

Structure-Based Discovery and Biological Assays of a Novel PRMT5 Inhibitor for Non-Small Cell Lung Cancer

Abstract
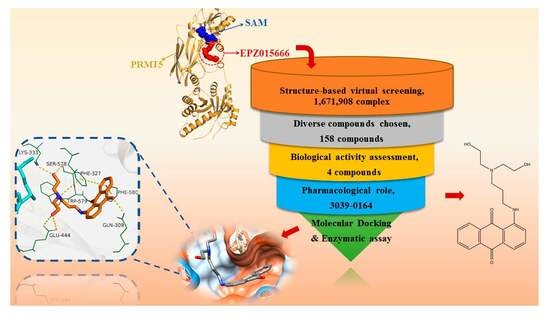
1. Introduction
2. Materials and Methods
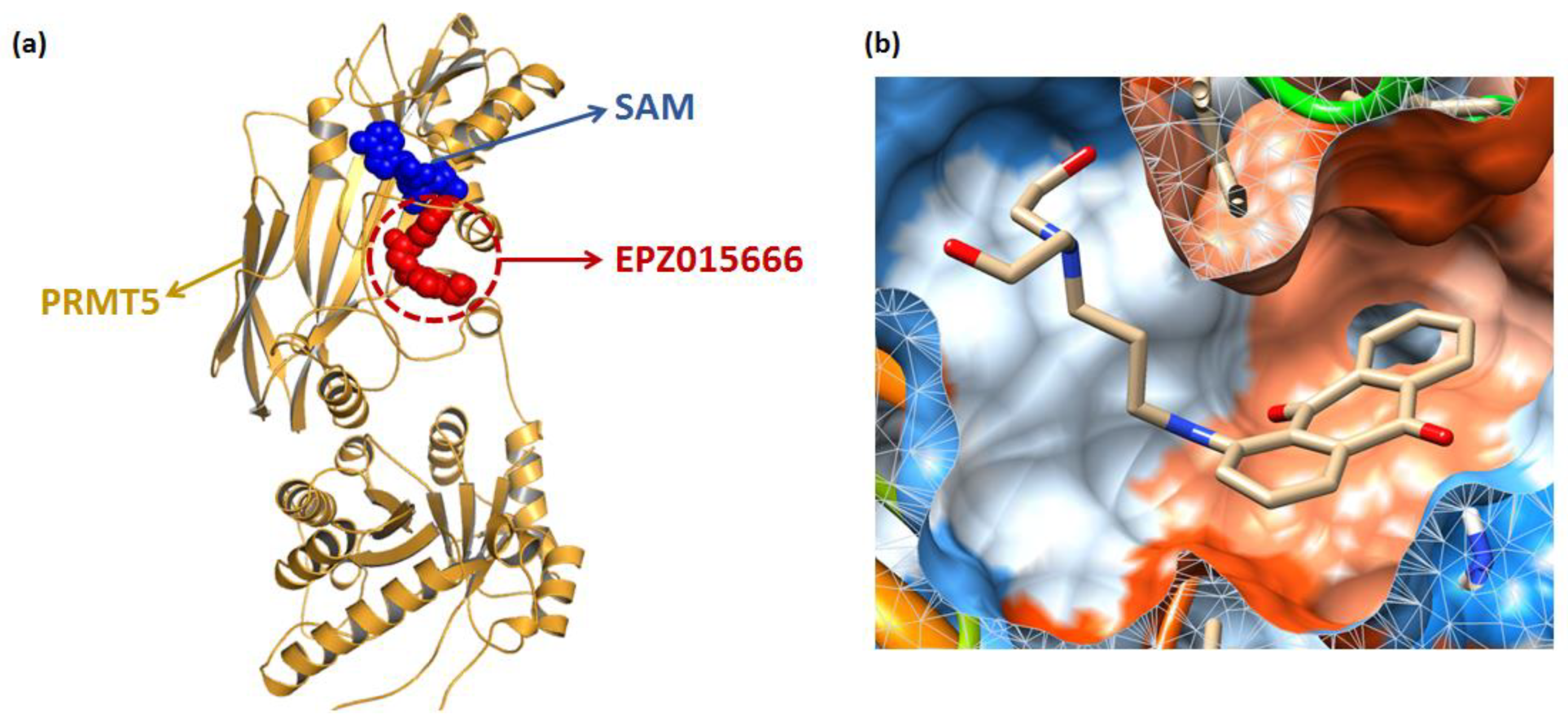
2.1. Virtual Screening Based on Protein Structure
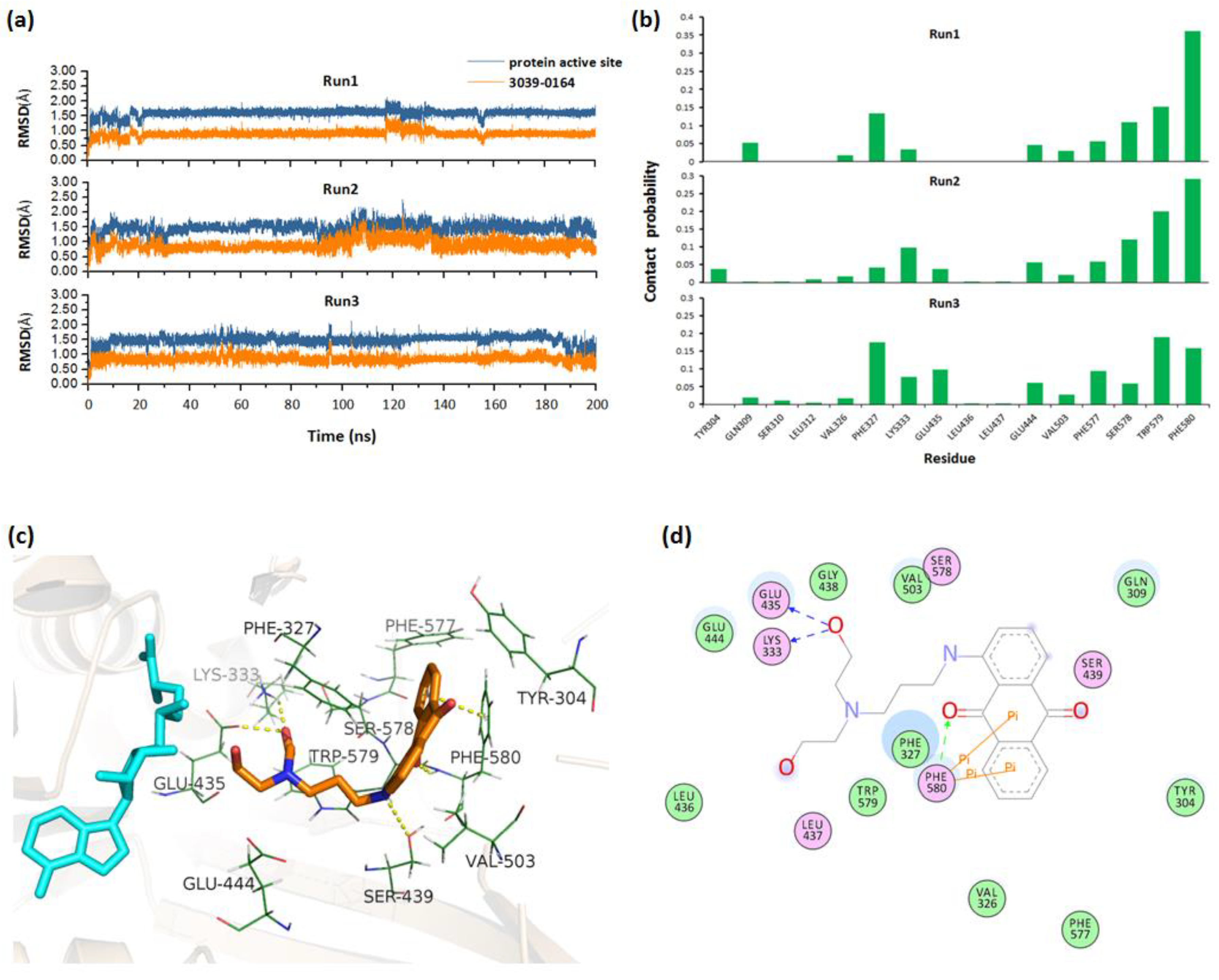
2.2. Molecular Docking and MD Simulations
2.3. Cell Culture and Cytotoxicity Test
2.4. In Vitro Enzymatic Assays
2.5. Western Blotting
2.6. Statistics
3. Results
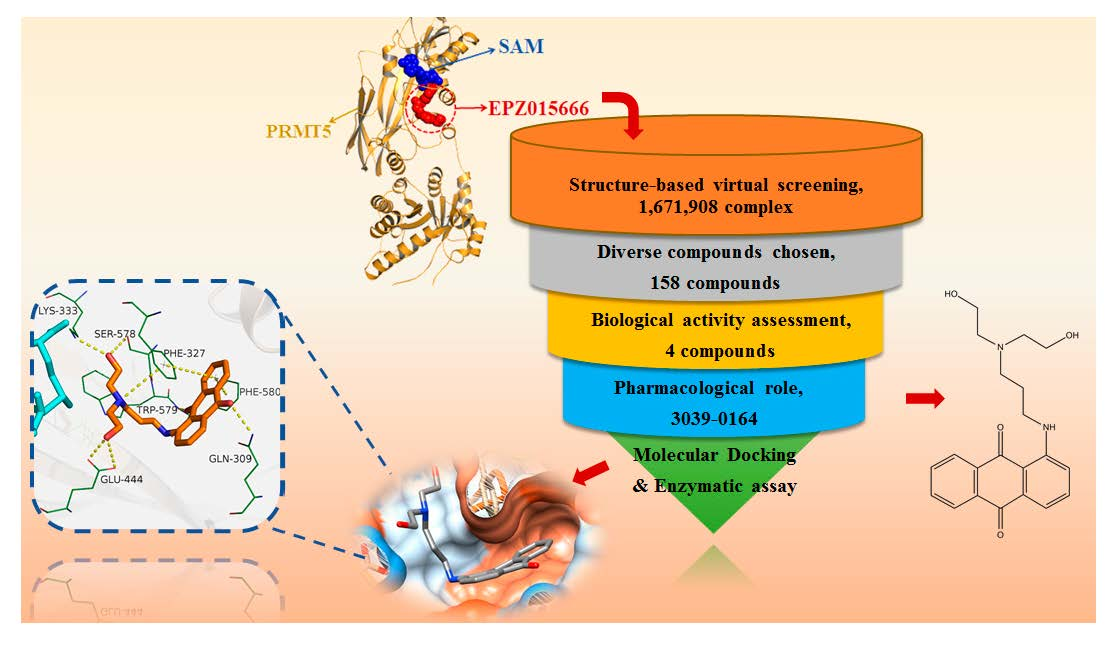
3.1. Screening of Candidate Compounds by Structure-Based Virtual Screening
3.2. Molecular Mechanism of 3039-0164 Binding to PRMT5
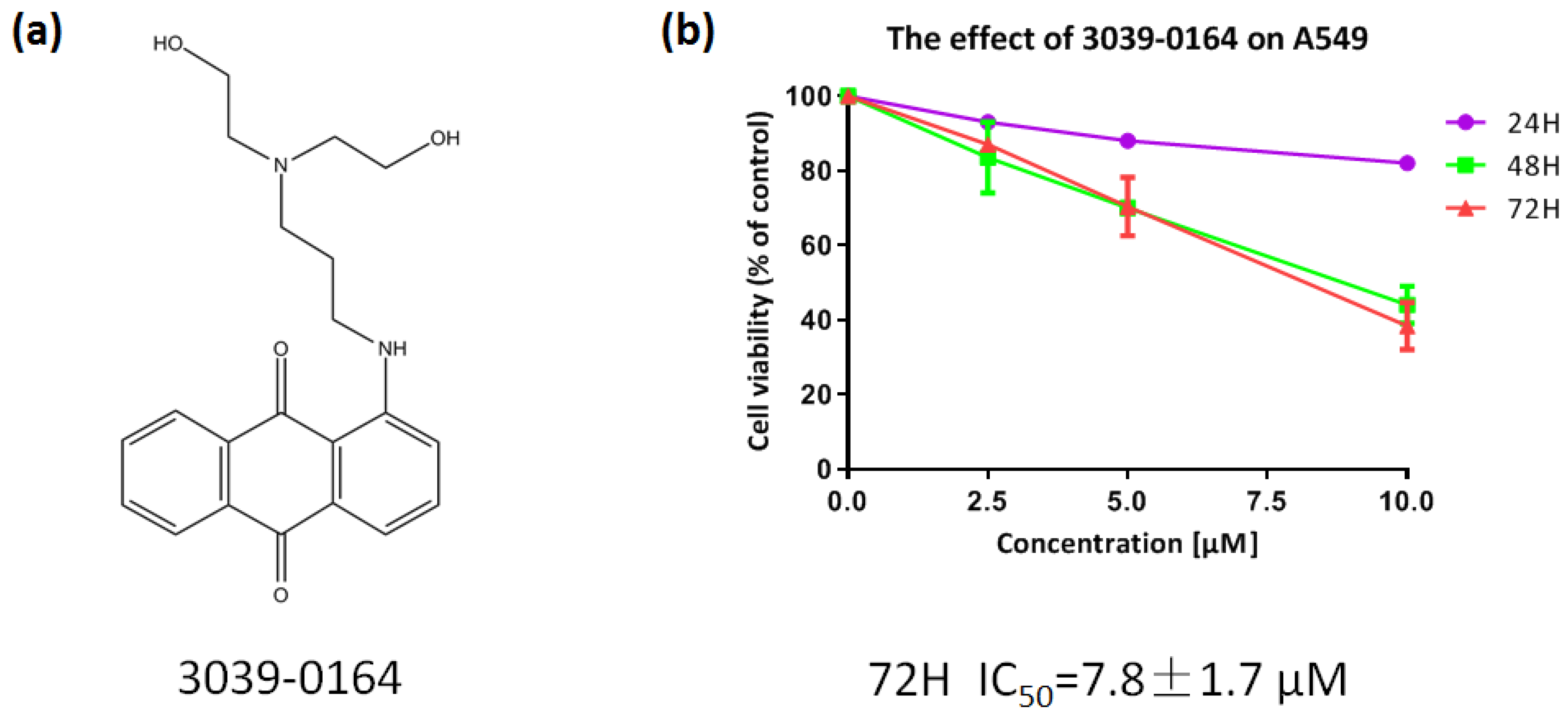
3.3. 3039-0164 Possesses Strong Cytotoxicity for A549 Cells
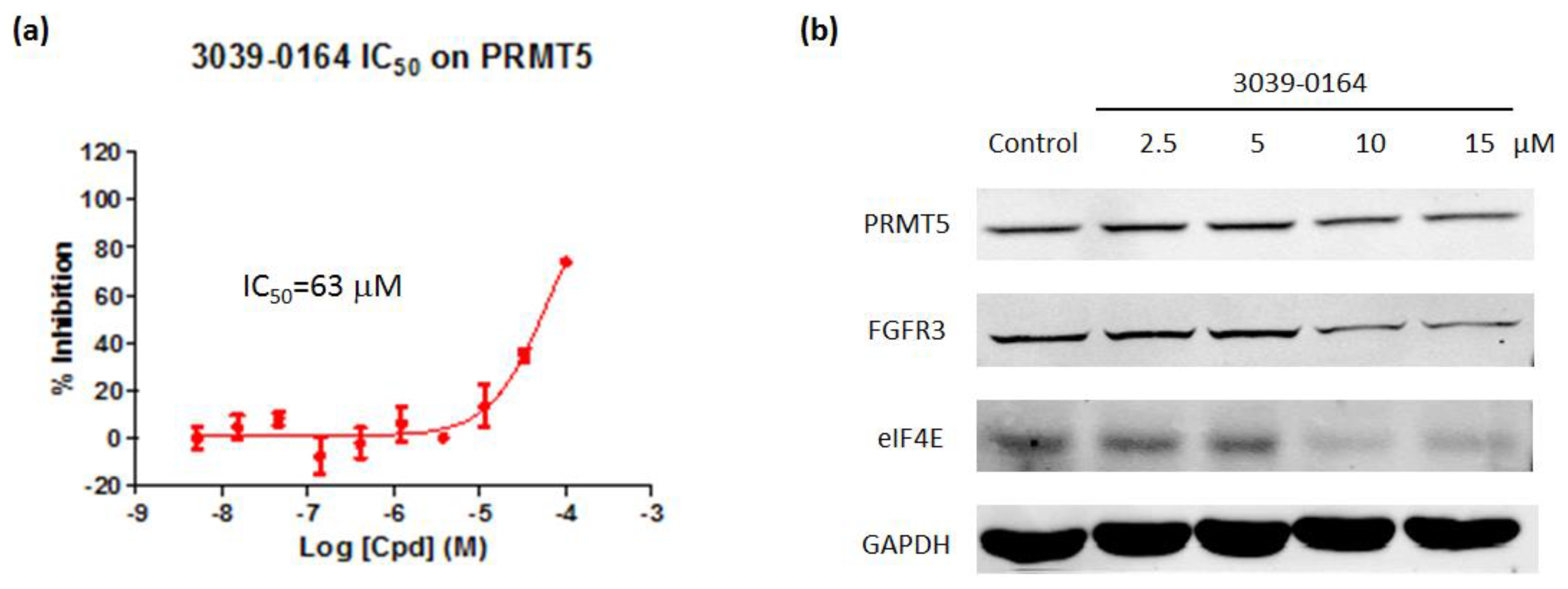
3.4. 3039-0164 Inhibits PRMT5 Methyltransferase Activity and the Expression of Its Downstream Target Genes
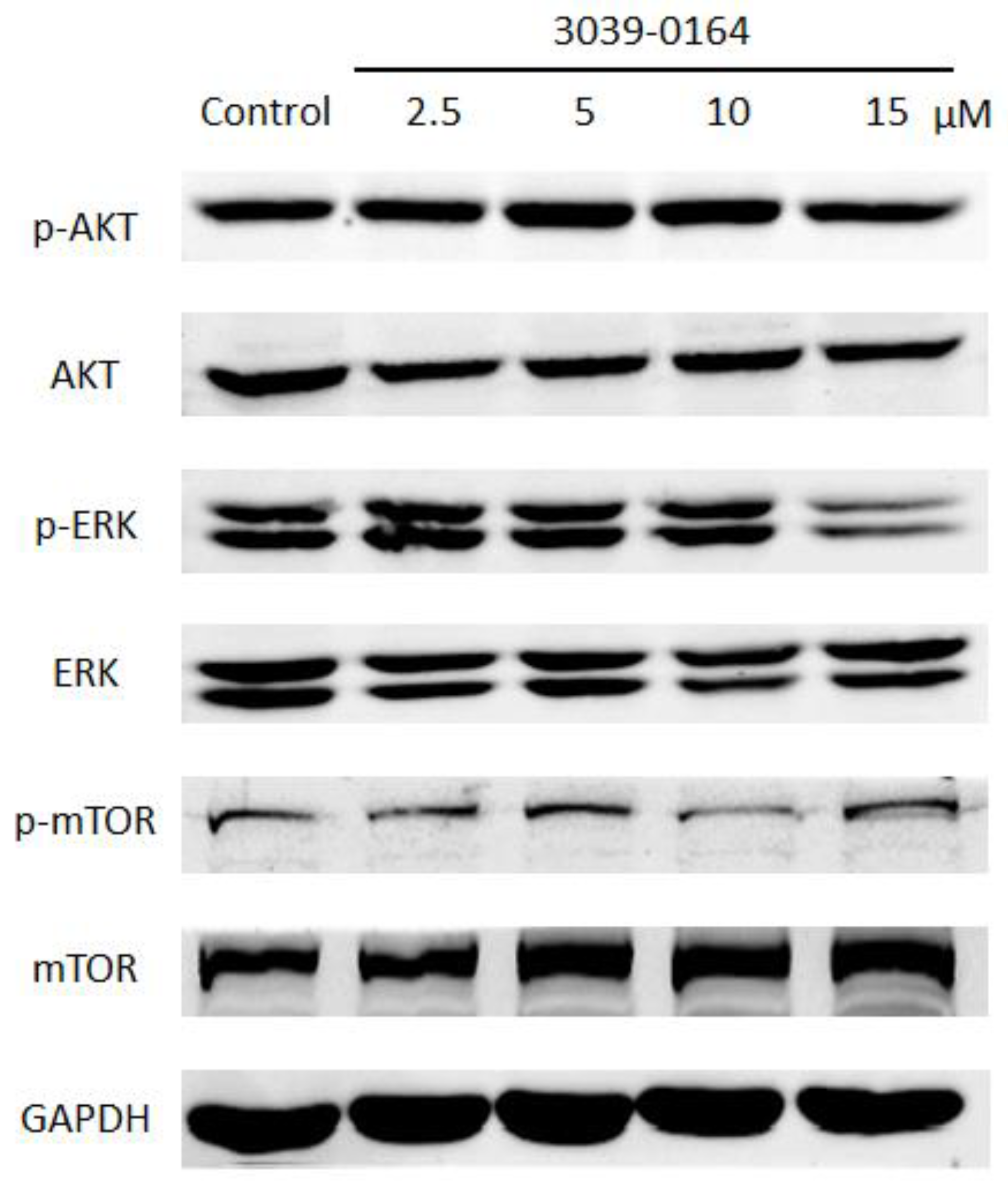
3.5. 3039-0164 Blocks the Activation of the FGFR3 Downstream Signaling Pathway
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, T.; Hassanabad, M.F.; Hassanabad, A.F. Non-small cell lung cancer: Emerging molecular targeted and immunotherapeutic agents. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188636. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.; Barbash, O.; Creasy, C. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wen, C.; Jiang, H.; Ma, S.; Liu, X. Protein Arginine Methyltransferase 5 Functions via Interacting Proteins. Front. Cell Dev. Biol. 2021, 9, 725301. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Blanc, R.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef]
- Gao, J.; Liu, R.; Feng, D.; Huang, W.; Huo, M.; Zhang, J.; Leng, S.; Yang, Y.; Yang, T.; Yin, X.; et al. Snail/PRMT5/NuRD complex contributes to DNA hypermethylation in cervical cancer by TET1 inhibition. Cell. Death Differ. 2021, 28, 2818–2836. [Google Scholar] [CrossRef]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, Y.; Hu, X.; Zheng, Y.; Chen, X. Targeting PRMT5/Akt signalling axis prevents human lung cancer cell growth. J. Cell. Mol. Med. 2019, 23, 1333–1342. [Google Scholar] [CrossRef]
- Sheng, X.; Wang, Z. Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer 2016, 16, 567. [Google Scholar] [CrossRef]
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.; Olsen, S.; Ostrowski, M.; Gan, W. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 2021, 12, 3444. [Google Scholar] [CrossRef]
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef]
- Janisiak, J.; Kopytko, P.; Tkacz, M.; Rogińska, D.; Perużyńska, M.; Machaliński, B.; Pawlik, A.; Tarnowski, M. Protein Arginine Methyltransferase (PRMT) Inhibitors-AMI-1 and SAH Are Effective in Attenuating Rhabdomyosarcoma Growth and Proliferation in Cell Cultures. Int. J. Mol. Sci. 2021, 22, 8023. [Google Scholar] [CrossRef]
- Mao, R.; Shao, J.; Zhu, K.; Zhang, Y.; Ding, H.; Zhang, C.; Shi, Z.; Jiang, H.; Sun, D.; Duan, W.; et al. Potent, Selective, and Cell Active Protein Arginine Methyltransferase 5 (PRMT5) Inhibitor Developed by Structure-Based Virtual Screening and Hit Optimization. J. Med. Chem. 2017, 60, 6289–6304. [Google Scholar] [CrossRef]
- Bajbouj, K.; Ramakrishnan, R.K.; Saber-Ayad, M.; Omar, H.A.; Sharif-Askari, N.S.; Shafarin, J.; Elmoselhi, A.B.; Ihmaid, A.; Ali, S.A.; Alalool, A.; et al. PRMT5 Selective Inhibitor Enhances Therapeutic Efficacy of Cisplatin in Lung Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6131. [Google Scholar] [CrossRef]
- Sachamitr, P.; Ho, J.; Ciamponi, F.; Ba-Alawi, W.; Coutinho, F.; Guilhamon, P.; Kushida, M.; Cavalli, F.; Lee, L.; Rastegar, N.; et al. PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Li, Y.; Huang, J.; Jiang, Z.; Wang, Y.; Liu, L.; Leung, E.L.H.; Yao, X. Identification of a Novel Protein Arginine Methyltransferase 5 Inhibitor in Non-small Cell Lung Cancer by Structure-Based Virtual Screening. Front. Pharmacol. 2018, 9, 173. [Google Scholar] [CrossRef]
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E.; et al. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Kuplast, K.; Majer, C.; Boriack-Sjodin, P.; Wigle, T.; Johnston, L.; Rioux, N.; Munchhof, M.; Jin, L.; Jacques, S.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shao, X.; Zhao, X.; Ji, Y.; Liu, X.; Li, P.; Zhang, M.; Wang, Q. Targeting protein arginine methyltransferase 5 in cancers: Roles, inhibitors and mechanisms. Biomed. Pharmacother. 2021, 144, 112252. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Liu, F.; Veazey, K.; Gao, G.; Das, P.; Neves, L.; Lin, K.; Zhong, Y.; Lu, Y.; Giuliani, V.; et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Song, C.; Wang, X.; Zhang, G.; Wang, Y.; Jiang, X.; Sun, Q.; Huang, L.; Xiang, R.; Hu, Y.; et al. Discovery of New SIRT2 Inhibitors by Utilizing a Consensus Docking/Scoring Strategy and Structure-Activity Relationship Analysis. J. Chem. Inf. Model 2017, 57, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, J.; Mao, L.; Zhang, Y.; Cui, W.; Duan, S.; Jiang, A.; Gao, Y.; Sang, Y.; Huang, G. EPZ015666, a selective protein arginine methyltransferase 5 (PRMT5) inhibitor with an antitumour effect in retinoblastoma. Exp. Eye Res. 2021, 202, 108286. [Google Scholar] [CrossRef]
- Schrödinger. Maestro Version 10.2., New York. 2015. Available online: https://www.schrodinger.com/products/maestro (accessed on 20 January 2022).
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.; Raguette, L.; Huang, H.; Migues, A.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.; Caldwell, J.; Kollman, P.; Case, D. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Roe, D.; Cheatham, T. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Ji, S.; Ma, S.; Wang, W.; Huang, S.; Wang, T.; Xiang, R.; Hu, Y.; Chen, Q.; Li, L.; Yang, S. Discovery of selective protein arginine methyltransferase 5 inhibitors and biological evaluations. Chem. Biol. Drug Des. 2017, 89, 585–598. [Google Scholar] [CrossRef]
- Sahihi, M.; Gaci, F.; Navizet, I. Identification of new alpha-synuclein fibrillogenesis inhibitor using in silico structure-based virtual screening. J. Mol. Graph. Model 2021, 108, 108010. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Y.; Zhong, L.; Yu, N.; Ouyang, L.; Fang, R.; Wang, Y.; He, Q. Structure-based discovery of neoandrographolide as a novel inhibitor of Rab5 to suppress cancer growth. Comput. Struct. Biotechnol. J. 2020, 18, 3936–3946. [Google Scholar] [CrossRef]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L. Machine-learning methods for ligand-protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef]
- Smil, D.; Eram, M.; Li, F.; Kennedy, S.; Szewczyk, M.; Brown, P.; Barsyte-Lovejoy, D.; Arrowsmith, C.; Vedadi, M.; Schapira, M. Discovery of a Dual PRMT5-PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. [Google Scholar] [CrossRef]
- Sulimov, V.; Kutov, D.; Sulimov, A. Advances in Docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- He, Z.; Jiao, H.; An, Q.; Zhang, X.; Zengyangzong, D.; Xu, J.; Liu, H.; Ma, L.; Zhao, W. Discovery of novel 4-phenylquinazoline-based BRD4 inhibitors for cardiac fibrosis. Acta Pharm. Sin. B 2022, 12, 291–307. [Google Scholar] [CrossRef]
- Cai, C.; Gu, S.; Yu, Y.; Zhu, Y.; Zhang, H.; Yuan, B.; Shen, L.; Yang, B.; Feng, X. PRMT5 Enables Robust STAT3 Activation via Arginine Symmetric Dimethylation of SMAD7. Adv. Sci. 2021, 8, 2003047. [Google Scholar] [CrossRef]
- Gu, Z.; Gao, S.; Zhang, F.; Wang, Z.; Ma, W.; Davis, R.; Wang, Z. Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem. J. 2012, 446, 235–241. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, S.; Zhu, R.; Hu, C.; Hou, J.; Li, Y.; Zhao, Q.; Shao, X.; Bu, Q.; Li, H.; et al. Targeting protein arginine methyltransferase 5 inhibits colorectal cancer growth by decreasing arginine methylation of eIF4E and FGFR3. Oncotarget 2015, 6, 22799–22811. [Google Scholar] [CrossRef]
- D’Abronzo, L.; Ghosh, P. eIF4E Phosphorylation in Prostate Cancer. Neoplasia 2018, 20, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Qi, F.; Lu, S.; Li, Y.; Han, W. Nicotine upregulates FGFR3 and RB1 expression and promotes non-small cell lung cancer cell proliferation and epithelial-to-mesenchymal transition via downregulation of miR-99b and miR-192. Biomed. Pharmacother. 2018, 101, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; An, X.; Xu, J.; Wang, Y.; Liu, L.; Leung, E.L.; Yao, X. Classical molecular dynamics and metadynamics simulations decipher the mechanism of CBP30 selectively inhibiting CBP/p300 bromodomains. Org. Biomol. Chem. 2018, 16, 6521–6530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Leung, E.L.H.; Chen, Y.; Yao, X. Selectively targeting individual bromodomain: Drug discovery and molecular mechanisms. Pharmacol. Res. 2021, 172, 105804. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Q.; Leung, E.L.H.; Li, Y.; Fan, X.; Wu, Q.; Yao, X.; Liu, L. Compound C620-0696, a new potent inhibitor targeting BPTF, the chromatin-remodeling factor in non-small-cell lung cancer. Front. Med. 2020, 14, 60–67. [Google Scholar] [CrossRef]
- Ibrahim, R.; Matsubara, D.; Osman, W.; Morikawa, T.; Goto, A.; Morita, S.; Ishikawa, S.; Aburatani, H.; Takai, D.; Nakajima, J.; et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Hum. Pathol. 2014, 45, 1397–1405. [Google Scholar] [CrossRef]
- Jing, P.; Zhao, N.; Ye, M.; Zhang, Y.; Zhang, Z.; Sun, J.; Wang, Z.; Zhang, J.; Gu, Z. Protein arginine methyltransferase 5 promotes lung cancer metastasis via the epigenetic regulation of miR-99 family/FGFR3 signaling. Cancer Lett. 2018, 427, 38–48. [Google Scholar] [CrossRef]
- Xiao, W.; Chen, X.; Liu, L.; Shu, Y.; Zhang, M.; Zhong, Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed. Pharmacother. 2019, 114, 108790. [Google Scholar] [CrossRef]
- Baqi, Y. Anthraquinones as a privileged scaffold in drug discovery targeting nucleotide-binding proteins. Drug Discov. Today 2016, 21, 1571–1577. [Google Scholar] [CrossRef]
- Siddamurthi, S.; Gutti, G.; Jana, S.; Kumar, A.; Singh, S. Anthraquinone: A promising scaffold for the discovery and development of therapeutic agents in cancer therapy. Future Med. Chem. 2020, 12, 1037–1069. [Google Scholar] [CrossRef]
- Volodina, Y.; Tikhomirov, A.; Dezhenkova, L.; Ramonova, A.; Kononova, A.; Andreeva, D.; Kaluzhny, D.; Schols, D.; Moisenovich, M.; Shchekotikhin, A.; et al. Thiophene-2-carboxamide derivatives of anthraquinone: A new potent antitumor chemotype. Eur. J. Med. Chem. 2021, 221, 113521. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | IC50 | PDB id | Chemical Structures | Mechanisms of Action | Cancer Types | Ref. |
|---|---|---|---|---|---|---|
| LLY-283 | 20 nM | 6CKC |  | Occupied the SAM pocket; inhibition of MDM4 splicing regulation; phenyl occupancy of the Phe327 side chain, which may make it highly selective for PRMT5. | Glioblastoma | [14,18] |
| AMI-1 | 8 µM | - |  | It has a double anion structure that binds to the SAM site; the interaction poly activity established by its sulfonic acid group makes it inhibit the test enzyme. | Rhabdomyosarcoma; lung cancer | [17] |
| T1551 | 34 µM | - |  | Through cation–π interactions with SAM, π–π interactions with Phe327, and hydrogen bonding with some residues. | Non-small cell lung cancer | [19] |
| EPZ015666 | 22 nM | 4 × 61 |  | Substrate competitive; the THIQ group interacts with the cation–π formed by the partially positively charged methyl group of SAM. | Multiple myeloma; retinoblastoma; Mantle cell lymphoma | [22,24,25] |
| JNJ-64619178 | 0.1 nM | 6RLQ |  | Occupies both SAM and substrate-binding sites; produces high affinity and, therefore, does not interact with MTAP-deficient cancer cell-specific complexes. | Acute myeloid leukemia; non-small cell lung cancer; pancreatic | [20] |
| MS4322 | 18 nM | - |  | PROTAC degraders; competes with EPZ015666 for PRMT5 substrate binding sites, and reduces PRMT5 expression. | Non-small cell lung cancer; cervical cancer; glioblastoma | [10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhang, M.; Wu, A.; Yao, X.; Wang, Q. Structure-Based Discovery and Biological Assays of a Novel PRMT5 Inhibitor for Non-Small Cell Lung Cancer. Molecules 2022, 27, 7436. https://doi.org/10.3390/molecules27217436
Chen Y, Zhang M, Wu A, Yao X, Wang Q. Structure-Based Discovery and Biological Assays of a Novel PRMT5 Inhibitor for Non-Small Cell Lung Cancer. Molecules. 2022; 27(21):7436. https://doi.org/10.3390/molecules27217436
Chicago/Turabian StyleChen, Yingqing, Mingyu Zhang, Anxin Wu, Xiaojun Yao, and Qianqian Wang. 2022. "Structure-Based Discovery and Biological Assays of a Novel PRMT5 Inhibitor for Non-Small Cell Lung Cancer" Molecules 27, no. 21: 7436. https://doi.org/10.3390/molecules27217436
APA StyleChen, Y., Zhang, M., Wu, A., Yao, X., & Wang, Q. (2022). Structure-Based Discovery and Biological Assays of a Novel PRMT5 Inhibitor for Non-Small Cell Lung Cancer. Molecules, 27(21), 7436. https://doi.org/10.3390/molecules27217436