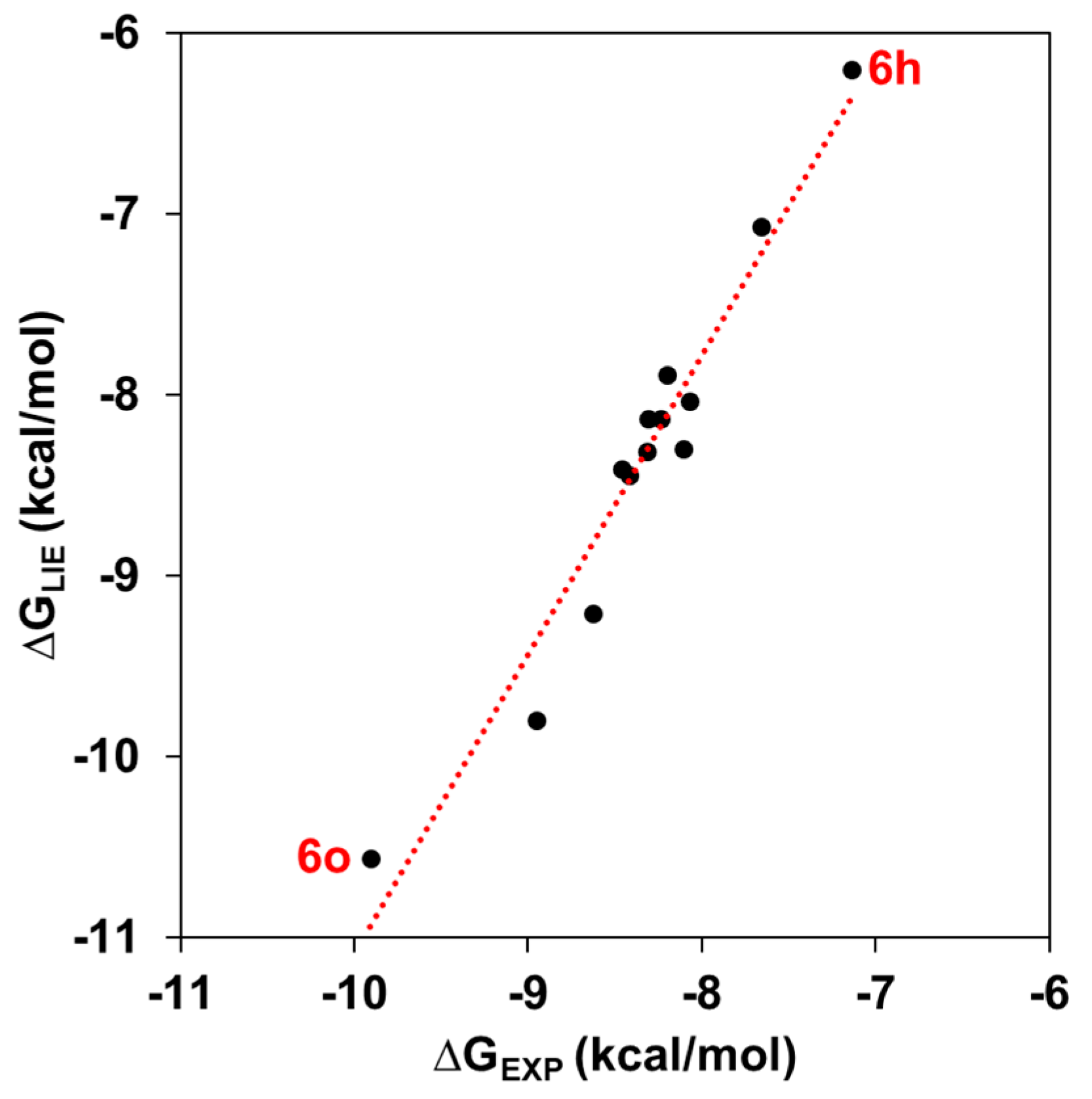
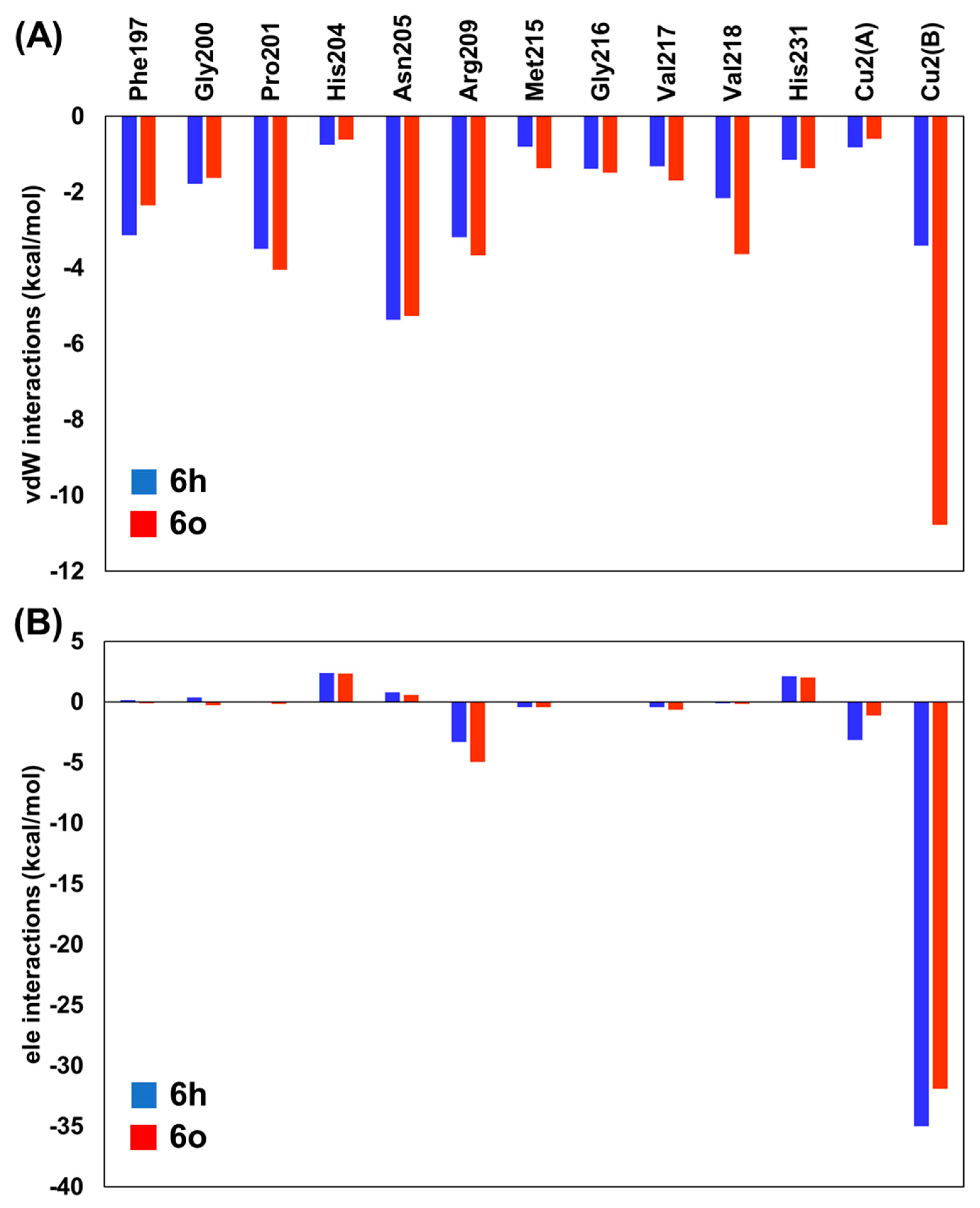
2.2. Binding Free Energy and Per-Residual Analysis
3. Materials and Methods
3.1. System Setup for Molecular Docking and MD Simulations
3.2. Binding Free Energy Calculations: LIE Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Mello, S.; Finlay, G.; Baguley, B.; Askarian-Amiri, M. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.M.; Kwon, E.-B.; Lee, B.; Kim, C.Y. Recent Trends in Controlling the Enzymatic Browning of Fruit and Vegetable Products. Molecules 2020, 25, 2754. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Hearing, V.J. The Protective Role of Melanin Against UV Damage in Human Skin†. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Del Bino, S.; Duval, C.; Bernerd, F. Clinical and Biological Characterization of Skin Pigmentation Diversity and Its Consequences on UV Impact. Int. J. Mol. Sci. 2018, 19, 2668. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef]
- Bose, A.; Petsko, G.A.; Eliezer, D. Parkinson’s Disease and Melanoma: Co-Occurrence and Mechanisms. J. Parkinsons. Dis. 2018, 8, 385–398. [Google Scholar] [CrossRef]
- Sánchez-Ferrer, Á.; Neptuno Rodríguez-López, J.; García-Cánovas, F.; García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Solano, F. On the Metal Cofactor in the Tyrosinase Family. Int. J. Mol. Sci. 2018, 19, 633. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A comprehensive review on tyrosinase inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Chang, T.-S. An Updated Review of Tyrosinase Inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Kligman, A.M. A New Formula for Depigmenting Human Skin. Arch. Dermatol. 1975, 111, 40. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.A.; Riley, P.A. Mechanistic aspects of the tyrosinase oxidation of hydroquinone. Bioorg. Med. Chem. Lett. 2014, 24, 2463–2464. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Wood, J.W. A possible mechanism of action for azelaic acid in the human epidermis. Arch. Dermatol. Res. 1990, 282, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Deri, B.; Kanteev, M.; Goldfeder, M.; Lecina, D.; Guallar, V.; Adir, N.; Fishman, A. The unravelling of the complex pattern of tyrosinase inhibition. Sci. Rep. 2016, 6, 34993. [Google Scholar] [CrossRef]
- Chen, W.-C.; Tseng, T.-S.; Hsiao, N.-W.; Lin, Y.-L.; Wen, Z.-H.; Tsai, C.-C.; Lee, Y.-C.; Lin, H.-H.; Tsai, K.-C. Discovery of Highly Potent Tyrosinase Inhibitor, T1, with Significant Anti-Melanogenesis Ability by zebrafish in vivo Assay and Computational Molecular Modeling. Sci. Rep. 2015, 5, 7995. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, S.M.; Emami, S. Kojic acid-derived tyrosinase inhibitors: Synthesis and bioactivity. Pharm. Biomed. Res. 2015, 1, 1–17. [Google Scholar] [CrossRef]
- Ashooriha, M.; Khoshneviszadeh, M.; Khoshneviszadeh, M.; Moradi, S.E.; Rafiei, A.; Kardan, M.; Emami, S. 1,2,3-Triazole-based kojic acid analogs as potent tyrosinase inhibitors: Design, synthesis and biological evaluation. Bioorg. Chem. 2019, 82, 414–422. [Google Scholar] [CrossRef]
- Lima, C.R.; Silva, J.R.A.; De Tássia Carvalho Cardoso, É.; Silva, E.O.; Lameira, J.; Do Nascimento, J.L.M.; do Socorro Barros Brasil, D.; Alves, C.N. Combined Kinetic Studies and Computational Analysis on Kojic Acid Analogs as Tyrosinase Inhibitors. Molecules 2014, 19, 9591–9605. [Google Scholar] [CrossRef]
- Martins, L.S.; Lameira, J.; Kruger, H.G.H.G.; Alves, C.N.; Silva, J.R.A. Evaluating the Performance of a Non-Bonded Cu2+ Model Including Jahn−Teller Effect into the Binding of Tyrosinase Inhibitors. Int. J. Mol. Sci. 2020, 21, 4783. [Google Scholar] [CrossRef]
- Brasil, E.M.; Canavieira, L.M.; Cardoso, É.T.C.; Silva, E.O.; Lameira, J.; Nascimento, J.L.M.; Eifler-Lima, V.L.; Macchi, B.M.; Sriram, D.; Bernhardt, P.V.; et al. Inhibition of tyrosinase by 4 H -chromene analogs: Synthesis, kinetic studies, and computational analysis. Chem. Biol. Drug Des. 2017, 90, 804–810. [Google Scholar] [CrossRef]
- Canavieira, L.M.; Brasil, E.M.; Silva, T.d.M.e.; Borges, R.d.S.; Silva, J.R.A.; Lameira, J.; Bernhardt, P.V.; Williams, C.M.; Alves, C.N. Experimental and theoretical approaches for the development of 4H-Chromene derivatives as inhibitors of tyrosinase. Mol. Simul. 2021, 47, 762–770. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Bijelic, A.; Pretzler, M.; Molitor, C.; Zekiri, F.; Rompel, A. The Structure of a Plant Tyrosinase from Walnut Leaves Reveals the Importance of “Substrate-Guiding Residues” for Enzymatic Specificity. Angew. Chemie Int. Ed. 2015, 54, 14677–14680. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, M.; Kanteev, M.; Isaschar-Ovdat, S.; Adir, N.; Fishman, A. Determination of tyrosinase substrate-binding modes reveals mechanistic differences between type-3 copper proteins. Nat. Commun. 2014, 5, 4505. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Rubio-Carrasquilla, M.; Valiente, P.A.; Moreno, E. Tailored Parameterization of the LIE Method for Calculating the Binding Free Energy of Vps34-Inhibitor Complexes. ACS Omega 2021, 6, 29525–29536. [Google Scholar] [CrossRef]
- Carlsson, J.; Boukharta, L.; Åqvist, J. Combining Docking, Molecular Dynamics and the Linear Interaction Energy Method to Predict Binding Modes and Affinities for Non-nucleoside Inhibitors to HIV-1 Reverse Transcriptase. J. Med. Chem. 2008, 51, 2648–2656. [Google Scholar] [CrossRef]
- Tam, N.M.; Pham, M.Q.; Ha, N.X.; Nam, P.C.; Phung, H.T.T. Computational estimation of potential inhibitors from known drugs against the main protease of SARS-CoV-2. RSC Adv. 2021, 11, 17478–17486. [Google Scholar] [CrossRef]
- Hoelz, L.V.B.; Leal, V.F.; Rodrigues, C.R.; Pascutti, P.G.; Albuquerque, M.G.; Muri, E.M.F.; Dias, L.R.S. Molecular dynamics simulations of the free and inhibitor-bound cruzain systems in aqueous solvent: Insights on the inhibition mechanism in acidic pH. J. Biomol. Struct. Dyn. 2016, 34, 1969–1978. [Google Scholar] [CrossRef]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Åqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225. [Google Scholar] [CrossRef]
- Bauer, P.; Barrozo, A.; Purg, M.; Amrein, B.A.; Esguerra, M.; Wilson, P.B.; Major, D.T.; Åqvist, J.; Kamerlin, S.C.L. Q6: A comprehensive toolkit for empirical valence bond and related free energy calculations. SoftwareX 2018, 7, 388–395. [Google Scholar] [CrossRef]
- Liao, Q.; Kamerlin, S.C.L.; Strodel, B. Development and Application of a Nonbonded Cu 2+ Model That Includes the Jahn–Teller Effect. J. Phys. Chem. Lett. 2015, 6, 2657–2662. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aided. Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Murcko, M.A. Computational methods to predict binding free energy in ligand-receptor complexes. J. Med. Chem. 1995, 38, 4953–4967. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided. Mol. Des. 1998, 27–35. [Google Scholar] [CrossRef]
- Vanga, S.R.; Sävmarker, J.; Ng, L.; Larhed, M.; Hallberg, M.; Åqvist, J.; Hallberg, A.; Chai, S.Y.; Gutiérrez-De-Terán, H. Structural Basis of Inhibition of Human Insulin-Regulated Aminopeptidase (IRAP) by Aryl Sulfonamides. ACS Omega 2018, 3, 4509–4521. [Google Scholar] [CrossRef]
- ChemAxon MarvinSketch. MarvinSketch, Version 22.18. 2022. Available online: https://macdownload.informer.com/marvinsketch/ (accessed on 4 November 2022).
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kitchen, D.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug. Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.; Maxwell, D.; Tirado-rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L.; Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79. [Google Scholar] [CrossRef]
- Schrödinger Schrödinger Release 2020-1: MacroModel; Schrödinger, LLC: New York, NY, USA, 2020.
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- King, G.; Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989, 91, 3647–3661. [Google Scholar] [CrossRef]
- Diaz, L.; Bujons, J.; Delgado, A.; Gutierrez-de-Terán, H.; Aqvist, J. Computational prediction of structure-activity relationships for the binding of aminocyclitols to beta-glucocerebrosidase. J. Chem. Inf. Model 2011, 51, 601–611. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KA Analog | MOLDOCK Scoring | IC50 * |
|---|---|---|
| 6a | −120.78 | 1.33 |
| 6b | −132.33 | 0.88 |
| 6c | −132.03 | 0.69 |
| 6d | −128.15 | 6.80 |
| 6e | −125.68 | 1.07 |
| 6f | −136.38 | 0.99 |
| 6g | −129.55 | 1.12 |
| 6h | −139.06 | 6.29 |
| 6i | −132.92 | 0.52 |
| 6j | −135.60 | 2.64 |
| 6k | −132.85 | 1.32 |
| 6l | −125.93 | 1.24 |
| 6m | −130.46 | 0.87 |
| 6n | −130.17 | 0.74 |
| 6o | −130.15 | 0.06 |
| 6p | −131.51 | 0.30 |
| System | Protein RMSD | Ligand RMSD |
|---|---|---|
| TYR-6a | 0.44 ± 0.05 | 0.47 ± 0.15 |
| TYR-6b | 0.50 ± 0.04 | 0.80 ± 0.20 |
| TYR-6c | 0.46 ± 0.07 | 0.53 ± 0.14 |
| TYR-6d | 0.40 ± 0.03 | 0.51 ± 0.16 |
| TYR-6e | 0.40 ± 0.05 | 0.60 ± 0.20 |
| TYR-6f | 0.44 ± 0.04 | 0.81 ± 0.24 |
| TYR-6g | 0.45 ± 0.03 | 0.79 ± 0.21 |
| TYR-6h | 0.43 ± 0.04 | 0.54 ± 0.13 |
| TYR-6i | 0.44 ± 0.05 | 0.51 ± 0.13 |
| TYR-6j | 0.43 ± 0.03 | 0.47 ± 0.12 |
| TYR-6k | 0.44 ± 0.04 | 0.62 ± 0.16 |
| TYR-6l | 0.43 ± 0.05 | 0.57 ± 0.13 |
| TYR-6m | 0.49 ± 0.06 | 0.55 ± 0.13 |
| TYR-6n | 0.47 ± 0.04 | 0.79 ± 0.19 |
| TYR-6o | 0.39 ± 0.04 | 0.49 ± 0.13 |
| TYR-6p | 0.46 ± 0.06 | 0.50 ± 0.13 |
| KA Analog | ∆GLIE | ∆GEXP | ||||
|---|---|---|---|---|---|---|
| 6a | −26.21 ± 0.01 | −26.70 ± 0.49 | −49.16 ± 0.98 | −84.09 ± 0.24 | −8.03 ± 0.45 | −8.07 |
| 6b | −26.31 ± 0.06 | −26.91 ± 0.10 | −46.90 ± 0.34 | −85.71 ± 0.63 | −8.13 ± 0.34 | −8.31 |
| 6c | −27.77 ± 0.02 | −25.57 ± 0.17 | −49.79 ± 0.41 | −84.45 ± 0.70 | −8.41 ± 0.40 | −8.46 |
| 6e | −26.24 ± 0.03 | −26.97 ± 0.31 | −46.53 ± 0.21 | −85.27 ± 0.76 | −7.89 ± 0.44 | −8.20 |
| 6f | −27.61 ± 0.02 | −25.32 ± 0.18 | −50.79 ± 0.90 | −82.86 ± 0.89 | −8.13 ± 0.56 | −8.24 |
| 6h | −28.48 ± 0.08 | −30.39 ± 0.45 | −51.28 ± 0.29 | −89.28 ± 0.78 | −6.20 ± 0.47 | −7.14 |
| 6i | −29.42 ± 0.04 | −29.93 ± 0.02 | −55.29 ± 0.51 | −89.10 ± 0.26 | −9.21 ± 0.20 | −8.63 |
| 6j | −30.14 ± 0.08 | −42.95 ± 0.30 | −55.83 ± 0.37 | −99.47 ± 0.97 | −7.07 ± 0.53 | −7.66 |
| 6l | −28.59 ± 0.03 | −28.16 ± 0.16 | −50.52 ± 0.68 | −86.78 ± 1.01 | −8.30 ± 0.56 | −8.11 |
| 6m | −31.90 ± 0.10 | −33.77 ± 0.61 | −57.72 ± 0.40 | −90.52 ± 0.99 | −8.31 ± 0.68 | −8.32 |
| 6n | −28.72 ± 0.09 | −29.18 ± 0.74 | −49.41 ± 0.79 | −88.77 ± 0.90 | −8.44 ± 0.76 | −8.42 |
| 6o | −29.99 ± 0.30 | −27.33 ± 0.05 | −61.99 ± 0.37 | −87.15 ± 0.24 | −10.56 ± 0.23 | −9.91 |
| 6p | −30.01 ± 0.20 | −27.97 ± 0.43 | −61.40 ± 0.91 | −86.21 ± 0.22 | −9.80 ± 0.44 | −8.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, L.S.; Gonçalves, R.W.A.; Moraes, J.J.S.; Alves, C.N.; Silva, J.R.A. Computational Analysis of Triazole-Based Kojic Acid Analogs as Tyrosinase Inhibitors by Molecular Dynamics and Free Energy Calculations. Molecules 2022, 27, 8141. https://doi.org/10.3390/molecules27238141
Martins LS, Gonçalves RWA, Moraes JJS, Alves CN, Silva JRA. Computational Analysis of Triazole-Based Kojic Acid Analogs as Tyrosinase Inhibitors by Molecular Dynamics and Free Energy Calculations. Molecules. 2022; 27(23):8141. https://doi.org/10.3390/molecules27238141
Chicago/Turabian StyleMartins, Lucas Sousa, Reinaldo W. A. Gonçalves, Joana J. S. Moraes, Cláudio Nahum Alves, and José Rogério A. Silva. 2022. "Computational Analysis of Triazole-Based Kojic Acid Analogs as Tyrosinase Inhibitors by Molecular Dynamics and Free Energy Calculations" Molecules 27, no. 23: 8141. https://doi.org/10.3390/molecules27238141
APA StyleMartins, L. S., Gonçalves, R. W. A., Moraes, J. J. S., Alves, C. N., & Silva, J. R. A. (2022). Computational Analysis of Triazole-Based Kojic Acid Analogs as Tyrosinase Inhibitors by Molecular Dynamics and Free Energy Calculations. Molecules, 27(23), 8141. https://doi.org/10.3390/molecules27238141