
Effect of Natural Adenylcyclase/cAMP/CREB Signalling Activator Forskolin against Intra-Striatal 6-OHDA-Lesioned Parkinson’s Rats: Preventing Mitochondrial, Motor and Histopathological Defects

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Behavioural Parameters
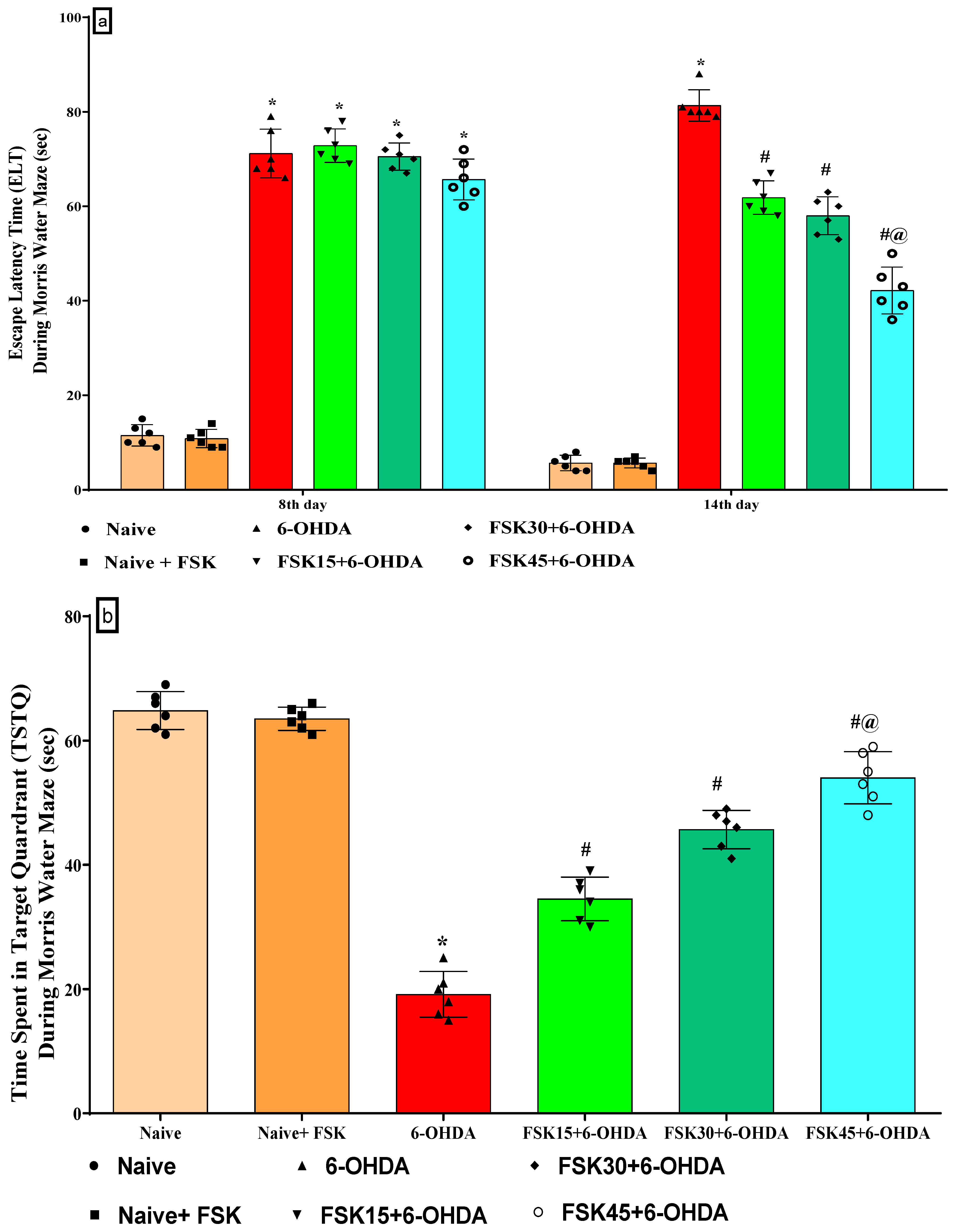
2.1.1. FSK Improved Memory and Cognition in an Experimental Model of PD
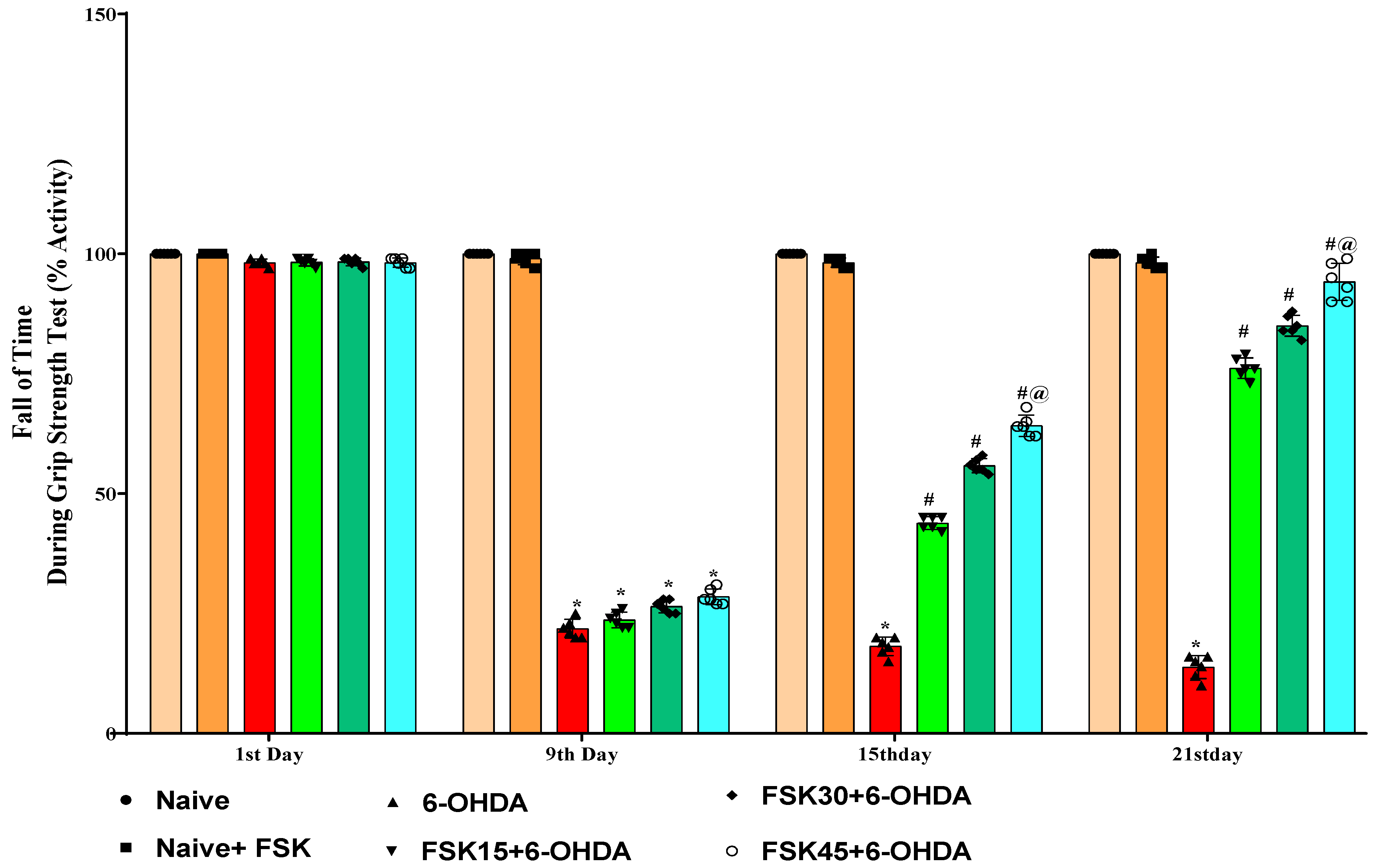
2.1.2. FSK Restored Muscle Grip Strength in the Experimental Model of PD
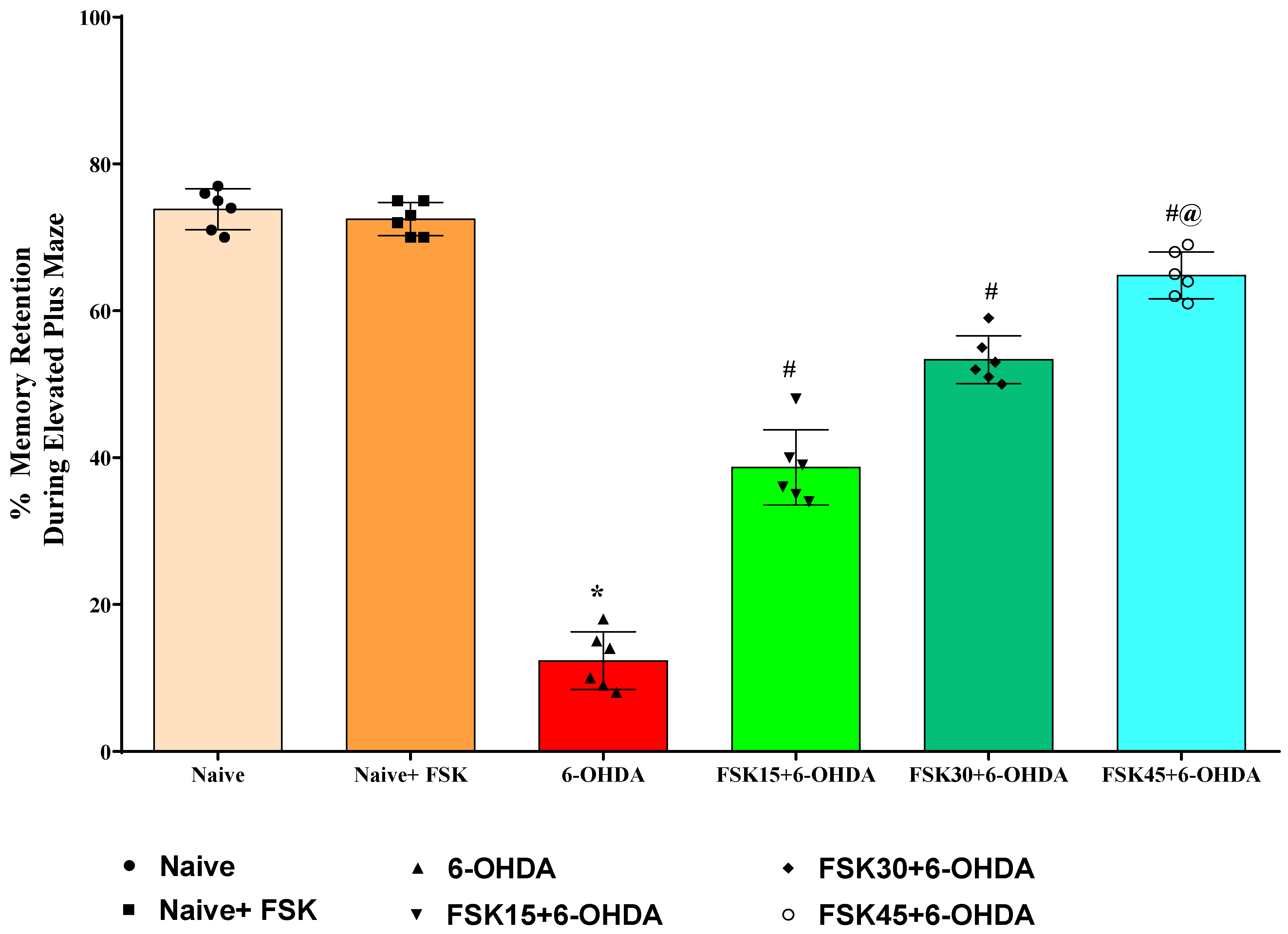
2.1.3. FSK Improved Memory Retention in the Experimental Model of PD
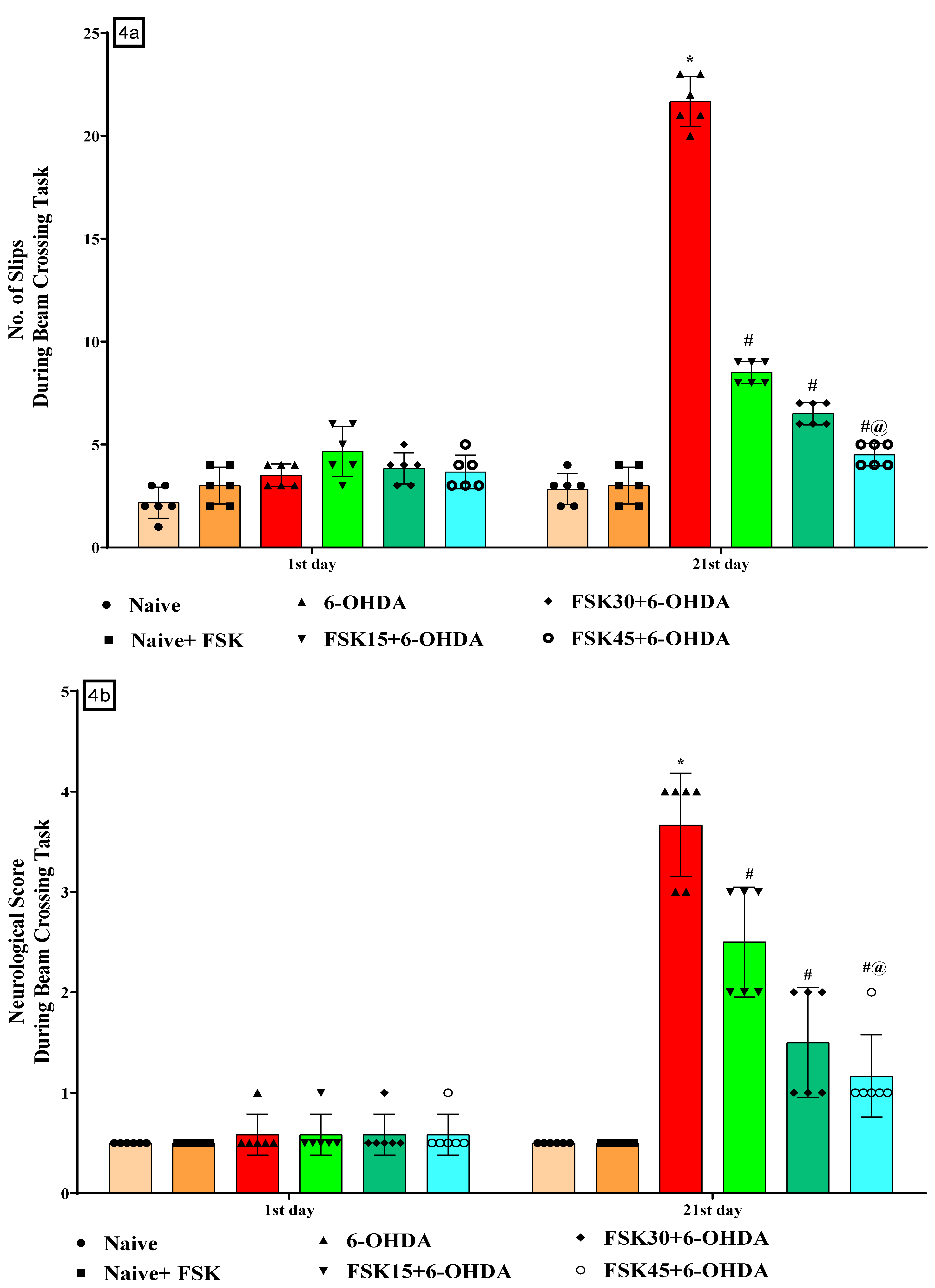
2.1.4. FSK Restored Neuromuscular Coordination in the Experimental Model of PD
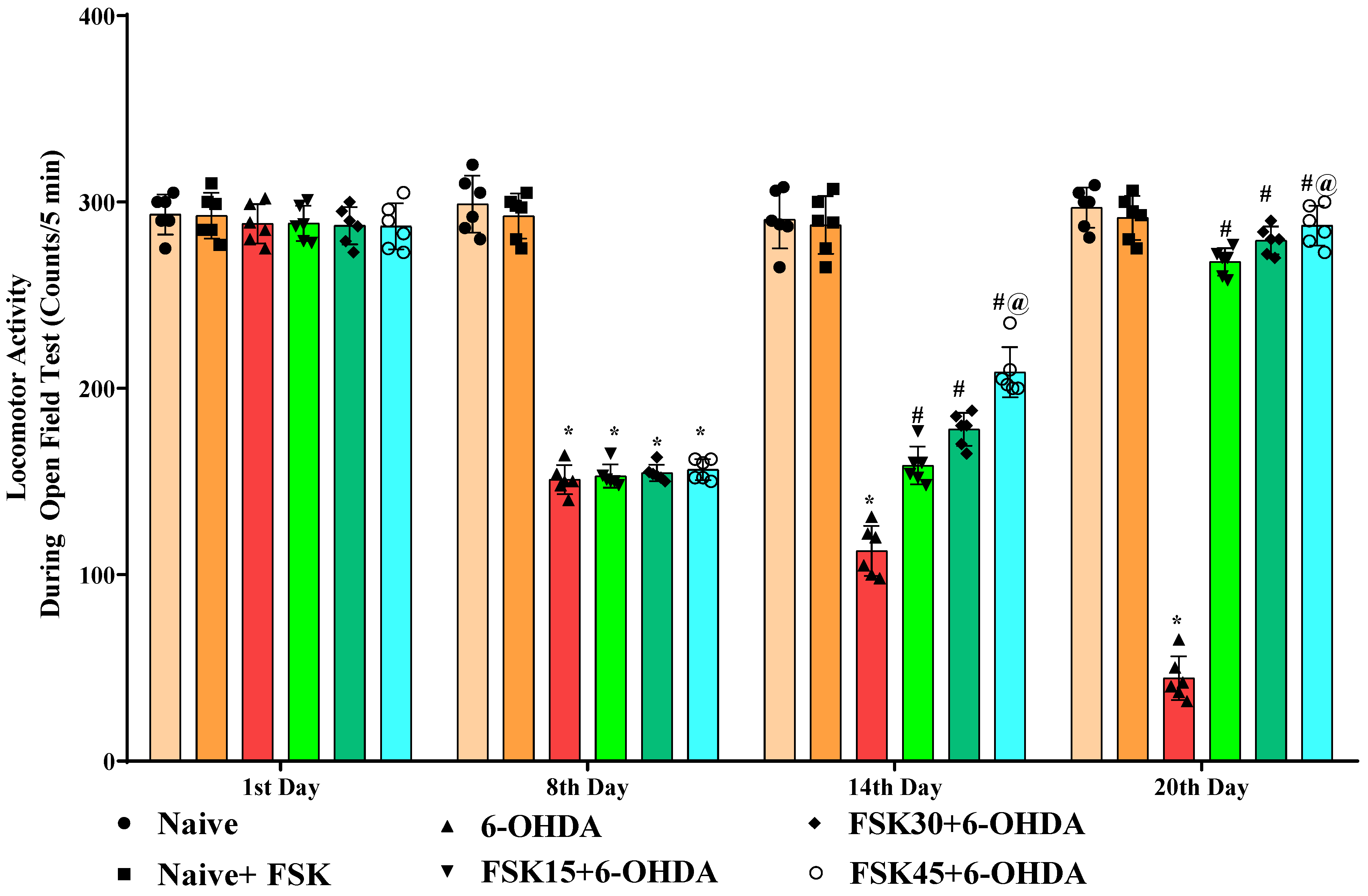
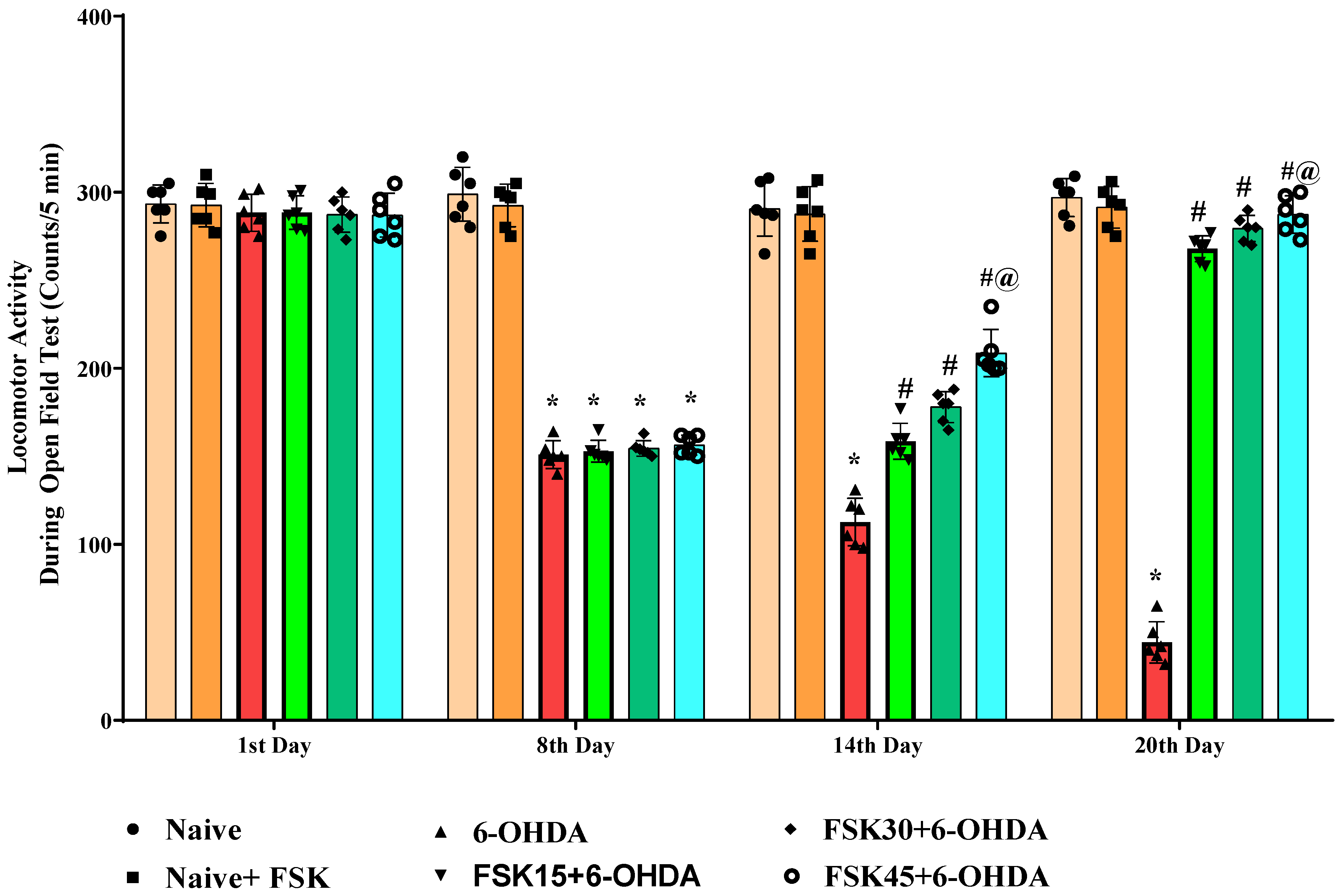
2.1.5. FSK Improved Locomotion in the Experimental Model of PD
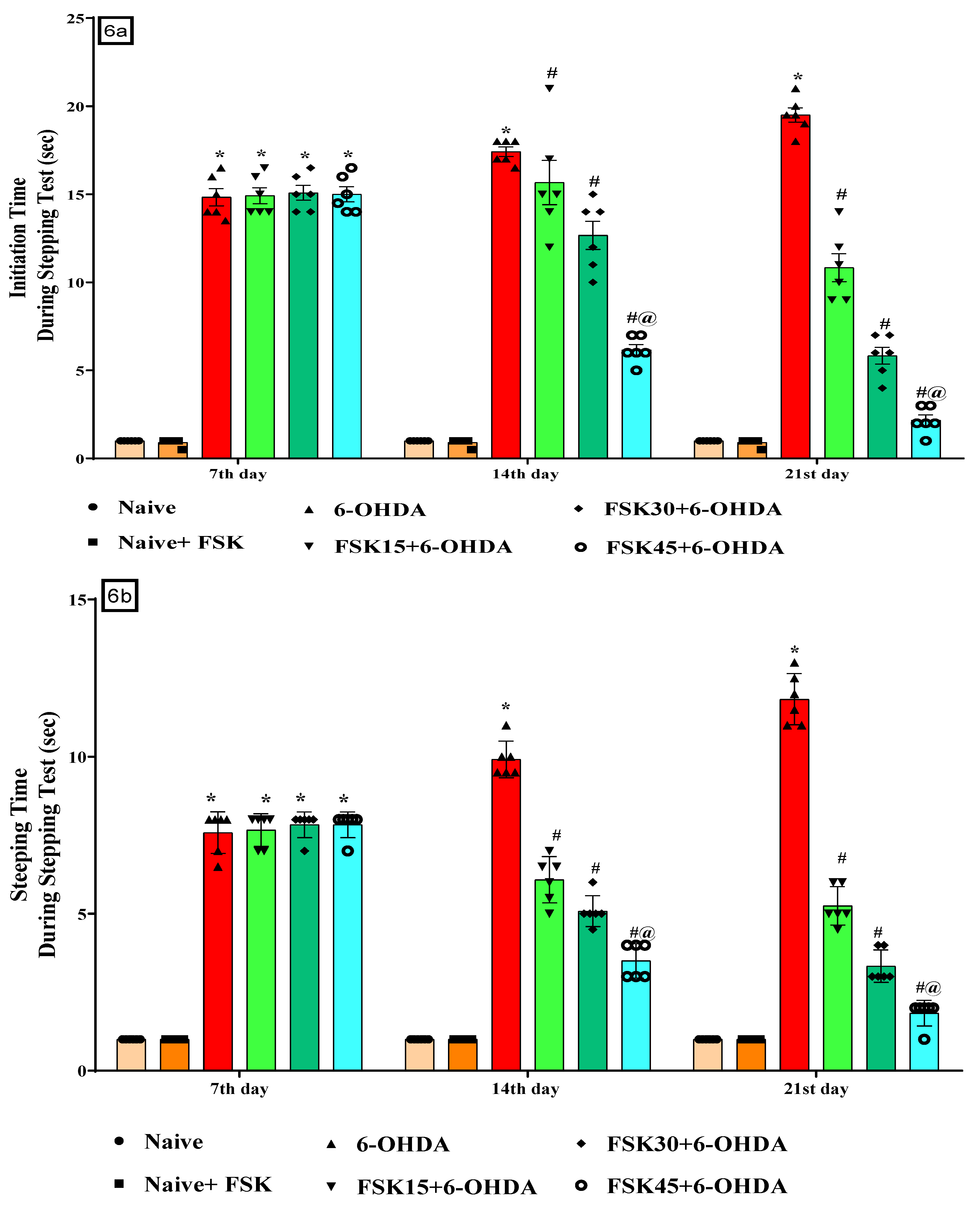
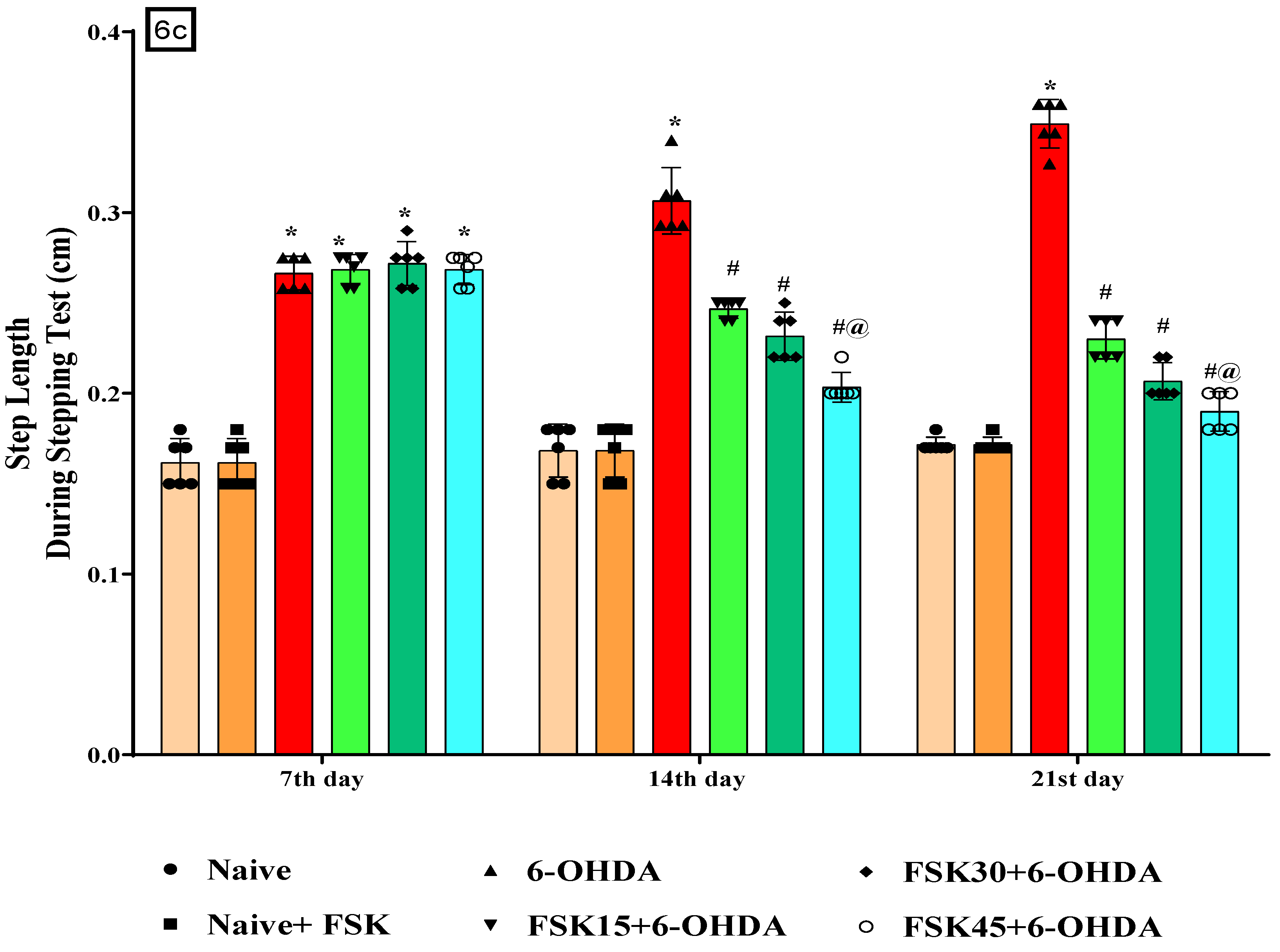
2.1.6. FSK Restored Voluntary Movement in the Experimental Model of PD
2.1.7. FSK Improved Motor Coordination in the Experimental Model of PD
2.2. Biochemical Parameters
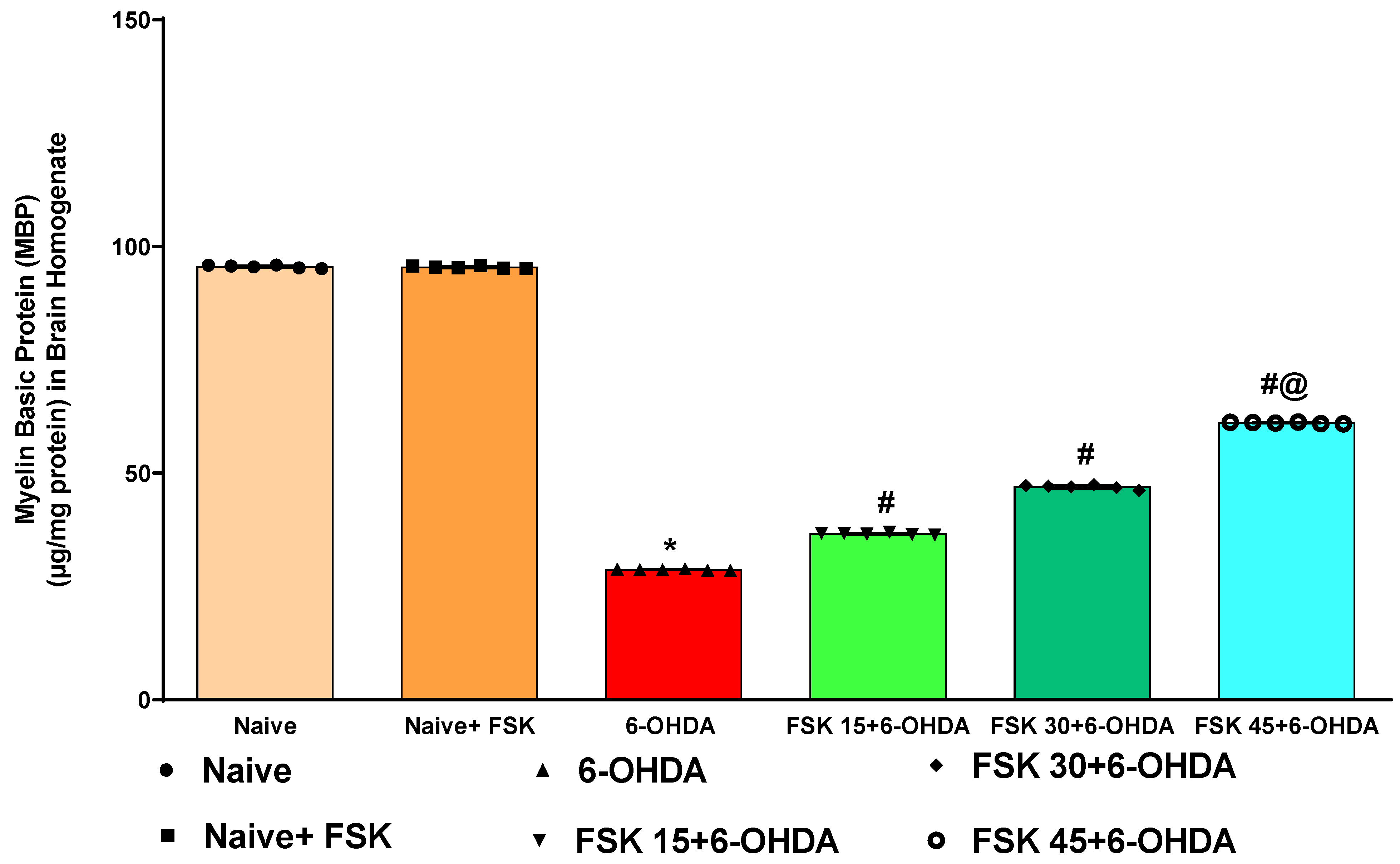
2.2.1. FSK Increases the Myelin Basic Protein (MBP) Level in the Experimental Model of PD
2.2.2. FSK Mediated the Restoration of Cellular and Molecular Alterations in the Experimental Model of PD
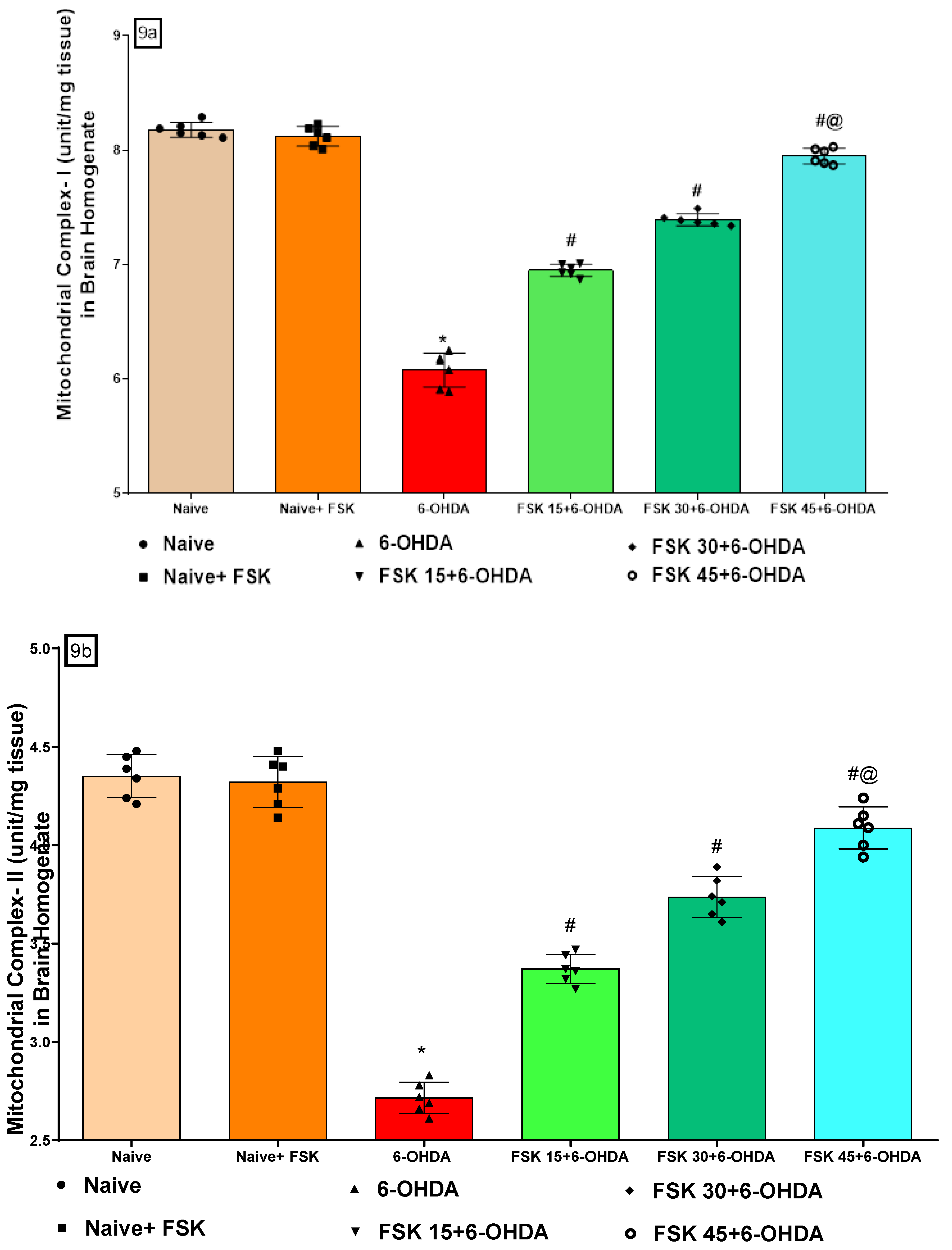
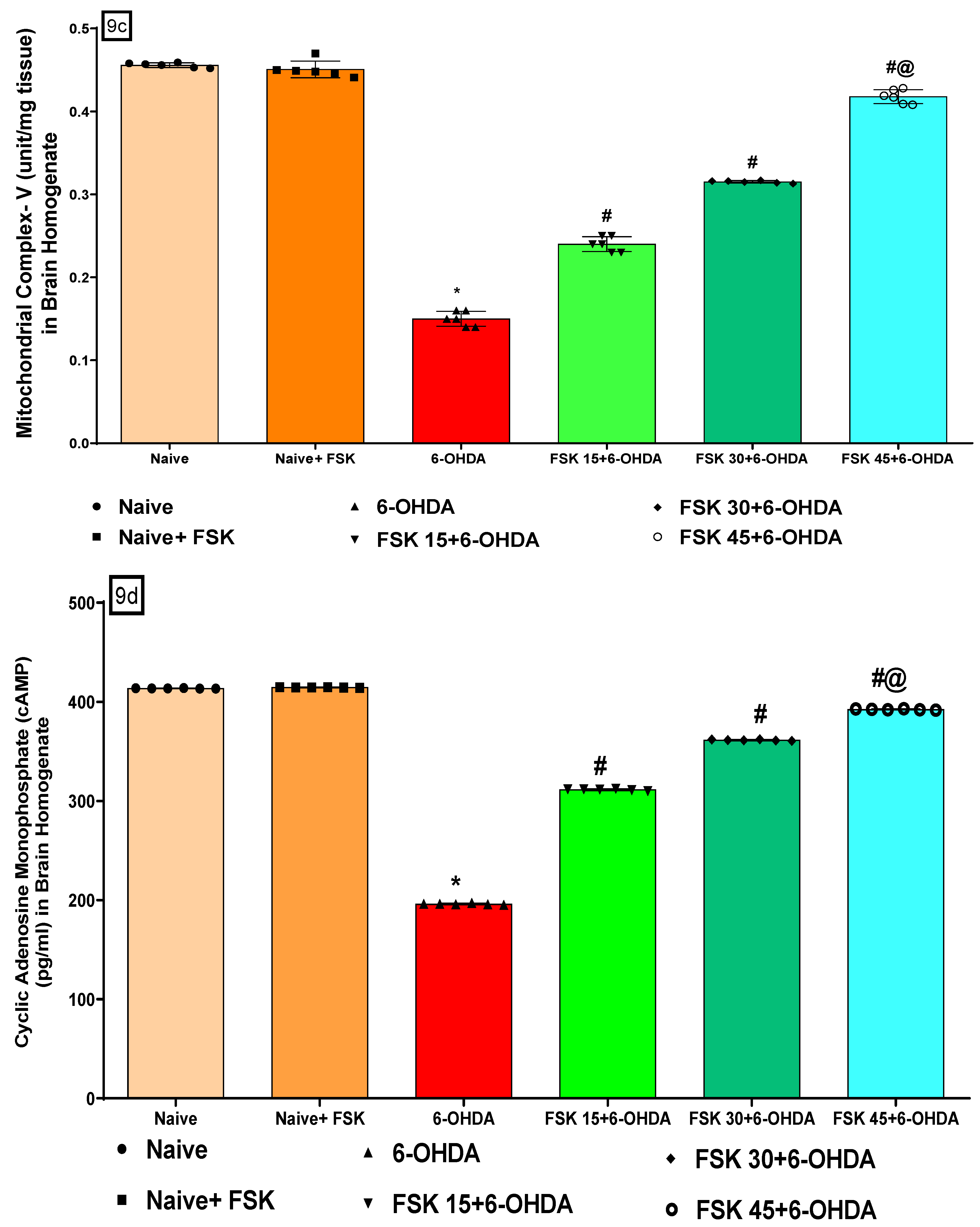
FSK Restored the ETC-Complexes (I, II, V) Mitochondrial Enzyme Levels in the Experimental Model of PD
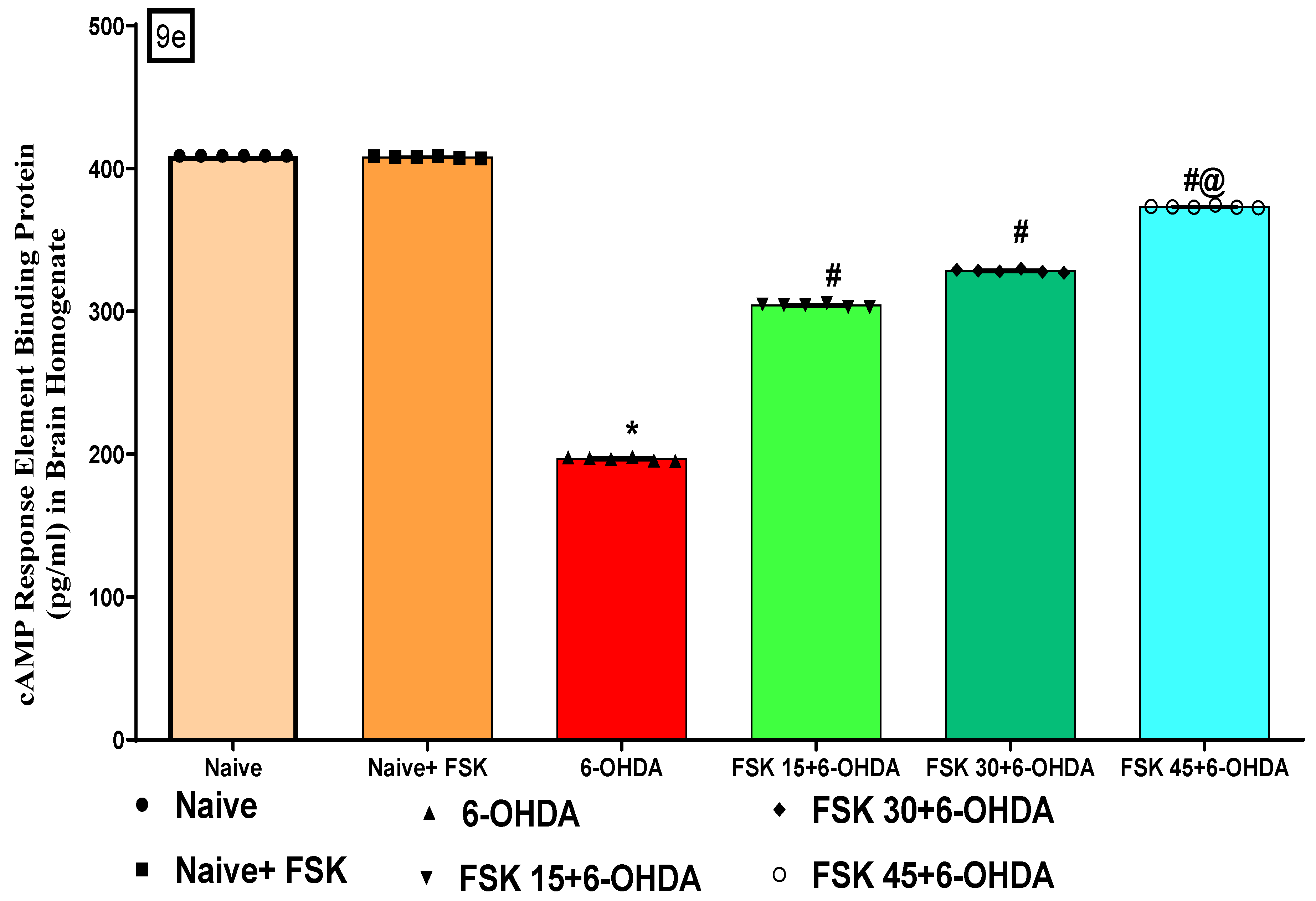
FSK Improved cAMP and CREB Protein Levels in the Experimental Model of PD
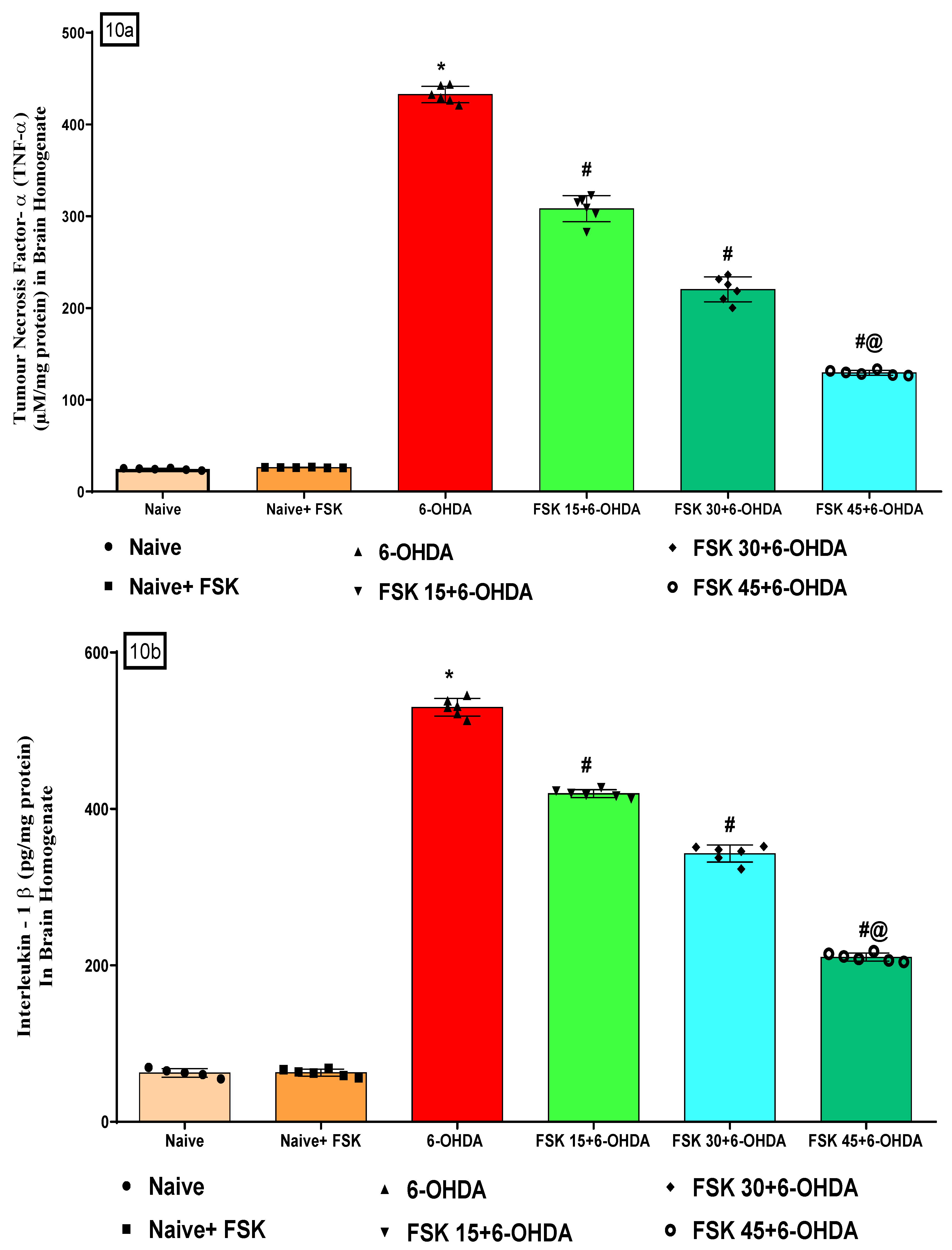
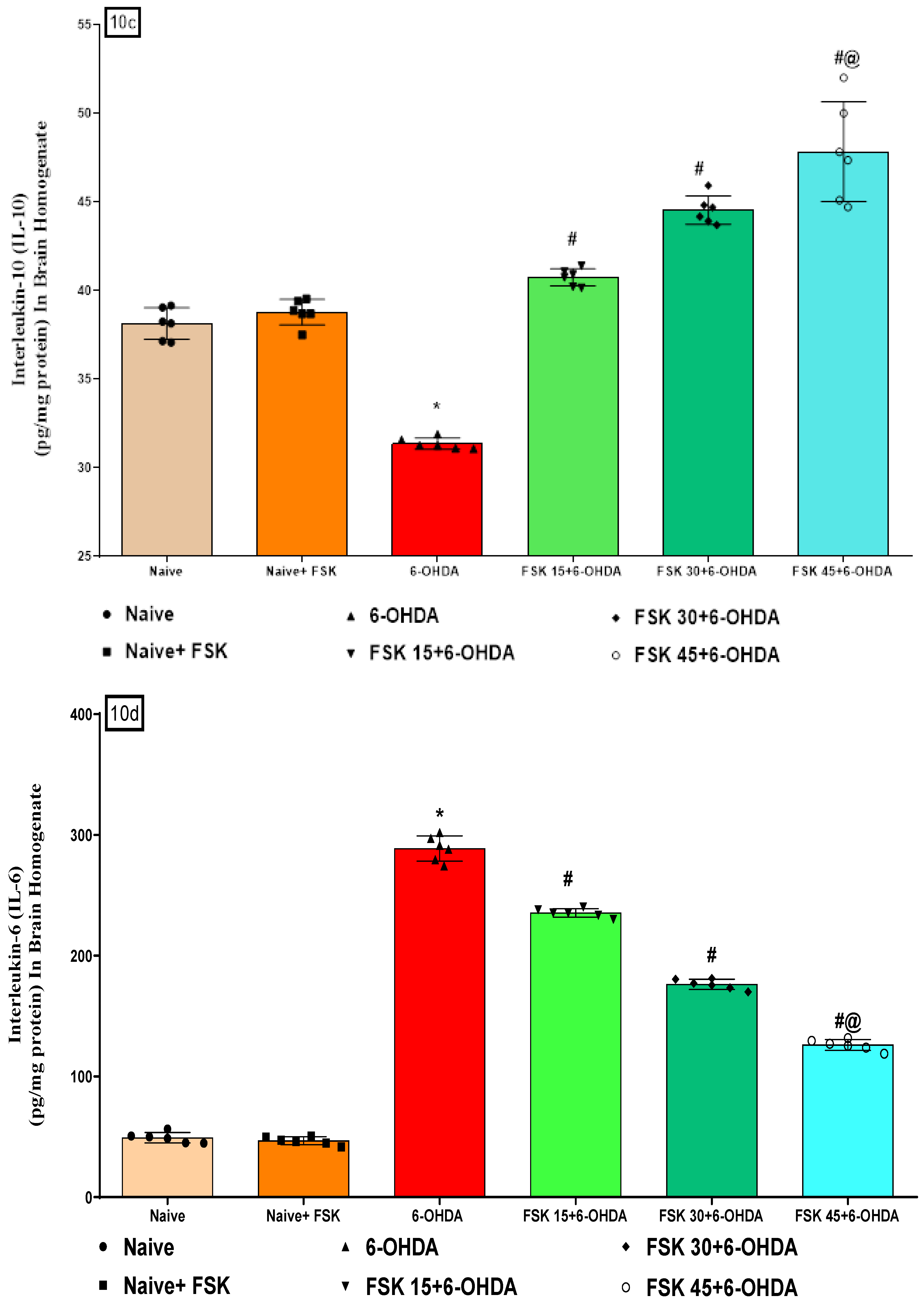
2.2.3. FSK Modulated Inflammatory Cytokines in the Experimental Model of PD
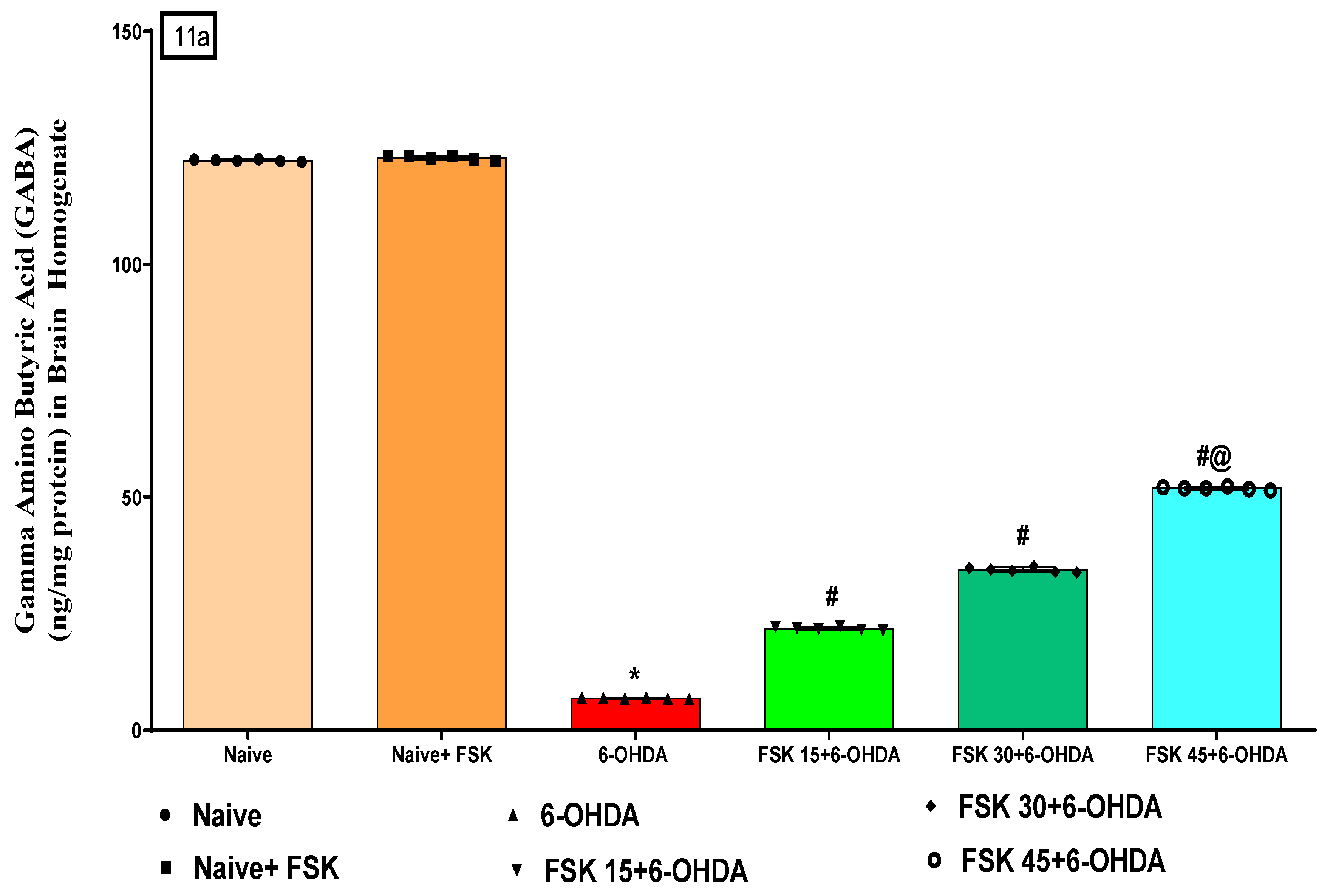
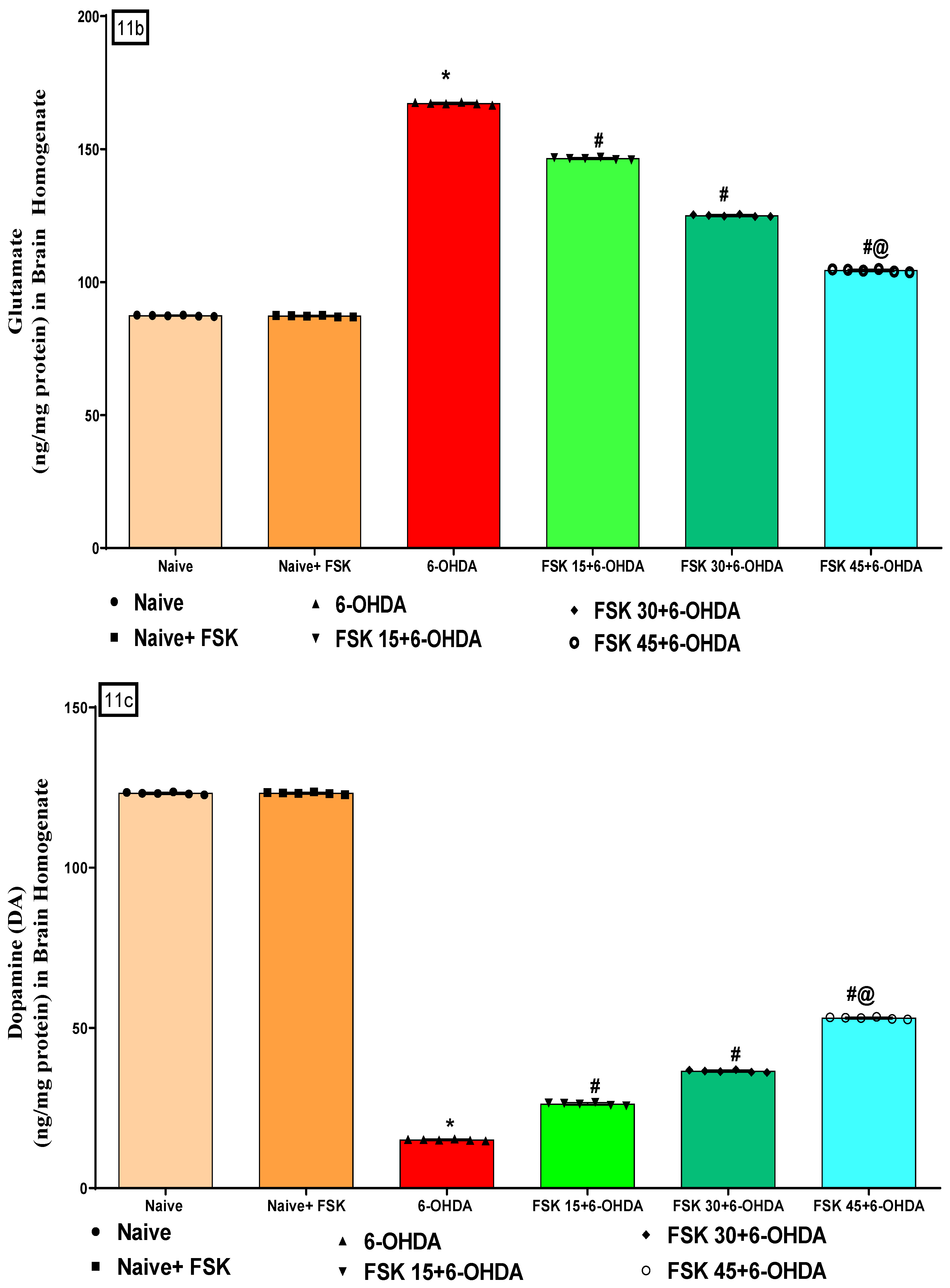
2.2.4. FSK Restored Neurotransmitters in the Experimental Model of PD
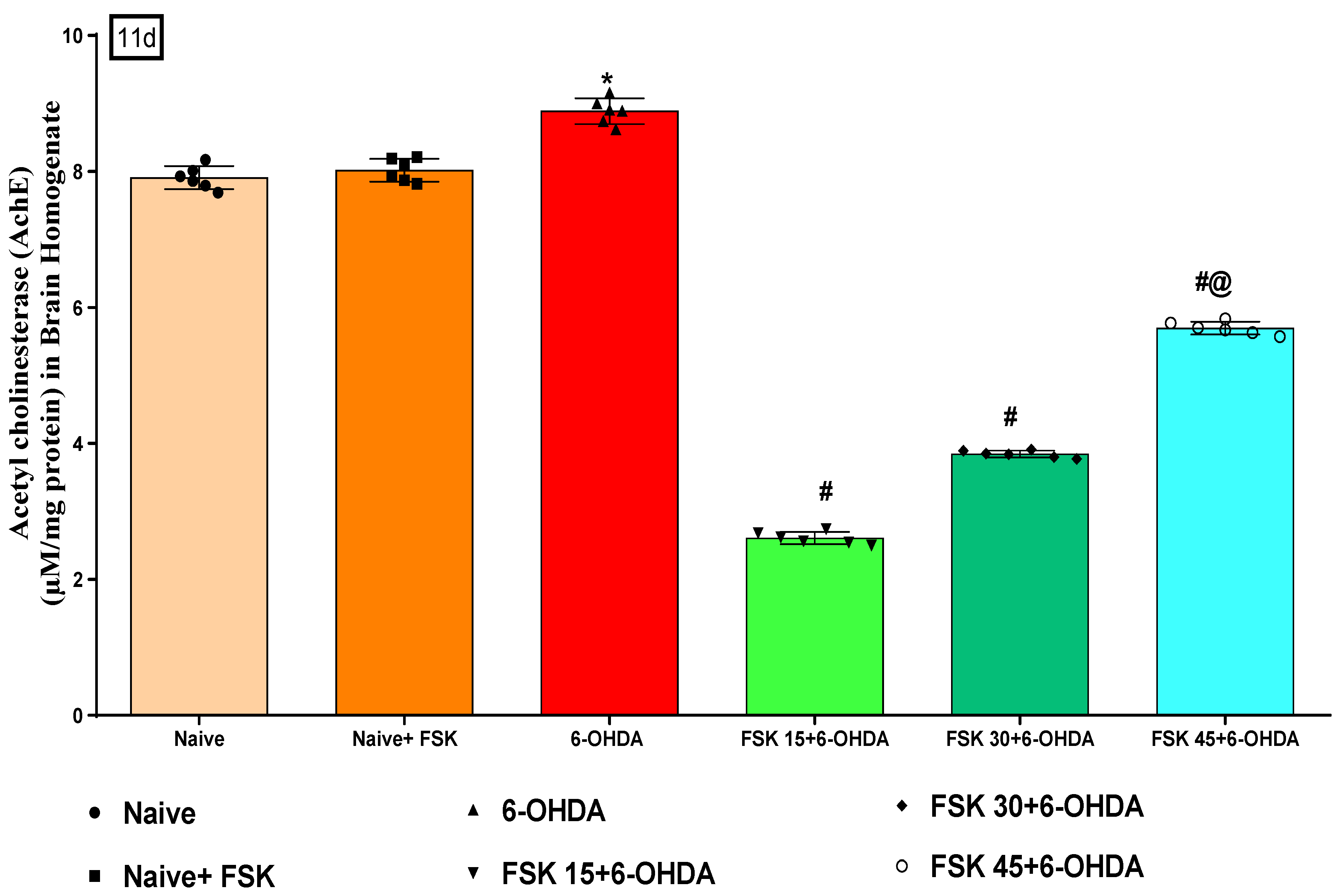
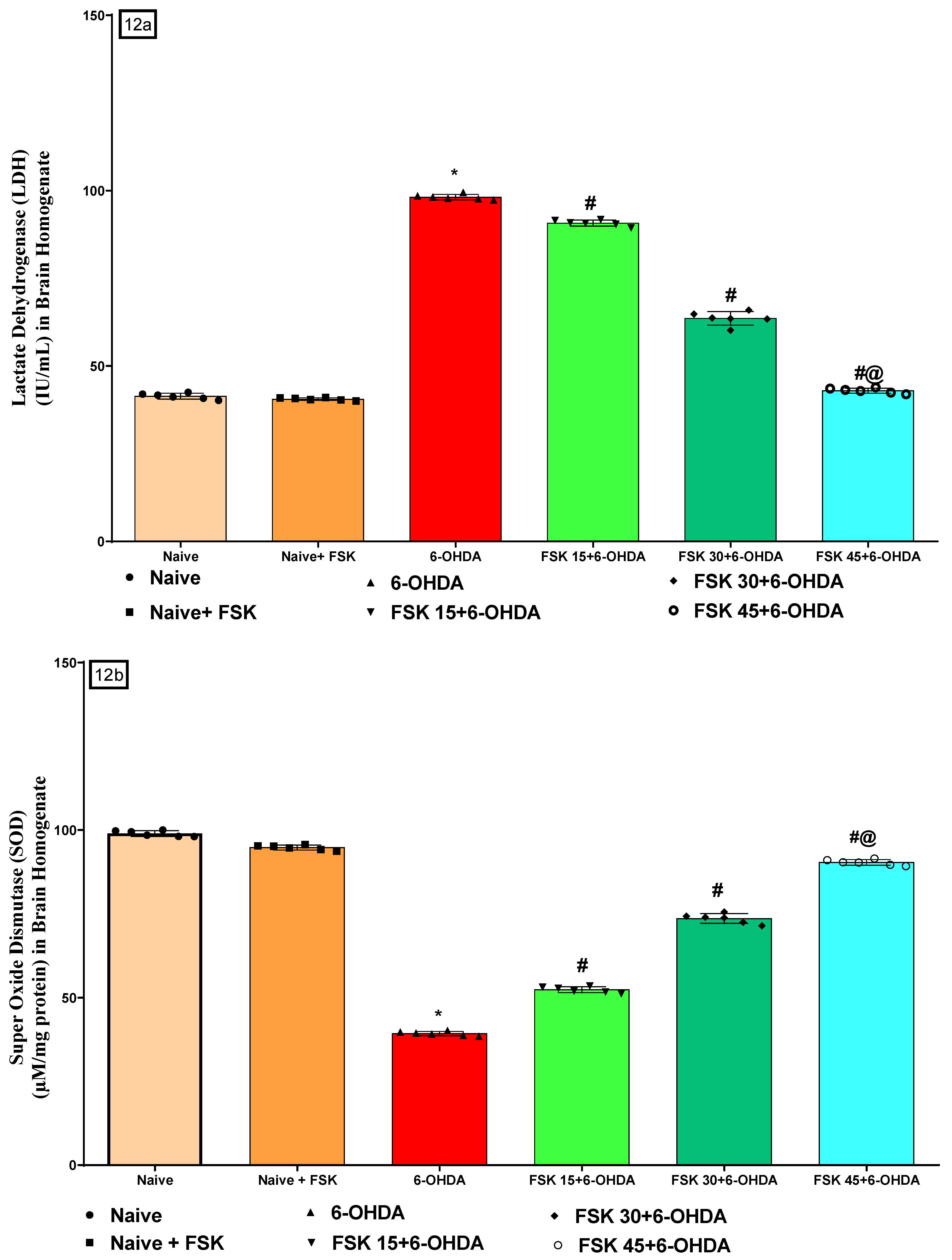
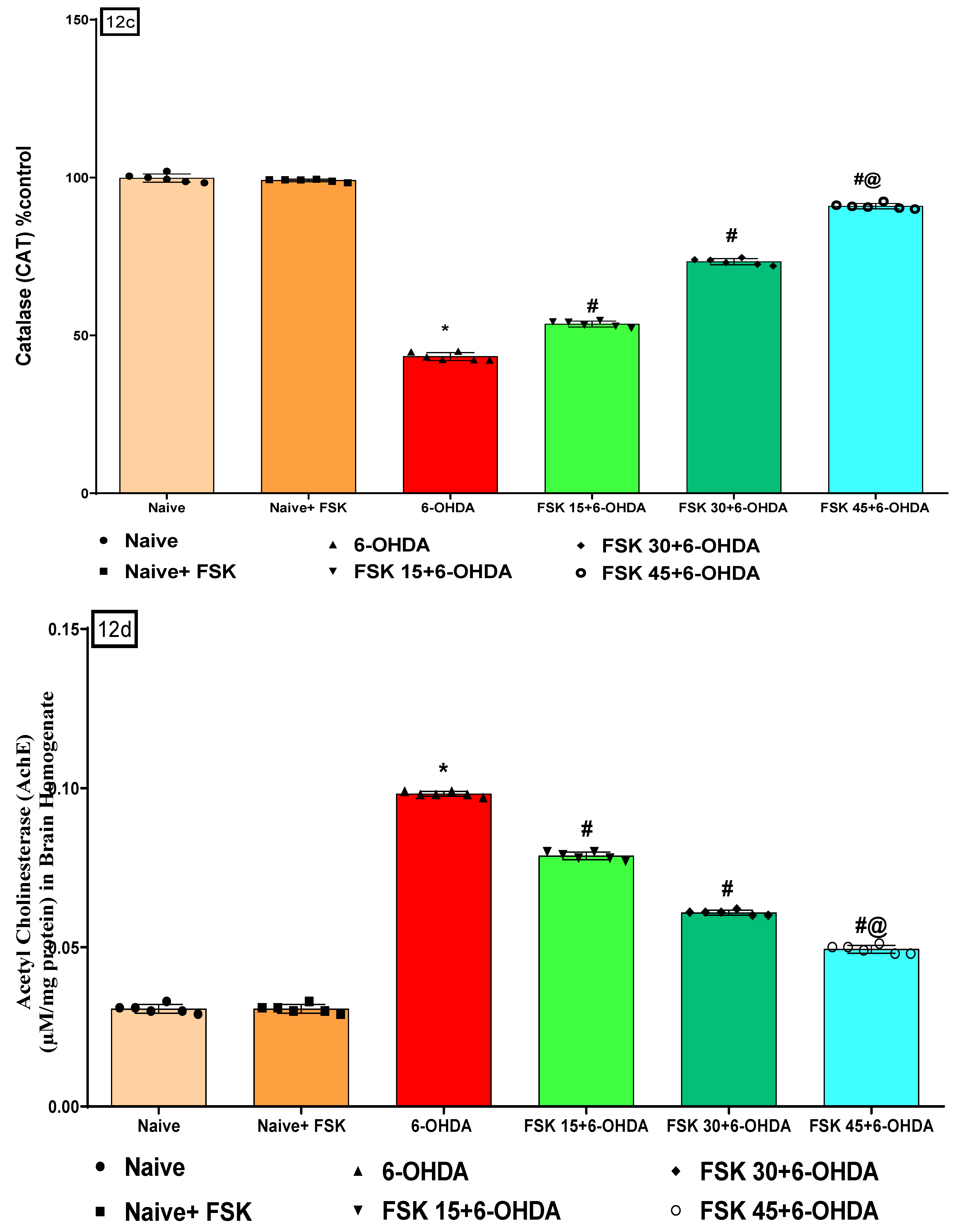
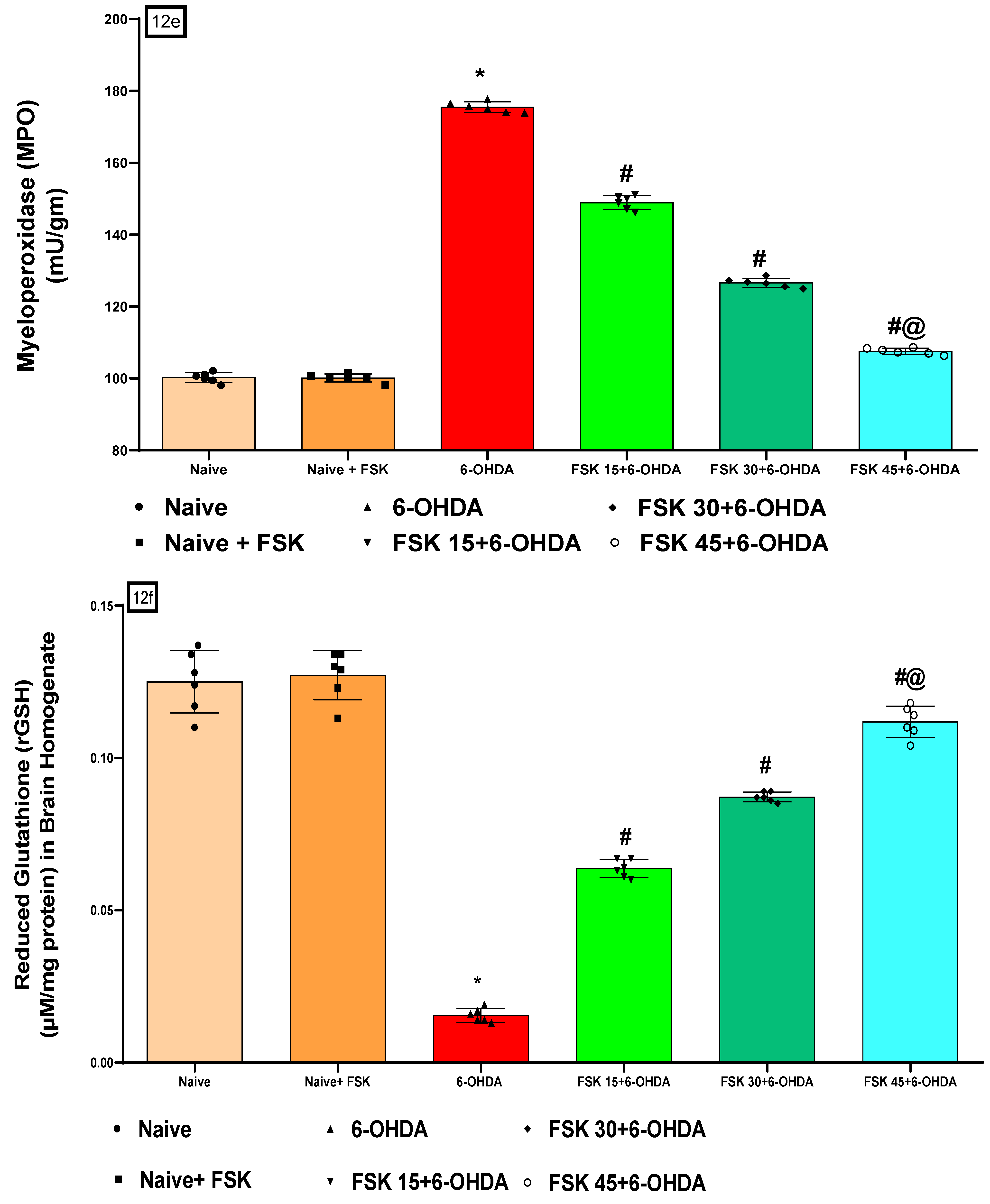
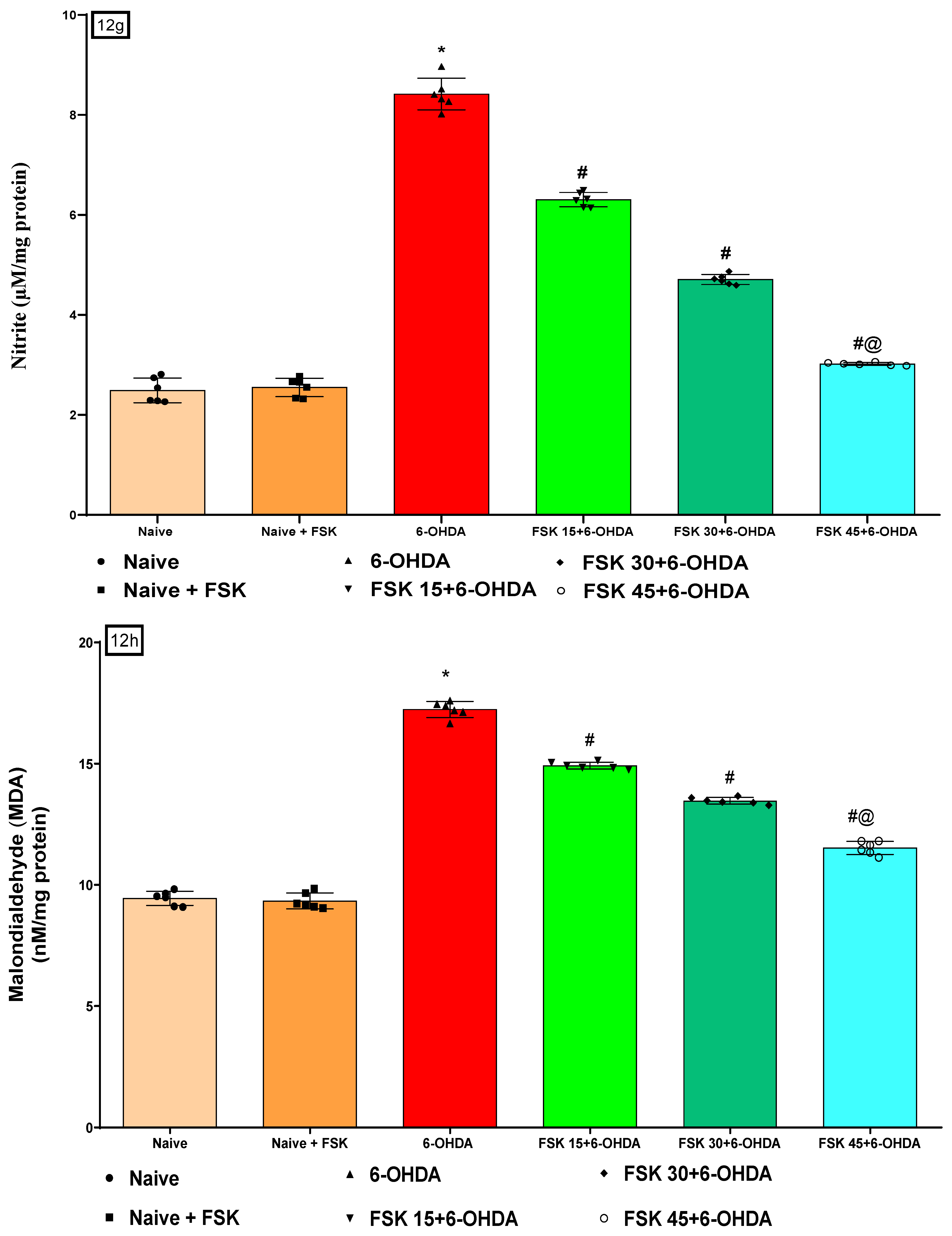
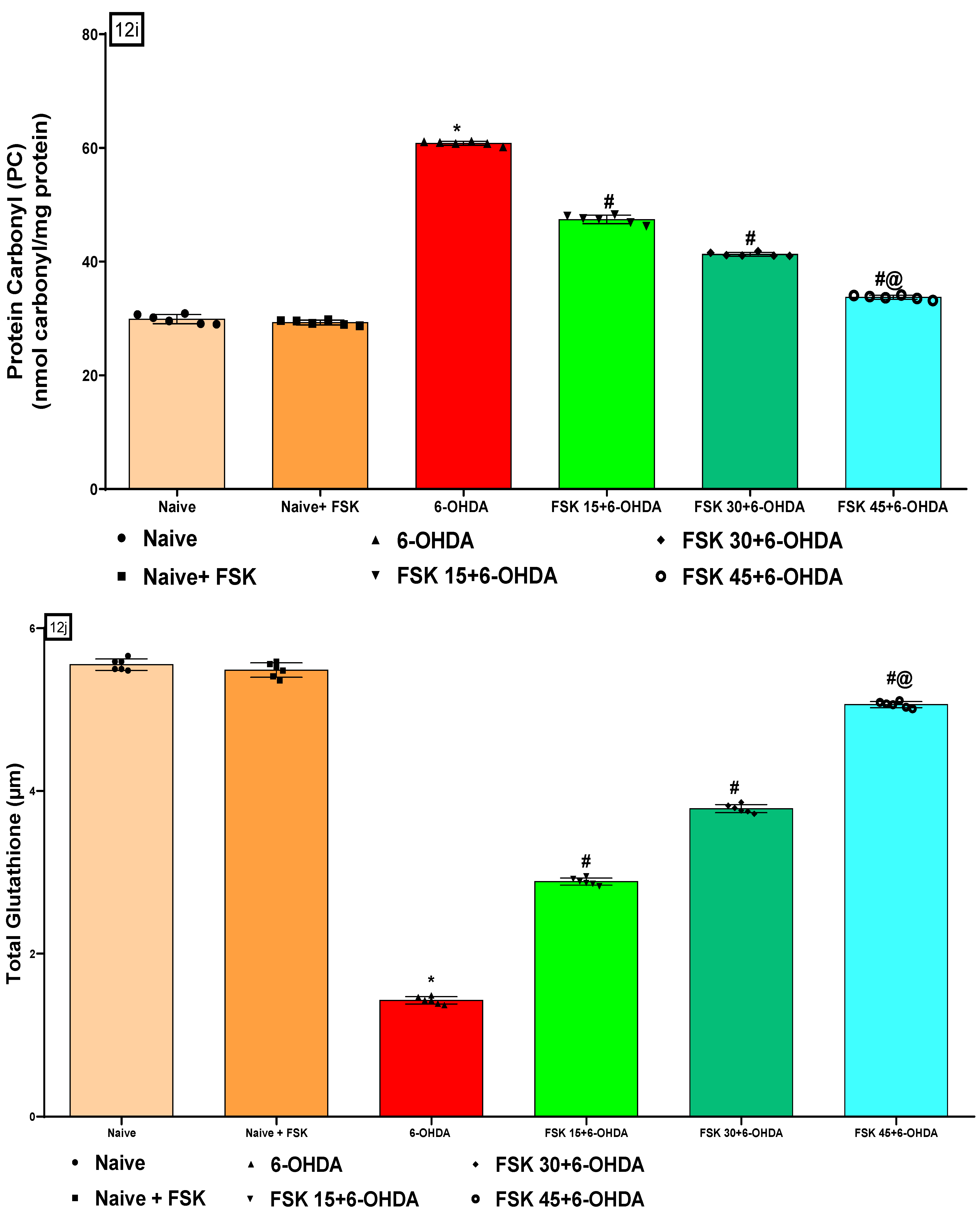
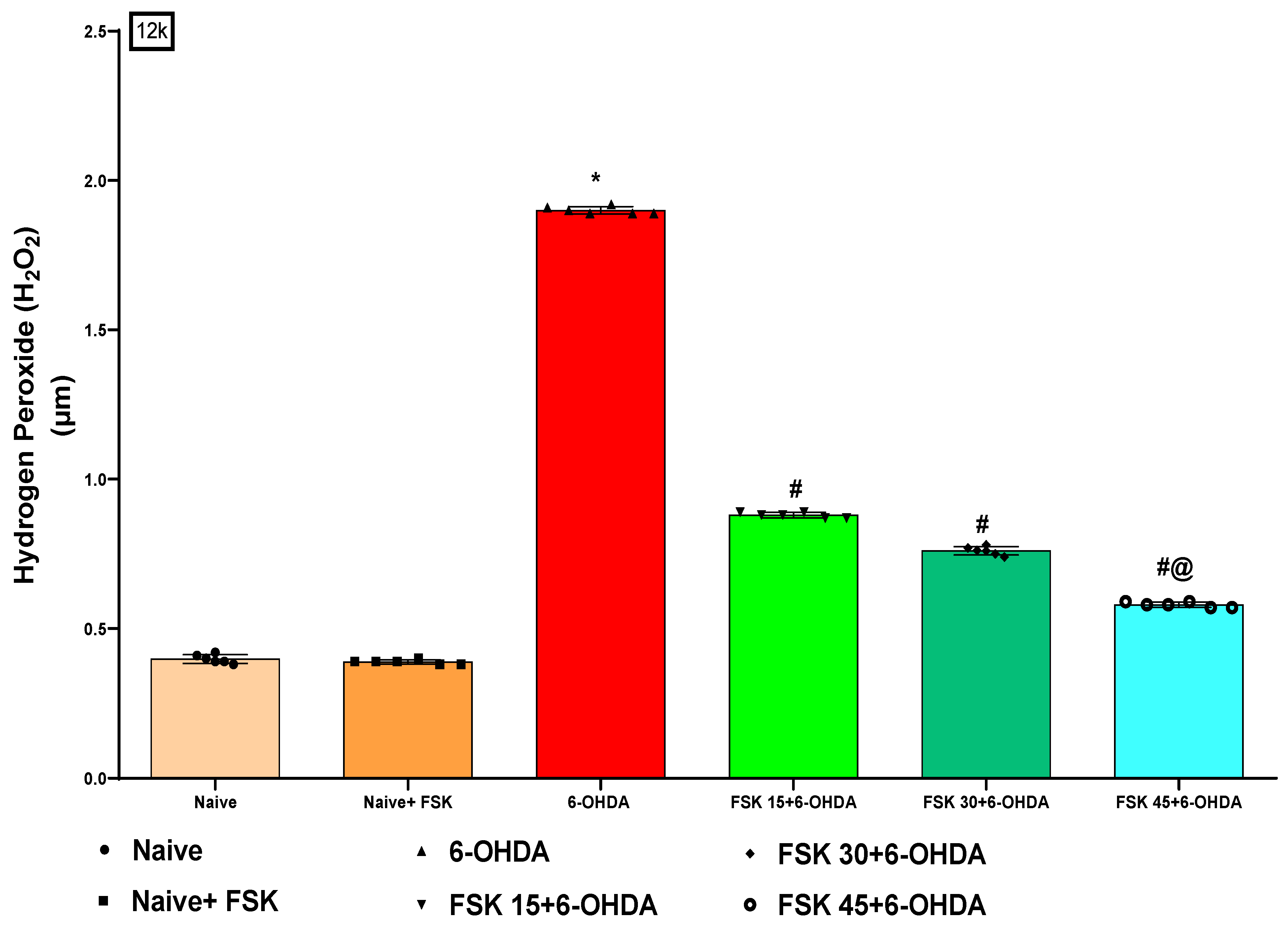
2.2.5. FSK Restored Anti-Oxidant Levels in the Experimental Model of PD
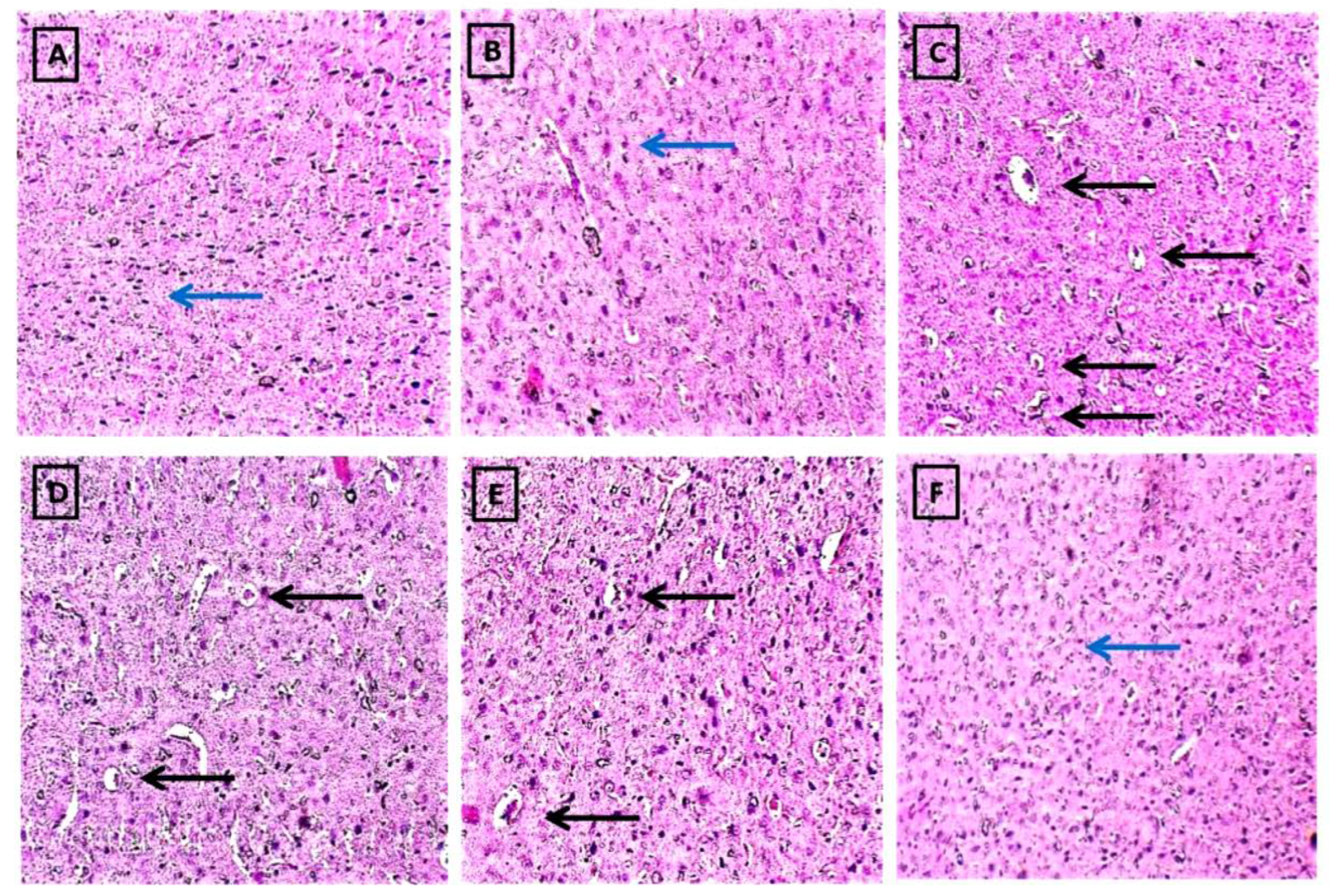
2.2.6. FSK Prevents Histopathological Alterations in a Striatal Brain Region in the Experimental Model of PD
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Drugs and Chemicals
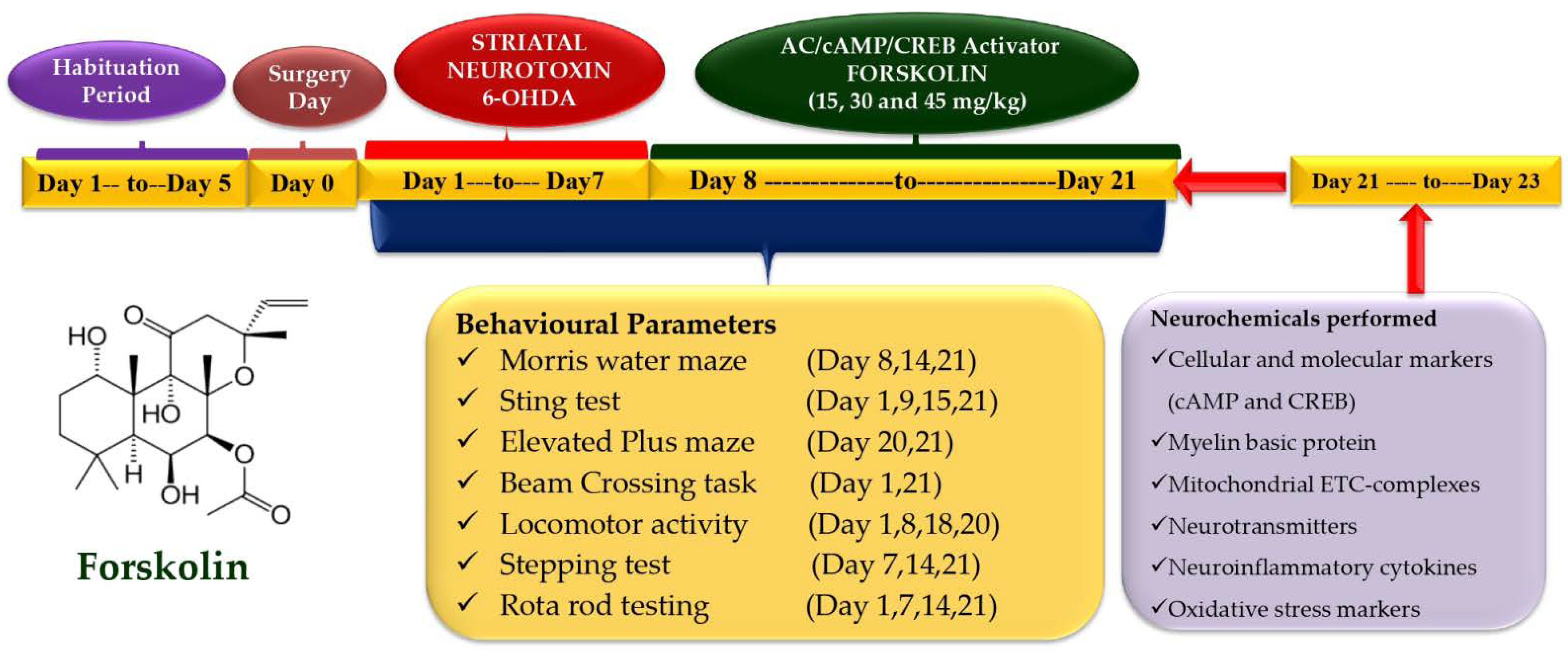
4.3. Experimental Protocol Schedule
4.4. Animal Model of 6-OHDA-Induced PD in Rats
4.5. Behavioural Parameters
4.5.1. Morris Water Maze (MWM) for Memory and Cognition
4.5.2. String Test for Grip Strength
4.5.3. Elevated Plus Maze Test (EPM) Task for Working Memory
4.5.4. Beam Crossing Task (BCT) for Neuromuscular Coordination
4.5.5. Actophotometer for Locomotion
4.5.6. Stepping Test for Akinesia
4.5.7. Rotarod Test for Grip and Neuromuscular Strength
4.6. Quantification of Biochemical Parameters
4.6.1. Estimation of Cellular and Molecular Markers
Analysis of Myelin Basic Protein (MBP) Level
4.6.2. Estimation of Mitochondrial ETC-Complexes’ Enzymes Activity
Preparation of Crude Mitochondrial Fraction from Rat Whole Brain Homogenate
Analysis of Complex-1 Enzyme Levels (NADPH Dehydrogenase)
Analysis of Complex-II Enzyme Level (Succinate Dehydrogenase/SDH)
Analysis of Complex-V Enzyme Level (ATPase)
Estimation of cAMP and CREB Levels
4.6.3. Estimation of Neuroinflammatory Cytokines
Analysis of TNF-α, IL-1β, IL-6 and IL-10 Levels
4.6.4. Neurotransmitter Measurement
Neuronal GABA and Glutamate Levels
Neuronal DA Level
Neuronal (Ach) Level
4.6.5. Assessment of Oxidative Stress Markers
Analysis of Lactate Dehydrogenase (LDH) Level
Analysis of Superoxide Dismutase (SOD) Level
Analysis of Catalase Level
Analysis of Acetylcholinesterase (AChE) Level
Analysis of Myeloperoxidase (MPO) Level
Analysis of Reduced Glutathione (GSH) Level
Analysis of Nitrite Level
Analysis of Malondialdehyde (MDA) Level
Analysis of Protein Carbonyl (PC) Level
Analysis of Total Glutathione Level
Analysis of Hydrogen Peroxide (H2O2) Level
4.7. Histopathological Analysis of Striatum
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| Aβ | Amyloid beta |
| AC | Adenylcyclase |
| ACh | Acetylcholine |
| AChE | Acetyl cholinesterase |
| ANOVA | Analysis of variance |
| ATP | Adenosine triphosphate |
| BCT | Beam crossing task |
| BDNF | Brain-derived neurotrophic factor |
| BG | Basal Ganglia |
| cAMP | Cyclic adenosine monophosphate |
| CAT | Catalase |
| CREB | cAMP-responsive element-binding protein |
| DA | Dopamine |
| DNA | Deoxy ribonucleic acid |
| DTNB | 5,5′-dithiobis-(2-nitrobenzoic acid) |
| ECD | Electron capture detector |
| EDTA | Ethylenediaminetetraacetic acid |
| ELISA | Enzyme-linked immunosorbent assay |
| ELT | Escape latency test |
| EPM | Elevated plus maze |
| ERK1/2 | Extracellular signal-regulated kinase 2 |
| ETC | Electron transport chain |
| FSK | Forskolin |
| GABA | Gamma- amino butyric acid |
| GSH | Glutathione |
| HD | Huntington’s disease |
| H2O2 | Hydrogen peroxide |
| HPLC | High-Performance Liquid Chromatography |
| IL-β | Interleukin beta |
| i-NOS | Inducible nitric oxide synthase |
| INT- p | Iodonitrotetrazolium |
| I.P. | Intraperitoneal |
| LA | Locomotor activity |
| LDH | Lactate dehydrogenase |
| LTP | Long-term memory potentiation |
| MBP | Myelin basic protein |
| MDA | Malondialdehyde |
| MPO | Myeloperoxidase |
| MPTP | Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MWM | Morris water maze |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NBW | Narrow beam walk |
| NGF | Neuronal growth factor |
| 6-OHDA | 6-Hydroxydopamine |
| PAF | Plasminogen activating factor |
| PC | Protein carbonyl |
| PD | Parkinson’s disease |
| PKA | Protein kinase A |
| PI3K/Akt | phosphatidylinositol 3-kinase and protein kinase B |
| PMS | phenyl methyl sulphide |
| PSA-NCAM | polysialylated neuronal cell adhesion molecule |
| P.O | by mouth |
| ROS | Reactive oxygen species |
| S.C | Subcutaneous |
| SDH | Succinate dehydrogenase |
| SLA | Spontaneous locomotor activity |
| SNcp | Substanianigra pars compacta |
| SOD | Superoxide dismutase |
| TCA | Trichloroacetic acid |
| TH | Tyrosine hydroxylase |
| TL | Transfer latency |
| TNF-α | Tumour necrosis factor-α |
| TSTQ | Time spent in the target quadrant zone |
References
- Greenland, J.C.; Barker, R.A. The differential diagnosis of Parkinson’s disease. Exon Publ. 2018, 21, 109–128. [Google Scholar]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Kathleen, A. Maguire-Zeiss. Protein Aggreg. Fibrillogenesis Cereb. Syst. Amyloid Dis. 2012, 65, 389. [Google Scholar]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s disease with the alpha-synuclein protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Surathi, P.; Jhunjhunwala, K.; Yadav, R.; Pal, P.K. Research in Parkinson’s disease in India: A review. Ann. Indian Acad. Neurol. 2016, 19, 9. [Google Scholar]
- Magrinelli, F.; Picelli, A.; Tocco, P.; Federico, A.; Roncari, L.; Smania, N.; Zanette, G.; Tamburin, S. Pathophysiology of motor dysfunction in Parkinson’s disease as the rationale for drug treatment and rehabilitation. Park. Dis. 2016, 2016, 9832839. [Google Scholar] [CrossRef]
- Pajares, M.; IRojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003. [Google Scholar]
- Zeng, X.S.; Geng, W.S.; Jia, J.J. Neurotoxin-induced animal models of Parkinson disease: Pathogenic mechanism and assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef]
- Perlbarg, V.; Lambert, J.; Butler, B.; Felfli, M.; Valabrègue, R.; Privat, A.L.; Lehéricy, S.; Petiet, A. Alterations of the nigrostriatal pathway in a 6-OHDA rat model of Parkinson’s disease evaluated with multimodal MRI. PLoS ONE 2018, 13, e0202597. [Google Scholar] [CrossRef]
- Antunes, M.S.; Goes, A.T.; Boeira, S.P.; Prigol, M.; Jesse, C.R. Protective effect of hesperidin in a model of Parkinson’s disease induced by 6-hydroxydopamine in aged mice. Nutrition 2014, 30, 1415–1422. [Google Scholar] [CrossRef]
- Mehan, S.; Kaur, G.; Dudi, R.; Rajput, M.; Kalra, S. Restoration of mitochondrial dysfunction in 6-hydroxydopamine induced Parkinson’s disease: A complete review. Open J. Park. Dis. Treat. 2017, 1, 001–026. [Google Scholar] [CrossRef]
- Hernandez-Baltazar, D.; Zavala-Flores, L.M.; Villanueva-Olivo, A. The 6-hydroxydopamine model and parkinsonian pathophysiology: Novel findings in an older model. Neurología 2017, 32, 533–539. [Google Scholar] [CrossRef]
- Antipova, V.; Wree, A.; Holzmann, C.; Mann, T.; Palomero-Gallagher, N.; Zilles, K.; Schmitt, O.; Hawlitschka, A. Unilateral botulinum neurotoxin-A injection into the striatum of C57BL/6 mice leads to a different motor behavior compared with rats. Toxins 2018, 10, 295. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Wu, X.; Sosunov, A.A.; McKhann, G.M.; Chen, J.X.; Yan, S.S. Cyclophilin D deficiency rescues Aβ-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim. (BBA)-Mol. Basis Dis. 2014, 1842, 2517–2527. [Google Scholar] [CrossRef]
- Mehan, S.; Khera, H.; Sharma, R. Neuroprotective Strategies of Blood-Brain Barrier Penetrant “Forskolin”(AC/cAMP/PK. In Recent Advances in Neurodegeneration; IntechOpen: London, UK, 2019; Volume 5. [Google Scholar]
- Lee, D. Global and local missions of cAMP signaling in neural plasticity, learning, and memory. Front. Pharmacol. 2015, 6, 161. [Google Scholar] [CrossRef]
- Chen, J.; Xu, J.; Huang, P.; Luo, Y.; Shi, Y.; Ma, P. The potential applications of traditional Chinese medicine in Parkinson’s disease: A new opportunity. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 149, 112866. [Google Scholar] [CrossRef]
- Vakilzadeh, G.; Khodagholi, F.; Ghadiri, T.; Darvishi, M.; Ghaemi, A.; Noorbakhsh, F.; Gorji, A.; Sharifzadeh, M. Protective effect of a cAMP analogue on behavioral deficits and neuropathological changes in cuprizone model of demyelination. Mol. Neurobiol. 2015, 52, 130–141. [Google Scholar] [CrossRef]
- Mehan, S.; Parveen, S.; Kalra, S. Adenylcyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders. Neural Regen. Res. 2017, 12, 290. [Google Scholar] [CrossRef]
- Salehi, B.; Staniak, M.; Czopek, K.; Stępień, A.; Dua, K.; Wadhwa, R.; Kumar Chellappan, D.; Sytar, O.; Brestic, M.; Ganesh Bhat, N.; et al. The Therapeutic Potential of the LabdaneDiterpenoidForskolin. Appl. Sci 2019, 9, 4089. [Google Scholar] [CrossRef]
- Puranam, K.; Rani, H.S. Forskolin: Its therapeutic applications. Int. J. Pharma Bio Sci. 2014, 5, 68–73. [Google Scholar]
- Alam, M.M.; Minj, E.; Yadav, R.K.; Mehan, S. Neuroprotective potential of adenylcyclase/cAMP/CREB and mitochondrial CoQ10 activator in amyotrophic lateral sclerosis rats. Curr. Bioact. Compd. 2021, 17, 53–69. [Google Scholar] [CrossRef]
- Kapoor, T.; Mehan, S.; Suri, M.; Sharma, N. Forskolin, an adenylcyclase/cAMP/CREB signaling activator restoring myelin-associated oligodendrocyte destruction in experimental ethidium bromide model of multiple sclerosis. Cells 2022, 11, 2771. [Google Scholar] [CrossRef]
- Gorji, H. Role of Adenylyl Cyclase Type 5 in the Regulation of the Dopamine D3 Receptor Phosphorylation. Doctoral Dissertation, University of Ottawa, Ottawa, ON, Canada.
- Han, X.R.; Wen, X.; Wang, Y.J.; Wang, S.; Shen, M.; Zhang, Z.F.; Fan, S.H.; Shan, Q.; Wang, L.; Li, M.Q.; et al. Effects of CREB1 gene silencing on cognitive dysfunction by mediating PKA-CREB signaling pathway in mice with vascular dementia. Mol. Med. 2018, 24, 18. [Google Scholar] [CrossRef]
- Mehan, S.; Rahi, S.; Tiwari, A.; Kapoor, T.; Rajdev, K.; Sharma, R.; Khera, H.; Kosey, S.; Kukkar, U. DudiRAdenylcyclase activator forskolin alleviates intracerebroventricular propionic acid-induced mitochondrial dysfunction of autistic rats. Neural Regen. Res. 2020, 15, 1140. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; ZTarunnoni, M.B.; Bekinschtein, P. Brain-derived neurotrophic factor: A key molecule for memory in the healthy and the pathological brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Dumaz, N.; Marais, R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways: Based on The Anniversary Prize of the GesellschaftfürBiochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 2005, 272, 3491–3504. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Kinane, J.; Oliver, R.P. Evidence that the appressorial development in barley powdery mildew is controlled by MAP kinase activity in conjunction with the cAMP pathway. Fungal Genet. Biol. 2003, 39, 94–102. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, H.; Song, Y.; Chen, S.Y.; Wang, Y.; Sun, Y.; Tang, C.; Du, J.; Huang, Y. Endogenous sulfur dioxide is a novel inhibitor of hypoxia-induced mast cell degranulation. J. Adv. Res. 2021, 29, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Yu, H.; Huang, C.; Zhong, Q.; Chen, Y.; Xie, J.; Zhou, Z.; Xu, J.; Wang, H. Inhibition of phosphodiesterase 4 by FCPR16 protects SH-SY5Y cells against MPP+-induced decline of mitochondrial membrane potential and oxidative stress. Redox Biol. 2018, 16, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Anis, E.; Zafeer, M.F.; Firdaus, F.; Islam, S.N.; Khan, A.A.; Hossain, M.M. Perillyl alcohol mitigates behavioural changes and limits cell death and mitochondrial changes in unilateral 6-OHDA lesion model of Parkinson’s disease through alleviation of oxidative stress. Neurotox. Res. 2020, 38, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis; Exon Publications: Brisbane, Australia, 2018; pp. 3–26. [Google Scholar]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, E.A.; Melzer, C.; Schönberger, A.; Hess, M.; Timmermann, L.; Eggers, C.; Tittgemeyer, M. Axonal degeneration in Parkinson’s disease–Basal ganglia circuitry and D2 receptor availability. NeuroImage Clin. 2019, 23, 101906. [Google Scholar] [CrossRef]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial uncoupling: A key controller of biological processes in physiology and diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Matsumoto, C.S.; Mizunami, M. Signaling pathways for long-term memory formation in the cricket. Front. Psychol. 2018, 9, 1014. [Google Scholar] [CrossRef]
- Hynie, S.; Šída, P.; Klenerová, V. Inhibitory and stimulatory effects of somatostatin and some of its derivatives on cAMP formation in rat striatum and hippocampus. Collect. Czechoslov. Chem. Commun. 2016, 4, 56–59. [Google Scholar]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 1–12. [Google Scholar] [CrossRef]
- Haense, C.; Kalbe, E.; Herholz, K.; Hohmann, C.; Neumaier, B.; Krais, R.; Heiss, W.D. Cholinergic system function and cognition in mild cognitive impairment. Neurobiol. Aging 2012, 33, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Perfeito, R.; Cunha-Oliveira, T.; Rego, A.C. Revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease—Resemblance to the effect of amphetamine drugs of abuse. Free Radic. Biol. Med. 2012, 53, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Z.; Guo, X.T.; Chen, J.W.; Zhao, Y.; Cong, X.; Jiang, Z.L.; Cao, R.F.; Cui, K.; Gao, S.S.; Tian, W.R. Saikosaponin-D attenuates heat stress-induced oxidative damage in LLC-PK1 cells by increasing the expression of anti-oxidant enzymes and HSP72. Am. J. Chin. Med. 2014, 42, 1261–1277. [Google Scholar] [CrossRef]
- Wang, Y.H.; Xuan, Z.H.; Tian, S.; He, G.R.; Du, G.H. Myricitrin attenuates 6-hydroxydopamine-induced mitochondrial damage and apoptosis in PC12 cells via inhibition of mitochondrial oxidation. J. Funct. Foods 2013, 5, 337–345. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Liu, L.X.; Du, D.; Wang, Z.Q.; Fang, Y.; Zheng, T.; Dong, Y.C.; Shi, Q.L.; Zhao, M.; Xiao, F.; Du, J. Differences in brain pathological changes between rotenone and 6-hydroxydopamine Parkinson’s disease models. Neural Regen. Res. 2018, 13, 1276. [Google Scholar] [CrossRef]
- Bala, R.; Khanna, D.; Mehan, S.; Kalra, S. Experimental evidence for the potential of lycopene in the management of scopolamine induced amnesia. RSC Adv. 2015, 5, 72881–72892. [Google Scholar] [CrossRef]
- Dudi, R.; Mehan, S. Neuroprotection of brain permeable Forskolin ameliorates behavioral, biochemical and histopatho-logical alterations in rat model of intracerebral hemorrhage. Pharmaspire 2018, 10, 68–86. [Google Scholar]
- Sahu, R.; Mehan, S.; Kumar, S.; Prajapati, A.; Alshammari, A.; Alharbi, M.; Assiri, M.A.; Narula, A.S. Effect of alpha-mangostin in the prevention of behavioural and neurochemical defects in methylmercury-induced neurotoxicity in experimental rats. Toxicol. Rep. 2022, 9, 977–998. [Google Scholar] [CrossRef]
- Sharma, A.; Mehan, S. Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 2021, 147, 105067. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Upadhayay, S.; Mehan, S. Inhibition of c-JNK/p38MAPK signaling pathway by Apigenin prevents neurobehavioral and neurochemical defects in ethidium bromide-induced experimental model of multiple sclerosis in rats: Evidence from CSF, blood plasma and brain samples. Phytomed. Plus 2021, 1, 100139. [Google Scholar] [CrossRef]
- BonaccorsoMarinelli, M.P.; Ledesma, M.J.; Nieto Grimalt, F.E.; Cabrera, R.J. Video Tracking Analysis System for Forelimb Akinesia test in the Rat Parkinson model. In Proceedings of the InVII Latin American Congress on Biomedical Engineering CLAIB 2016, Bucaramanga, Colombia, 26–28 October 2016; Springer: Singapore, 2016; pp. 745–748. [Google Scholar]
- Sharma, N.; Upadhayay, S.; Shandilya, A.; Sahu, R.; Singh, A.; Rajkhowa, B.; Mehan, S. Neuroprotection by solanesol against ethidium bromide-induced multiple sclerosis-like neurobehavioral, molecular, and neurochemical alterations in experimental rats. Phytomed. Plus 2021, 1, 100051. [Google Scholar] [CrossRef]
- Shandilya, A.; Mehan, S.; Kumar, S.; Sethi, P.; Narula, A.S.; Alshammari, A.; Alharbi, M.; Alasmari, A.F. Activation of IGF-1/GLP-1 Signalling via 4-Hydroxyisoleucine Prevents Motor Neuron Impairments in Experimental ALS-Rats Exposed to Methylmercury-Induced Neurotoxicity. Molecules 2022, 27, 3878. [Google Scholar] [CrossRef] [PubMed]
- Rajkhowa, B.; Mehan, S.; Sethi, P.; Prajapati, A.; Suri, M.; Kumar, S.; Bhalla, S.; Narula, A.S.; Alshammari, A.; Alharbi, M.; et al. Activating SIRT-1 Signalling with the Mitochondrial-CoQ10 Activator Solanesol Improves Neurobehavioral and Neurochemical Defects in Ouabain-Induced Experimental Model of Bipolar Disorder. Pharmaceuticals 2022, 15, 959. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, K.; Siddiqui, E.M.; Jadaun, K.S.; Mehan, S. Neuroprotective potential of solanesol in a combined model of intracerebral and intraventricular hemorrhage in rats. IBRO Rep. 2020, 8, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Minj, E.; Upadhayay, S.; Mehan, S. Nrf2/HO-1 signaling activator acetyl-11-keto-beta Boswellic acid (AKBA)-Mediated Neuroprotection in Methyl Mercury-induced experimental model of ALS. Neurochem. Res. 2021, 46, 2867–2884. [Google Scholar] [CrossRef]
- Khera, R.; Mehan, S.; Bhalla, S.; Kumar, S.; Alshammari, A.; Alharbi, M.; Sadhu, S.S. Guggulsterone mediated JAK/STAT and PPAR-gamma modulation prevents neurobehavioral and neurochemical abnormalities in propionic acid-induced experimental model of autism. Molecules 2022, 27, 889. [Google Scholar] [CrossRef]
- Gupta, R.; Mehan, S.; Sethi, P.; Prajapati, A.; Alshammari, A.; Alharbi, M.; Al-Mazroua, H.A.; Narula, A.S. Smo-Shh agonist Purmorphamine prevents neurobehavioral and neurochemical defects in 8-OH-DPAT-induced experimental model of obsessive-compulsive disorder. Brain Sci. 2022, 12, 342. [Google Scholar] [CrossRef]
- Agarwal, V.; Kaushik, A.S.; Rehman, M.; Chaudhary, R.; Jawaid, T.; Kamal, M.; Mishra, V. Interleukin-6 expression and its modulation by diacerein in a rat model of chronic stress induced cardiac dysfunction. Heliyon 2021, 7, e08522. [Google Scholar] [CrossRef]
- Yadav, R.K.; Mehan, S.; Sahu, R.; Kumar, S.; Khan, A.; Makeen, H.A.; Al Bratty, M. Protective effects of apigenin on methylmercury-induced behavioral/neurochemical abnormalities and neurotoxicity in rats. Hum. Exp. Toxicol. 2022, 41, 09603271221084276. [Google Scholar] [CrossRef] [PubMed]
- Rahi, S.; Gupta, R.; Sharma, A.; Mehan, S. Smo-Shh signaling activator purmorphamine ameliorates neurobehavioral, molecular, and morphological alterations in an intracerebroventricular propionic acid-induced experimental model of autism. Hum. Exp. Toxicol. 2021, 40, 1880–1898. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Parveen, S.; Mehan, S.; Khanna, D.; Kalra, S. Neuroprotective effect of ellagic acid against chronically scopolamine induced Alzheimer’s type memory and cognitive dysfunctions: Possible behavioural and biochemical evidences. Int. J. Preven. Med. Res. 2015, 1, 45–64. [Google Scholar]
- Deshmukh, R.; Sharma, V.; Mehan, S.; Sharma, N.; Bedi, K.L. Amelioration of intracerebroventricularstreptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine—A PDE1 inhibitor. Eur. J. Pharmacol. 2009, 620, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Khera, R.; Rahi, S.; Mehan, S.; Makeen, H.A.; Khormi, Y.H.; Rehman, M.U.; Khan, A. Neuroprotective effect of α-mangostin in ameliorating propionic acid-induced experimental model of autism in Wistar rats. Brain Sci. 2021, 11, 288. [Google Scholar] [CrossRef]
- Feng, Q.; Wang, Y.I.; Yang, Y. Neuroprotective effect of interleukin-6 in a rat model of cerebral ischemia. Exp. Ther. Med. 2015, 9, 1695–1701. [Google Scholar] [CrossRef]
- Sharma, R.; Rahi, S.; Mehan, S. Neuroprotective potential of solanesol in intracerebroventricular propionic acid induced experimental model of autism: Insights from behavioral and biochemical evidence. Toxicol. Rep. 2019, 6, 1164–1175. [Google Scholar] [CrossRef]
- Kumar, N.; Sharma, N.; Khera, R.; Gupta, R.; Mehan, S. Guggulsterone ameliorates ethidium bromide-induced experimental model of multiple sclerosis via restoration of behavioral, molecular, neurochemical and morphological alterations in rat brain. Metab. Brain Dis. 2021, 36, 911–925. [Google Scholar] [CrossRef]
- Upadhayay, S.; Mehan, S.; Prajapati, A.; Sethi, P.; Suri, M.; Zawawi, A.; Almashjary, M.N.; Tabrez, S. Nrf2/HO-1 Signaling Stimulation through Acetyl-11-Keto-Beta-Boswellic Acid (AKBA) Provides Neuroprotection in Ethidium Bromide-Induced Experimental Model of Multiple Sclerosis. Genes 2022, 13, 1324. [Google Scholar] [CrossRef]
- Liu, Z.; Kumar, M.; Devi, S.; Kabra, A. The mechanisms of cucurbitacin E as a neuroprotective and memory-enhancing agent in a cerebral hypoperfusion rat model: Attenuation of oxidative stress, inflammation, and excitotoxicity. Front. Pharmacol. 2021, 12, 794933. [Google Scholar] [CrossRef]
- Scalcon, V.; Folda, A.; Lupo, M.G.; Tonolo, F.; Pei, N.; Battisti, I.; Ferri, N.; Arrigoni, G.; Bindoli, A.; Holmgren, A.; et al. Mitochondrial depletion of glutaredoxin 2 induces metabolic dysfunction-associated fatty liver disease in mice. Redox Biol. 2022, 51, 102277. [Google Scholar] [CrossRef] [PubMed]
- Gorąca, A.; Asłanowicz-Antkowiak, K. Prophylaxis with α-lipoic acid against lipopolysaccharide-induced brain injury in rats. Arch. Immunol. Ther. Exp. 2009, 57, 141–146. [Google Scholar] [CrossRef] [PubMed]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alharbi, M.; Alshammari, A.; Kaur, G.; Kalra, S.; Mehan, S.; Suri, M.; Chhabra, S.; Kumar, N.; Alanazi, W.A.; Alshanwani, A.R.; et al. Effect of Natural Adenylcyclase/cAMP/CREB Signalling Activator Forskolin against Intra-Striatal 6-OHDA-Lesioned Parkinson’s Rats: Preventing Mitochondrial, Motor and Histopathological Defects. Molecules 2022, 27, 7951. https://doi.org/10.3390/molecules27227951
Alharbi M, Alshammari A, Kaur G, Kalra S, Mehan S, Suri M, Chhabra S, Kumar N, Alanazi WA, Alshanwani AR, et al. Effect of Natural Adenylcyclase/cAMP/CREB Signalling Activator Forskolin against Intra-Striatal 6-OHDA-Lesioned Parkinson’s Rats: Preventing Mitochondrial, Motor and Histopathological Defects. Molecules. 2022; 27(22):7951. https://doi.org/10.3390/molecules27227951
Chicago/Turabian StyleAlharbi, Metab, Abdulrahman Alshammari, Gurpreet Kaur, Sanjeev Kalra, Sidharth Mehan, Manisha Suri, Swesha Chhabra, Nitish Kumar, Wael A. Alanazi, Aliah R. Alshanwani, and et al. 2022. "Effect of Natural Adenylcyclase/cAMP/CREB Signalling Activator Forskolin against Intra-Striatal 6-OHDA-Lesioned Parkinson’s Rats: Preventing Mitochondrial, Motor and Histopathological Defects" Molecules 27, no. 22: 7951. https://doi.org/10.3390/molecules27227951
APA StyleAlharbi, M., Alshammari, A., Kaur, G., Kalra, S., Mehan, S., Suri, M., Chhabra, S., Kumar, N., Alanazi, W. A., Alshanwani, A. R., AL-Ghamdi, A. H., Narula, A. S., & Kalfin, R. (2022). Effect of Natural Adenylcyclase/cAMP/CREB Signalling Activator Forskolin against Intra-Striatal 6-OHDA-Lesioned Parkinson’s Rats: Preventing Mitochondrial, Motor and Histopathological Defects. Molecules, 27(22), 7951. https://doi.org/10.3390/molecules27227951