3. Experimental
Melting points are uncorrected, and were measured with a Kofler hot-stage. Optical rotations were determined at room temperature using a Perkin-Elmer 241 polarimeter. 1H and 13C NMR spectra were recorded with a Bruker DRX 360 (1H: 360 MHz, 13C: 90 MHz) or a Bruker DRX 400 (1H: 400 MHz, 13C: 100 MHz) spectrometer. The 1D ROESY and HSQMBC measurements were recorded on a Bruker Avance II 500 (1H: 500 MHz, 13C: 125 MHz) spectrometer. Chemical shifts were referenced to Me4Si (1H) or to the residual solvent signals (13C). Mass spectra were recorded with a Bruker micrOTOF-Q and Thermo Accela LTQ XL or a Bruker maXis II UHR ESI-TOF (HRMS) spectrometer.
Thin layer chromatography was performed using DC-Alurolle Kieselgel F254 (Merck) plates and the spots were visualized under UV light (λ = 254 nm) and by gentle heating after staining the plate (staining solutions: 1% of anisaldehyde and 5% of cc. sulfuric acid in ethanol or 6% of vanilline and 1% of cc. sulfuric acid in ethanol). For the detection of bromine containing compounds, the plates were sprayed with a fluorescein solution (0.01% in ethanol) followed by a solution of hydrogen peroxide (1:1 mixture of 30% aqueous H2O2 and glacial acetic acid). Bromides appeared as a pink spot after gentle heating of the plate. For column chromatography, Kieselgel 60 (Merck, particle size: 0.063–0.200 mm) type silica gel was used. Anhydrous chloroform was prepared from commercial chloroform stabilized with 1% of EtOH. It was kept on anhydrous CaCl2 overnight, then filtered and distilled from P2O5 and the collected distillate was stored over molecular sieves (4Å). Other solvents were dried by conventional methods.
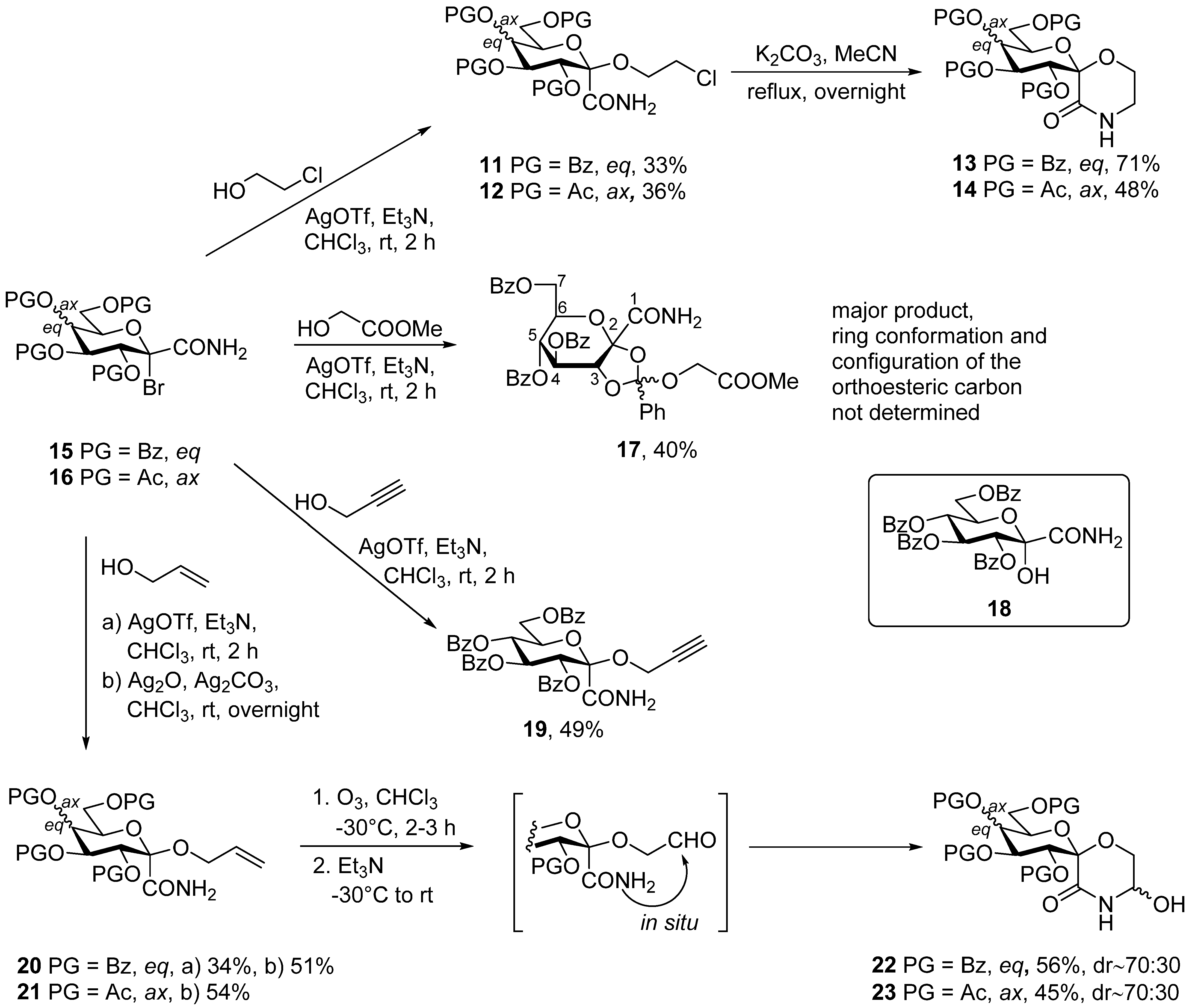
3.1. General Method I. for the Synthesis of C-(2,3,4,6-Tetra-O-Acyl-1-Alkoxy-D-Glycopyranosyl)Formamides (11, 12, 17, 19, 20, 21)
In a flame dried round bottom flask, a bromoamide 15 or 16, (0.5 mmol) was dissolved in anhydrous chloroform (5 mL). Triethylamine (2 equiv.), the corresponding alcohol (20 equiv.) and silver triflate (2 equiv.) were added and the flask was covered in tin foil. After 2 h of stirring at room temperature, no starting material could be detected by TLC. Evaporation of the solvent gave a syrup, which was purified by column chromatography.
3.2. General Method II. for the Synthesis of C-(2,3,4,6-Tetra-O-Acyl-1-Alkoxy-D-Glycopyranosyl)Formamides (17, 20)
In a flame dried round bottom flask, a bromoamide 15 or 16, (0.5 mmol) was dissolved in anhydrous chloroform (5 mL). Silver oxide (2 equiv.), silver carbonate (2 equiv.) and the corresponding alcohol (20 equiv.) were added and the mixture was stirred overnight. After TLC showed complete conversion, the mixture was filtered through a celite pad. Evaporation of the solvent gave a syrup, which was purified by column chromatography.
3.3. General Method III. for the Ring Closure Reactions of C-(2,3,4,6-Tetra-O-Acyl-1-(2-Chloroethoxy)-α-D-Glycopyranosyl)Formamides (11, 12) into Spiro-Morpholin-3-Ones (13, 14)
2-Chloroethyl glycoside (11 or 12) was dissolved in dry acetonitrile (~1 mL/50 μmol), and flame dried potassium carbonate (2 equiv.) was added. The mixture was stirred at reflux temperature until TLC showed full conversion. Insoluble materials were removed by filtration. The solvent was removed by evaporation and the residue was purified by column chromatography.
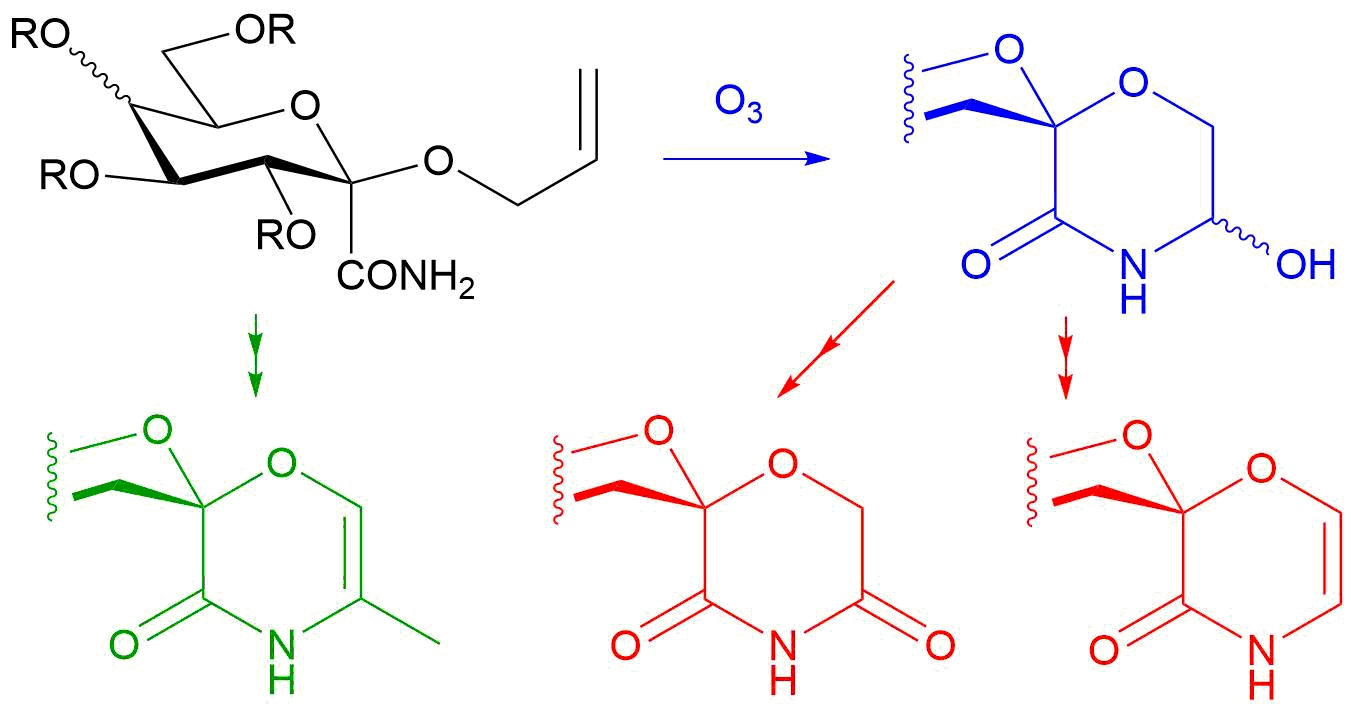
3.4. General Method IV. for the Ozonolysis of C-(2,3,4,6-Tetra-O-Acyl-1-Allyloxy-D-Glycopyranosyl)Formamides (20, 21) and 2-Allyloxy-2-Phenylacetamide (24) to Yield 5-Hydroxymorpholin-3-Ones (22, 23, 25)
In a flame dried round bottom flask, an α-allyloxyamide (20, 21, 24, 0.8–1.4 mmol) was dissolved in dry chloroform (~1 mL/80 μmol), and the solution was cooled to −30 °C. Ozone was bubbled through the reaction mixture for 2–3 h. The progress of the reaction was monitored by TLC: a sample (1–2 drops) was taken from the mixture, diluted with chloroform and 1–2 drops of triethylamine were added then applied to a TLC plate. After total consumption of the starting material, the ozone flow was stopped, triethylamine (2 equiv.) was added and the mixture was allowed to reach room temperature. After evaporation of the solvent, the product was obtained by column chromatography (hexane/acetone 2:1).
3.5. General Method V. for the Synthesis of Morpholinediones (27, 28, 31)
In a flame dried round bottom flask, a 5-hydroxymorpholinone derivative (22, 23, 25, 0.5–0.8 mmol) was dissolved in dry chloroform (1 mL/0.1 mmol), the solution was cooled to 0 °C then pyridine (5 equiv.) and chromium(VI) oxide (5 equiv.) were added to the stirred solution. The solution was slowly allowed to warm up and stirred at room temperature overnight. The next day, TLC showed full conversion, and a brown precipitate appeared. The solution was diluted with chloroform (20 mL), filtered and concentrated. A few milliliters of toluene was also added and evaporated to remove most of the pyridine. The crude product was purified by column chromatography (hexane/acetone 4:1 → 3:1).
3.6. General Method VI. for the Synthesis of Unsaturated Morpholinones (29, 30, 32)
A 5-hydroxymorpholinone derivative (22, 23, 25, 0.2–0.5 mmol) was dissolved in dry chloroform (~1 mL/35 μmol), and p-toluenesulfonic acid (0.2 equiv.) was added. The solution was stirred and refluxed for 2–3 h, after which time TLC showed complete conversion. The solvent was evaporated, and the crude product was purified by column chromatography (hexane/acetone 3:1).
3.7. General Method VII. for the Removal of O-Acyl Groups to Yield Deprotected Compounds (34–42)
An O-peracylated spiro-morpholine derivative (13, 14, 22, 23, 27–30, 33, 0.1–0.7 mmol) was dissolved in dry methanol (1 mL/~30–40 μmol), and pH was adjusted to ~10–11 with 1M NaOMe/MeOH solution (approx. 10–15 drops). The reaction mixture was stirred at room temperature for 1–4 h until TLC showed complete conversion. The solution was neutralized using acidic ion exchange resin (Amberlyst 15®) until pH ~6–7 was reached, the resin was filtered and the filtrate was concentrated. The products were purified by column chromatography (chloroform/methanol 8:1–3:1, triethylamine (0.5% V/V) was added to the eluents).
3.8. C-(2,3,4,6-Tetra-O-Benzoyl-1-(2-Chloroethoxy)-α-D-Glucopyranosyl)Formamide (11)
Prepared according to General method I. from 15 (300 mg, 0.43 mmol) and 2-chloroethanol. Column chromatography (hexane/acetone 3:1 → 2:1) gave 100 mg (33%), colorless oil. Rf = 0.34 (hexane/acetone 3:1). [α]D = +54 (c = 0.50, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.07 (2H, Ar), 7.95 (4H, Ar), 7.83 (2H, Ar), 7.59–7.25 (12H, Ar), 6.80 (1H, d, J = 3.2 Hz, NH), 6.63 (1H, t, J = 9.3 Hz, H-3), 5.92 (1H, brs, NH), 5.83 (1H, t, J = 9.8 Hz, H-4), 5.77 (1H, d, J = 9.2 Hz, H-2), 5.09 (1H, dt, J = 10.1, 3.2 Hz, H-5), 4.73 (1H, dd, J = 12.4, 2.8 Hz, H-6a), 4.42 (1H, dd, J = 12.4, 3.7 Hz, H-6b), 4.13–4.03 (2H, m, OCH2), 3.68–3.56 (2H, m CH2Cl). 13C NMR (100 MHz, CDCl3) δ (ppm): 169.3 (CONH2), 166.2, 165.4 (2), 165.2 (4 × C=O), 133.7–128.4 (Ar), 97.7 (C-1), 72.7, 71.9, 69.7, 68.9 (C-2–C-5), 63.1, 62.5 (C-6, OCH2), 43.1 (CH2Cl). HRMS (positive mode, m/z): 724.1557 (calculated for C37H32ClNO11Na: 724.1556).
3.9. C-(2,3,4,6-Tetra-O-Acetyl-1-(2-Chloroethoxy)-α-D-Galactopyranosyl)Formamide (12)
Prepared according to General method I. from 16 (200 mg, 0.44 mmol) and 2-chloroethanol. Column chromatography (hexane/acetone 3:1 → 2:1) gave 100 mg (36%), colorless oil. Rf = 0.34 (hexane/acetone 3:1). [α]D = +70 (c = 0.51, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 6.74 (1H, d, J = 3.1 Hz, NH), 5.90 (1H, dd, J = 10.5, 3.3 Hz, H-3), 5.86 (brs, 1H, NH), 5.53 (1H, dd, J = 3.3, 1.2 Hz, H-4), 5.47 (1H, d, J = 10.5 Hz, H-2), 4.87 (1H, t, J = 6.9 Hz, H-5), 4.10 (2H, d, J = 6.6 Hz, OCH2), 4.06 (1H, dd, J = 11.2, 5.2 Hz, H-6a), 3.98 (1H, dd, J = 10.7, 5.4 Hz, H-6b), 3.71–3.62 (2H, m, CH2Cl), 2.17, 2.08, 2.04, 1.98 (4 × 3H, s, 4 × OCOCH3). 13C NMR (90 MHz, CDCl3) δ (ppm): 170.5, 170.1, 170.0, 169.8, 169.3 (5 × C=O), 97.7 (C-1), 71.6, 69.9, 67.9, 66.9 (C-2–C-5), 62.7, 61.5 (C-6, OCH2), 43.3 (CH2Cl), 20.9, 20.8, 20.7, 20.7 (4 × OCOCH3).
3.10. (1′S)-1′,5′-Anhydro-2′,3′,4′,6′-Tetra-O-Benzoyl-D-Glucitol-Spiro-[1′,2]-Morpholin-3-One (13)
Prepared according to General method III. from 11 (100 mg, 0.14 mmol). Reaction time: 30 h. Column chromatography (hexane/acetone 3:1) gave 60 mg (71%) colorless, amorphous solid. Rf = 0.42 (hexane/acetone 3:1). [α]D = +17 (c = 0.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.06–7.81 (8H, m Ar), 7.57–7.25 (12H, Ar), 6.70 (1H, t, J = 9.8 Hz, H-3′), 6.50 (1H, d, J = 3.9 Hz, NH), 5.81 (1H, t, J = 9.8 Hz, H-4′), 5.68 (1H, d, J = 9.9 Hz, H-2′), 5.0 (1H, dt, J = 10.0, 3.5 Hz, H-5′), 4.65 (1H, dd, J = 12.3, 2.9 Hz, H-6′a), 4.44 (1H, dd, J = 12.3, 4.1 Hz, H-6′b), 4.26 (1H, td, J = 11.9, 3.0 Hz, H-6a), 3.88 (1H, dd, J = 11.9, 3.8 Hz, H-6b), 3.49 (1H, td, J = 12.1, 4.2 Hz, H-5a), 3.23 (1H, dt, J = 12.2, 3.2 Hz, H-5b). 13C NMR (100 MHz, CDCl3) δ (ppm): 166.4, 165.7, 165.5, 165.4, 165.2 (5 × C=O), 133.5–128.3 (Ar), 97.5 (C-1′), 73.8, 72.4, 72.4, 69.5 (C-2′–C-5′), 63.1 (C-6′), 59.3 (C-6), 41.5 (C-5). HSQMBC NMR: 3JH-2′, CO = 5.0 Hz; HRMS (positive mode, m/z): 688.1790 (calculated value for C37H31NO11Na: 688.1789).
3.11. (1′S)-2′,3′,4′,6′-Tetra-O-Acetyl-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-Morpholin-3-One (14)
Prepared according to General method III. from 12 (100 mg, 0.22 mmol). Reaction time: 24 h. Column chromatography (hexane/acetone 3:1) gave 44 mg (48%), colorless, amorphous solid. Rf = 0.35 (hexane/acetone 3:1). [α]D = +29 (c = 0.40, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 7.18 (1H, brs, NH), 5.93 (1H, dd, J = 10.6, 3.5 Hz, H-3′), 5.52 (1H, dd, J = 3.5, 1.1 Hz, H-4′), 5.38 (1H, d, J = 10.6 Hz, H-2′), 4.75 (1H, ddd, J = 7.6, 6.2, 1.4 Hz, H-5′), 4.23 (1H, td, J = 12.0, 3.1 Hz, H-6a), 4.18–4.08 (2H, m, H-6b, H-6′a), 3.91 (1H, dd, J = 11.9, 4.0 Hz, H-6′b), 3.58 (1H, td, J = 12.2, 4.2 Hz, H-5a), 3.27 (1H, dt, J = 12.4, 3.6 Hz, H-5b), 2.17, 2.09, 2.04, 1.96 (4 × 3H, s, 4 × OCOCH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.5, 170.4, 170.1, 169.6 (4 × C=O), 165.6 (C-3), 97.4 (C-1′), 71.5, 70.4, 70.1, 67.4 (C-2′–C-5′), 61.6, 59.2 (C-6, C-6′), 41.3 (C-5), 20.9, 20.8 (2), 20.8 (4 × OCOCH3).
3.12. 4,5,7-Tri-O-Benzoyl-2,3-O-[α-(2-Methoxy-2-Oxoethoxy)Benzylidene]-α-D-Gluco-Hept-2-Ulopyranosonamide (17)
Prepared according to General method I. from 15 (300 mg, 0.43 mmol) and methyl glycolate. Column chromatography (hexane/ethyl acetate 7:3 → 6:4) gave 122 mg (40%), colorless oil. Preparation according to General method II. from 15 (300 mg, 0.43 mmol) and methyl glycolate gave 102 mg (34%). Rf = 0.28 (hexane/ethyl acetate 1:1). [α]D = +45 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.04–7.20 (20H, m, Ar), 7.10 (1H, s, NH), 5.78 (1H, dd, J = 3.1, 1.0 Hz, H-2), 5.67 (1H, s, NH), 5.55 (1H, dt, J = 8.6, 1.2 Hz, H-5), 5.24 (1H, dd, J = 3.1, 1.3 Hz, H-4), 4.60 (1H, dd, J = 12.2, 2.9 Hz, H-7a), 4.44 (1H, dd, J = 12.2, 4.9 Hz, H-7b), 4.13 (1H, m, H-5), 4.11 (1H, d, J = 16.0 Hz, CH2COOCH3), 4.04 (1H, d, J = 16.0 Hz, CH2COOCH3), 3.71 (3H, s, CH2COOCH3). 13C NMR (90 MHz, CDCl3) δ (ppm): 169.6, 168.4, 166.2, 165.2, 164.6 (5 × C=O), 134.1–122.3 (Ar), 102.1 (C-2), 74.3, 69.6, 68.7, 67.9 (C-3–C-6), 63.9, 62.1 (C-7, CH2COOCH3), 52.1 (COOCH3). HRMS (positive mode, m/z): 734.1840 (calculated value for C38H33NO13Na: 734.1844).
3.13. C-(2,3,4,6-Tetra-O-Benzoyl-1-Propargyloxy-α-D-Glucopyranosyl)Formamide (19)
Prepared according to General method I. from 15 (150 mg, 0.21 mmol) and propargyl alcohol. Column chromatography (hexane/acetone 9:1 → 4:1) gave 71 mg (49%), colorless oil. Rf = 0.42 (hexane/acetone 3:1). [α]D = +38 (c = 0.30, CHCl3); 1H NMR (360 MHz, CDCl3) δ (ppm): 8.07–7.84 (8H, m, Ar), 7.59–7.28 (12H, m, Ar), 6.76 (1H, d, J = 3.1 Hz, NH), 6.57 (1H, t, J = 9.1 Hz, H-3), 5.84–5.78 (3H, m, H-2, H-4, NH), 5.11 (1H, dt, J = 9.6, 3.4 Hz, H-5), 4.71 (1H, dd, J = 12.3, 2.9 Hz, H-6a), 4.54 (2H, dd, J = 2.6, 1.3 Hz, -OCH2-C≡CH), 4.46 (1H, dd, J = 12.4, 3.9 Hz, H-6b), 2.42 (1H, t, J = 2.4 Hz, -OCH2-C≡CH). 13C NMR (90 MHz, CDCl3) δ (ppm): 168.9, 166.2, 165.5, 165.4, 165.0 (5 × C=O), 133.7–128.4 (Ar), 97.7 (C-1), 78.6 (-OCH2-C≡CH), 75.1 (-OCH2-C≡CH), 72.8, 71.8, 69.6, 68.8 (C-2–C-5), 62.7 (C-6), 51.6 (-OCH2-C≡CH).
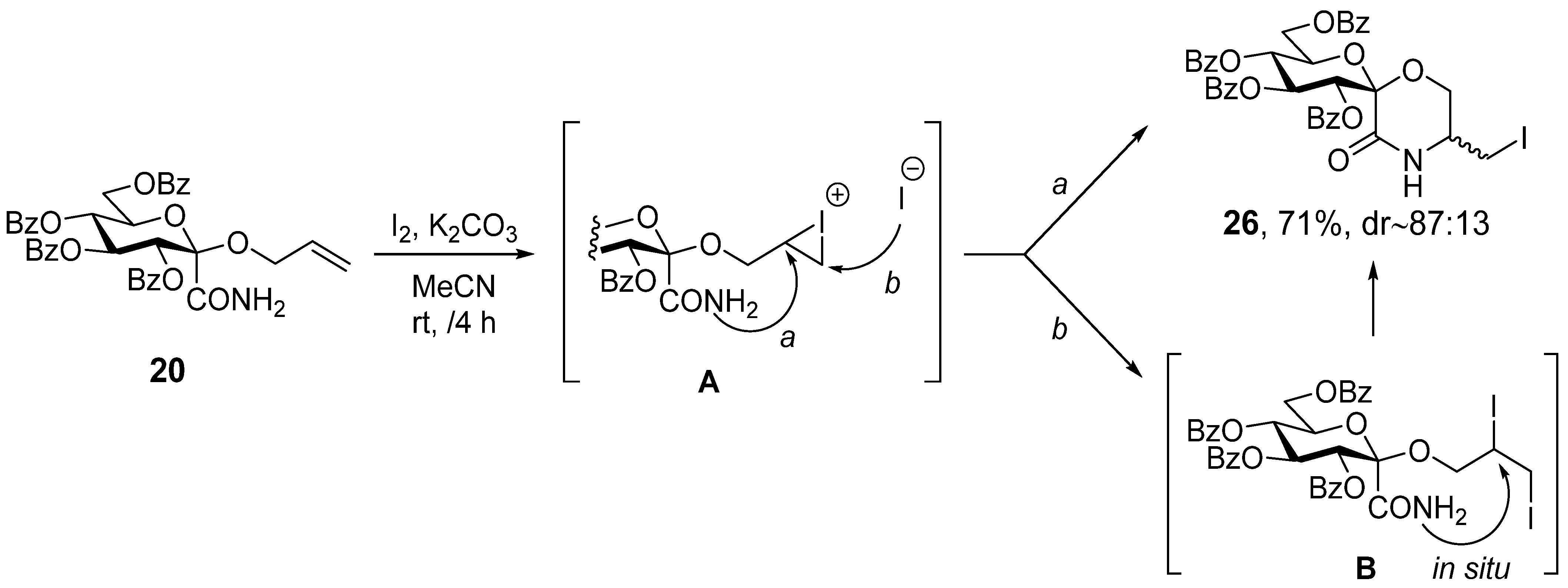
3.14. C-(1-Allyloxy-2,3,4,6-Tetra-O-Benzoyl-α-D-Glucopyranosyl)Formamide (20)
Prepared according to General method I. from 15 (100 mg, 0.14 mmol) and allyl alcohol to give 45 mg (43%) white powder. Preparation according to General method II. gave 540 mg (51%). Rf = 0.16 (hexane/acetone 3:1). [α]D = +49 (c = 0.48, CHCl3); 1H NMR (360 MHz, CDCl3) δ (ppm): 8.08–7.85 (8H, m, Ar), 7.58–7.25 (12H, m, Ar), 6.79 (1H, d, J = 3.2 Hz, NH), 6.60 (1H, t, J = 9.1 Hz, H-3), 6.12 (1H, d, J = 3.2 Hz, NH), 5.92–5.81 (3H, m, H-2, H-4, -OCH2-CH=CH2), 5.24 (1H, dd, J = 17.3, 1.5 Hz, -OCH2-CH=CH2), 5.14 (1H, dd, J = 10.9, 1.3 Hz, -OCH2-CH=CH2), 5.13 (1H, m, H-5), 4.73 (1H, dd, J = 12.3, 2.8 Hz, H-6a), 4.42 (1H, dd, J = 12.3, 3.60 Hz, H-6b), 4.36 (2H, d, J = 5.7 Hz, -OCH2-CH=CH2). 13C NMR (90 MHz, CDCl3) δ (ppm): 169.7 (CONH2), 166.2, 165.5, 165.4, 165.1 (4 × C=O), 133.6–128.3 (Ar and -OCH2-CH=CH2), 117.9 (-OCH2-CH=CH2), 97.7 (C-1), 72.5, 72.2, 69.7, 68.9 (C-2–C-5), 64.7, 62.7 (C-6, OCH2-CH=CH2).
3.15. C-(2,3,4,6-Tetra-O-Acetyl-1-Allyloxy-α-D-Galactopyranosyl)Formamide (21)
Prepared according to General method I. from 16 (1.0 g, 2.20 mmol) and allyl alcohol. Yield: 513 mg (54%), colorless, white powder. Rf = 0.48 (hexane/acetone = 2:1). [α]D = +56 (c = 0.44, CHCl3); 1H NMR (400 MHz, CDCl3) δ (ppm): 6.72 (1H, d, J = 3.1 Hz, NH), 6.25 (1H, d, J = 3.0 Hz, NH), 5.89 (1H, dd, J = 10.4, 3.4 Hz, H-3), 5.96–5.86 (1H, m, -OCH2-CH=CH2), 5.55 (1H, d, J = 10.5 Hz, H-2), 5.52 (1H, dd, J = 3.2, 1.2 Hz, H-4), 5.29 (1H, dd, J = 17.2, 1.5 Hz, -OCH2-CH=CH2), 5.20 (1H, dd, J = 10.1, 1.4 Hz, -OCH2-CH=CH2), 4.88 (1H, td, J = 7.7, 6.8, 1.2 Hz, H-5), 4.34 (1H, dd, J = 12.0, 5.7 Hz, -OCH2-CH=CH2), 4.24 (1H, dd, J = 12.0, 6.1 Hz, -OCH2-CH=CH2), 4.13 (1H, dd, J = 11.2, 7.0 Hz, H-6a), 4.08 (1H, dd, J = 11.2, 6.4 Hz, H-6b), 2.17, 2.07, 2.04, 1.97 (4 × 3H, s, 4 × OCOCH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.5, 170.0, 169.9, 169.8 (2) (4 × C=O), 133.8 (-OCH2-CH=CH2), 117.9 (-OCH2-CH=CH2), 97.6 (C-1), 71.3, 70.0, 67.5, 65.7 (C-2–C-5), 63.9, 61.5 (C-6, -OCH2-CH=CH2), 20.8, 20.8, 20.7, 20.70 (4 × OCOCH3).
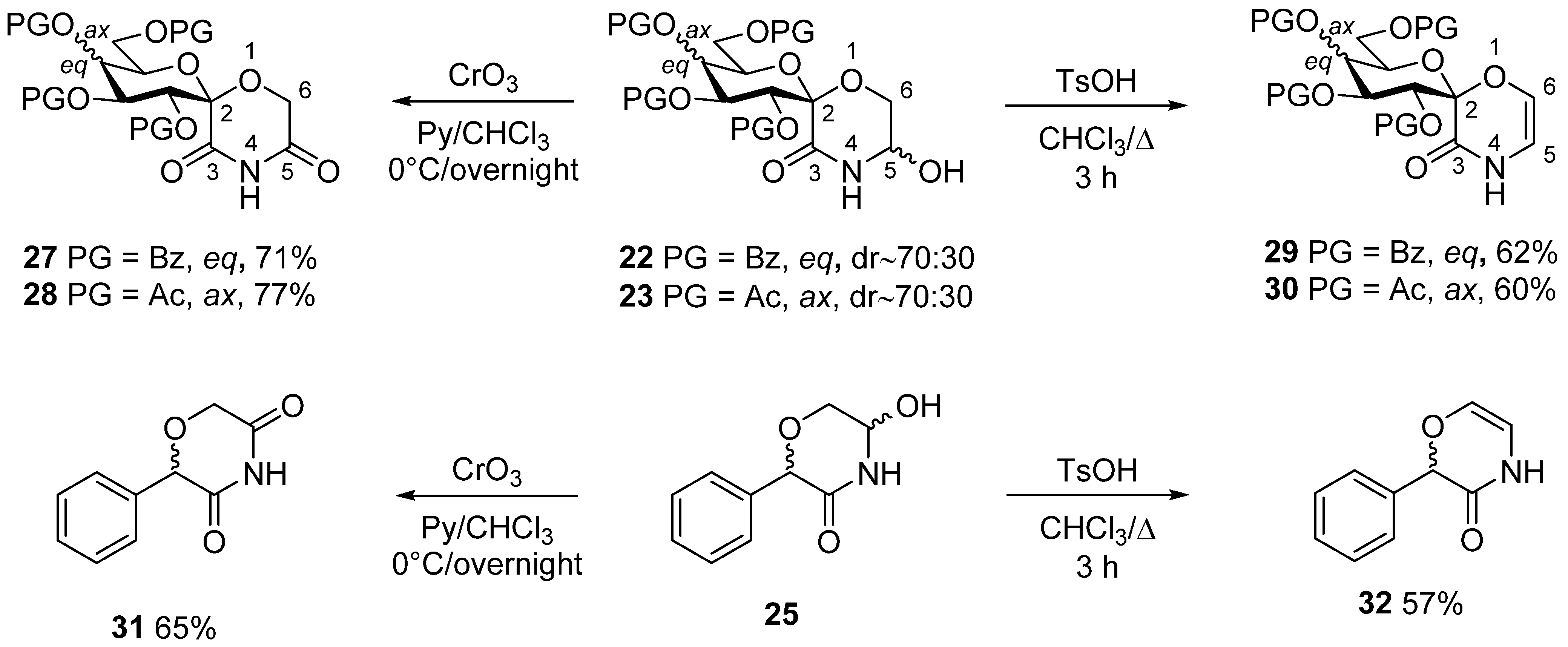
3.16. (1′S)-1′,5′-Anhydro-2′,3′,4′,6′-Tetra-O-Benzoyl-D-Glucitol-Spiro-[1′,2]-5-Hydroxymorpholin-3-One (22)
Prepared according to General method IV. from 20 (540 mg, 0.79 mmol). Yield: 294 mg (56%), colorless, fine powder, diastereomeric ratio (based on the 1H NMR integrals): ~70:30. Rf = 0.28 (hexane/acetone 2:1). (Only the peaks of the major diastereomer are listed here, because not all signals of the minor component were separately visible) 1H NMR (400 MHz, CDCl3) δ (ppm): 8.06–7.83 (8H, m, Ar), 7.56–7.05 (12H, Ar), 6.74 (1H, t, J = 9.8 Hz, H-3′), 5.83 (1H, t, J = 9.8 Hz, H-4′), 5.71 (1H, d, J = 10.0 Hz, NH), 5.20 (1H, dd, J = 8.2, 6.0 Hz, H-5′), 4.95 (1H, d, J = 9.8 Hz, H-2′), 4.75 (1H, brs, OH), 4.70 (1H, dd, J = 13.1, 3.1 Hz, H-6′a), 4.45 (1H, td, J = 12.4, 3.8 Hz, H-5), 4.43 (1H, dd, J = 12.3, 3.7 Hz, H-6′b), 4.06 (1H, dd, J = 11.3, 8.3 Hz, H-6a), 3.97 (1H, dd, J = 11.7, 4.8 Hz, H-6b). 13C NMR (100 MHz, CDCl3) δ (ppm): 166.8, 166.3, 165.8, 165.4, 165.1 (5 × C=O), 133.6–128.4 (Ar), 96.3 (C-1′), 74.0, 73.8, 72.4, 71.9, 69.3 (C-2′–C-5′, C-5), 64.1, 62.9 (C-6, C-6′).
3.17. (1′S)-2′,3′,4′,6′-Tetra-O-Acetyl-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-5-Hydroxymorpholin-3-One (23)
Prepared according to General method IV. from 21 (600 mg, 1.39 mmol). Yield: 270 mg (45%), colorless, fine powder, diastereomeric ratio (based on the 1H NMR integrals): ~70:30. Rf = 0.21 (hexane/acetone 2:1). (Only the peaks of the major diastereomer are listed here, because not all signals of the minor component were separately visible) 1H NMR (400 MHz, CDCl3) δ (ppm): 7.54 (1H, d, J = 6.1 Hz, NH), 5.93 (1H, dd, J = 10.6, 3.5 Hz, H-3′), 5.48 (1H, dd, J = 3.4, 1.1 Hz, H-4′), 5.38 (1H, d, J = 10.6 Hz, H-2′), 5.17 (1H, dd, J = 8.2, 5.2 Hz, H-5′), 4.66 (1H, dt, J = 7.0, 1.3 Hz, H-5), 4.33 (1H, brs, OH), 4.14 (2H, dd, J = 6.6, 3.3 Hz, H-6a,b), 4.00 (1H, dd, J = 11.8, 4.9 Hz, H-6′a), 3.92 (1H, dd, J = 11.7, 8.5 Hz, H-6′b), 2.18, 2.06, 2.04, 1.97 (4 × 3H, s, 4 × OCOCH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.8, 170.5, 170.5, 169.5 (4 × C=O), 165.4 (C-3), 96.3 (C-1′), 73.9, 71.1, 70.1, 70.0, 67.3 (C-2′–C-5′, C-5), 63.9, 61.6 (C-6, C-6′), 20.9, 20.8 (3) (4 × OCOCH3).
3.18. 5-Hydroxy-2-Phenylmorpholin-3-One (25)
Prepared according to General method IV. from 24 (2.0 g, 10.4 mmol, for its preparation see supporting information). Column chromatography (hexane/acetone 3:1) of the crude product gave 769 mg (38%) pale yellow oil. Rf = 0.25 (hexane/acetone 3:2). The product contained all 4 possible stereoisomers (two pairs of enantiomers). Two series of peaks are present in the spectra, in a ratio of 5:1. Only the peaks of the major components are listed here, because not all signals of the minor products were separately visible. 1H NMR (400 MHz DMSO-d6) δ (ppm): 8.56 (1H, s, NH), 7.38–7.30 (5H, m, Ar), 6.17 (1H, d, J = 7.8 Hz, OH), 5.03 (1H, s, H-2), 4.94 (1H, dddd, J = 7.7, 5.7, 3.7, 2.4 Hz, H-5), 3.84 (1H, dd, J = 11.8, 3.6 Hz, H-6a), 3.46 (1H, dd, J = 11.8, 5.4 Hz, H-6b). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 168.3 (C-3), 137.4, 128.2, 128.1 (2), 127.9 (2) (Ar), 77.7, 73.3 (C-2, C-5), 66.2 (C-6). HRMS (positive mode, m/z): 216.0630 (calculated value for C10H11NO3Na: 216.0631).
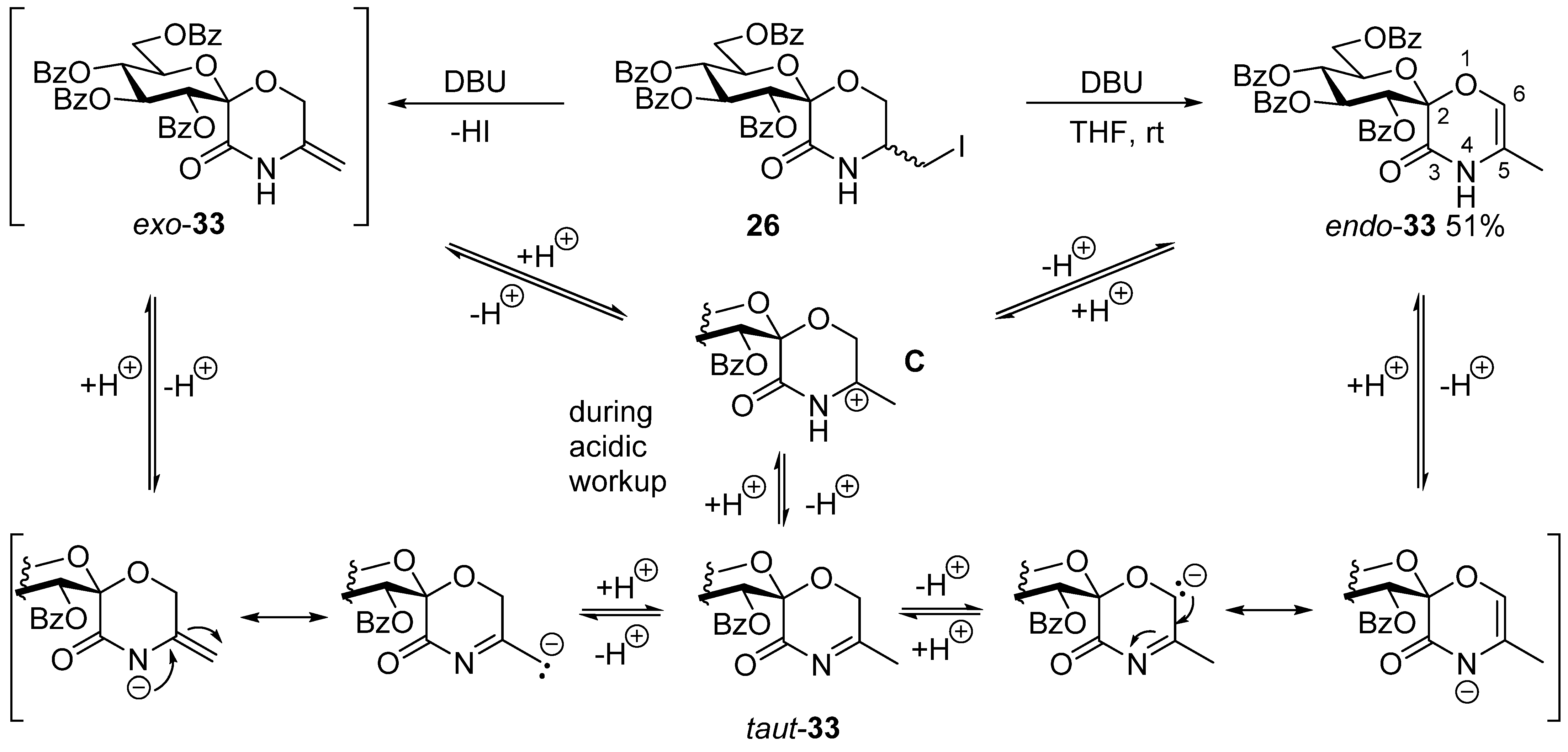
3.19. (1′S)-1′,5′-Anhydro-2′,3′,4′,6′-Tetra-O-Benzoyl-D-Glucitol-Spiro-[1′,2]-5-Iodomethyl-Morpholin-3-One (26)
In a flame dried round bottom flask, allyloxyamide 20 (0.72 g, 1.06 mmol) and iodine (0.81 g, 3 equiv.) were dissolved in dry acetonitrile (20 mL), and dried potassium carbonate (0.44 g, 3 equiv.) was added. The suspension was stirred at room temperature for 4 h, after which TLC showed complete conversion. The mixture was diluted with chloroform (40 mL), extracted with a ~5% aqueous solution of sodium sulfite and twice with water. The organic phase was dried over magnesium sulfate, filtered and concentrated. The product was obtained after column chromatography (hexane/acetone 3:1). Yield: 605 mg (71%), pale yellow oil. Rf = 0.38 (hexane/acetone 2:1). (Only the peaks of the major diastereomer are listed here, because not all signals of the minor component were separately visible 1H NMR (400 MHz CDCl3) δ (ppm): 8.06–7.81 (8H, m, Ar), 7.56–7.22 (13H, Ar, NH), 6.74 (1H, t, J = 9.7 Hz, H-3′), 5.79 (1H, t, J = 9.8 Hz, H-4′), 5.72 (1H, d, J = 9.7 Hz, H-2′), 4.98 (1H, dt, J = 9.5, 3.7 Hz, H-5′), 4.64 (1H, dd, J = 11.9, 2.3 Hz, H-6′a), 4.48 (1H, dd, J = 12.2, 4.4 Hz, H-6′b), 4.01–3.92 (2H, m, H-6a,b), 3.68–3.62 (1H, m, H-5), 3.13 (1H, dd, J = 10.7, 4.9 Hz, -CH2I), 3.30 (1H, dd, J = 10.8, 6.7 Hz, -CH2I). 13C NMR (100 MHz, CDCl3) δ (ppm): 166.3, 165.7, 165.7, 165.4, 165.1 (4 × C=O), 133.6–128.3 (Ar), 96.6 (C-1′), 73.4, 72.4, 72.3, 69.5 (C-2′–C-5′), 64.1, 63.1 (C-6, C-6′), 51.9 (C-5), 2.9 (-CH2I).
3.20. (1′S)-1′,5′-Anhydro-2′,3′,4′,6′-Tetra-O-Benzoyl-D-Glucitol-Spiro-[1′,2]-Morpholine-3,5-Dione (27)
Prepared according to General method V. from 22 (500 mg, 0.73 mmol). Yield: 354 mg (71%), colorless foam. Rf = 0.50 (hexane/acetone 2:1). [α]D = +21 (c = 0.51, CHCl3); 1H NMR (360 MHz CDCl3) δ (ppm): 8.29 (1H, s, NH), 8.02–7.80 (8H, m, Ar), 7.80–7.25 (12H, m, Ar), 6.71 (1H, t, J = 9.8 Hz, H-3′), 5.85 (1H, d, J = 10.1 Hz, H-2′), 5.82 (1H, t, J = 9.7 Hz, H-4′), 4.73 (1H, d, J = 17.0 Hz, H-6a), 4.69 (1H, dd, J = 12.3, 2.7 Hz, H-6′a), 4.50 (1H, ddd, J = 9.9, 4.3, 2.9 Hz, H-5′), 4.44 (1H, dd, J = 12.7, 4.6 Hz, H-6′b), 4.41 (1H, d, J = 17.0 Hz, H-6b). 13C NMR (90 MHz, CDCl3) δ (ppm): 167.7, 166.2, 165.6, 165.3, 164.9, 164.7 (6 × C=O), 133.8–128.4 (Ar), 95.8 (C-1′), 72.5, 72.4, 71.7, 69.0 (C-2′–C-5′), 62.3, 62.1 (C-6, C-6′).
3.21. (1′S)-2′,3′,4′,6′-Tetra-O-Acetyl-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-Morpholine-3,5-Dione (28)
Prepared according to General method V. from 23 (500 mg, 1.16 mmol). Yield: 384 mg (77%), colorless, foam. Rf = 0.50 (hexane/acetone 2:1). [α]D = +24 (c = 0.45, CHCl3); 1H NMR (400 MHz CDCl3) δ (ppm): 8.79 (1H, s, NH), 5.91 (1H, dd, J = 10.7, 3.5 Hz, H-3′), 5.55 (1H, dd, J = 3.4, 1.0 Hz, H-4′), 5.51 (1H, d, J = 10.7, H-2′), 4.68 (1H, d, J = 17.0 Hz, H-6a), 4.41 (1H, d, J = 17.0 Hz, H-6b), 4.30 (1H, td, J = 6.5, 1.0 Hz, H-5′), 4.18 (1H, dd, J = 11.3, 6.2 Hz, H-6′a), 4.13 (1H, dd, J = 11.1, 6.9 Hz, H-6′b), 2.19, 2.08, 2.05, 1.99 (4 × 3H, s, 4 × OCOCH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.5, 170.2, 169.9, 169.4, 168.1, 164.5 (6 × C=O), 95.6 (C-1′), 71.4, 69.4, 69.1, 66.9 (C-2′–C-5′), 61.8, 61.2 (C-6, C-6′), 20.3, 20.7 (2), 20.6 (4 × OCOCH3).
3.22. (1′S)-1′,5′-Anhydro-2′,3′,4′,6′-Tetra-O-Benzoyl-D-Glucitol-Spiro-[1′,2]-(2H-1,4-Oxazin-3[4H]-One) (29)
Prepared according to General method VI. from 22 (290 mg, 0.42 mmol) Yield: 174 mg (62%), colorless powder. Rf = 0.46 (hexane/acetone 2:1). [α]D = +29 (c = 0.50, CHCl3); 1H NMR (400 MHz DMSO-d6) δ (ppm): 10.40 (1H, d, J = 4.8 Hz, NH), 7.96–7.72 (8H, m, Ar), 7.67–7.34 (12H, m, Ar), 6.68 (1H, t, J = 9.6 Hz, H-3′), 6.36 (1H, d, J = 3.8 Hz, H-6), 6.00 (1H, t, J = 4.4 Hz, H-5), 5.78 (1H, t, J = 9.6 Hz, H-4′), 5.73 (1H, d, J = 9.9 Hz, H-2′), 4.52–4.45 (3H, m, H-5′, H-6′a, H-6′b). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 165.3, 164.9, 164.7, 164.4 (4 × C=O), 158.4 (C-3), 134.0–128.2 (Ar), 125.8 (C-6), 106.7 (C-5), 97.1 (C-1′), 72.6, 72.5, 72.0, 68.8 (C-2′–C-5′), 62.5 (C-6′).
3.23. (1′S)-2′,3′,4′,6′-Tetra-O-Acetyl-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-(2H-1,4-Oxazin-3[4H]-One) (30)
Prepared according to General method VI. from 23 (150 mg, 0.35 mmol). Yield: 86 mg (60%), colorless powder. Rf = 0.51 (hexane/acetone 2:1). [α]D = +40 (c = 0.33, CHCl3); 1H NMR (400 MHz CDCl3) δ (ppm): 7.78 (1H, d, J = 5.0 Hz, NH), 6.19 (1H, dd, J = 4.3, 1.4 Hz, H-6), 6.01 (1H, dd, J = 10.7, 3.6 Hz, H-5), 5.82 (1H, t, J = 4.8 Hz, H-3′), 5.55–5.52 (2H, m, H-2, H-4′), 4.31 (1H, dt, J = 6.6, 1.3 Hz, H-5′), 4.16 (1H, dd, J = 10.5, 5.4 Hz, H-6′a), 4.11 (1H, dd, J = 10.5, 6.9 Hz, H-6′b), 2.19, 2.08, 2.03, 1.98 (4 × 3H, s, 4 × OCOCH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.4, 170.3, 169.4, 169.7 (4 × C=O), 159.3 (C-3), 126.8 (C-6), 105.6 (C-5), 98.1 (C-1′), 71.9, 70.2, 69.7, 67.1 (C-2′–C-5′), 61.2 (C-6′), 20.9, 20.8, 20.8, 20.7 (4 × OCOCH3).
3.24. 2-Phenylmorpholine-3,5-Dione (31)
Prepared according to General method V. from 25 (310 mg, 1.60 mmol). Column chromatography (hexane/acetone 3:1) gave 198 mg (65%) pale yellow oil. Rf = 0.27 (hexane/acetone 2:1). 1H NMR (400 MHz CDCl3) δ (ppm): 9.04 (1H, s, NH), 7.43–7.37 (5H, m, Ar), 5.28 (1H, s, H-2), 4.32 (2H, d, J = 2.8 Hz, H-6a, H-6b). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.2, 170.1 (C-3, C-5), 132.9, 129.5, 128.9 (2), 127.7 (2) (Ar), 78.1 (C-2), 64.9 (C-6). HRMS (positive mode, m/z): [M+Na]+ was detected only as a small intensity peak, presumably a methanol molecule was added to the substrate under the MS conditions, therefore we detected the [M+MeOH+Na]+ as base peak. [M+Na]+: 214.0474 (calculated value for C10H9NO3Na: 214.0475) and [M+MeOH+Na]+: 246.0736 (calculated value for C11H13NO4Na: 246.0737).
3.25. 2-Phenyl-2H-1,4-Oxazin-3(4H)-One (32)
Prepared according to General method VI. from 25 (340 mg, 1.76 mmol) 5-hydroxy-2-phenylmorpholin-3-one and 45 mg (15 mol%) pTsOH. Yield: 176 mg (57%), pale yellow oil. Rf = 0.41 (hexane/acetone 2:1). 1H NMR (360 MHz CDCl3) δ (ppm): 8.40 (1H, s, NH), 7.47–7.37 (5H, m, Ar), 6.19 (1H, dd, J = 4.2, 1.2 Hz, H-6), 5.68 (1H, t, J = 4.4 Hz, H-5), 5.46 (1H, s, H-2). 13C NMR (90 MHz, CDCl3) δ (ppm): 165.4 (C-3), 135.5, 129.1, 129.0, 128.7 (2), 127.2 (2) (Ar, C-6), 106.5 (C-5), 78.5 (C-2). HRMS (positive mode, m/z): only the dimer [2M+Na]+ peak was detected at 373.1145 m/z (calculated value for C20H18N2O4Na: 373.1154).
3.26. (1′S)-1′,5′-Anhydro-2′,3′,4′,6′-Tetra-O-Benzoyl-D-Glucitol-Spiro-[1′,2]-(5-Methyl-2H-1,4-Oxazin-3[4H]-One) (33)
In a flame dried round bottom flask, 26 (140 mg, 0.17 mmol) was dissolved in dry THF (5 mL). While stirring, DBU (2 equiv., 52 μL) was added and the solution was stirred at room temperature. The conversion was monitored by TLC (hexane/acetone 2:1). Full conversion was reached after 2 h with minimal amounts of decomposition products. The mixture was quenched with one drop of glacial acetic acid, and the product was obtained after column chromatography (hexane/acetone 3:1). Yield: 60 mg (51%), colorless oil. Rf = 0.33 (hexane/acetone 2:1). [α]D = +32 (c = 0.31, CHCl3); 1H NMR (400 MHz CDCl3) δ (ppm): 8.28 (1H, s, NH), 8.03–7.93 (8H, m, Ar), 7.56–7.21 (12H, m, Ar), 6.82 (1H, t, J = 9.7 Hz, H-3′), 5.98 (1H, s, H-6), 5.85 (1H, d, J = 9.9 Hz, H-2′), 5.80 (1H, t, J = 9.8 Hz, H-4′), 4.60–4.53 (2H, m, H-5′, H-6′a), 4.48 (1H, dd, J = 11.7, 4.6 Hz, H-6′b), 1.79 (3H, s, -CH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.7, 165.4, 165.1 (4 × C=O), 160.1 (C-3), 133.5–128.4 (Ar), 122.4 (C-6), 114.3 (C-5), 97.2 (C-1′), 72.7, 72.6, 72.5, 69.6 (C-2′–C-5′), 63.0 (C-6′), 13.6 (-CH3). HRMS (positive mode, m/z): 700.1781 (calculated for C38H31NO11Na: 700.1789).
3.27. (1′S)-1′,5′-Anhydro-D-Glucitol-Spiro-[1′,2]-Morpholin-3-One (34)
Prepared according to General method VII. from 13 (60 mg, 90 μmol). Reaction time: 4 h. Column chromatography (CHCl3/MeOH 4:1 +0.5% Et3N) gave 16 mg (71%) colorless powder. Rf = 0.32 (CHCl3/MeOH 3:1), [α]D = +27 (c = 0.25, MeOH). Melting point: 178–181 °C. 1H NMR (CD3OD, 400 MHz) δ (ppm): 4.26 (1H, dt, J = 11.9, 3.3 Hz, H-6a), 4.24 (1H, t, J = 9.2 Hz, H-3′), 4.04 (1H, ddd, J = 9.9, 5.7, 2.3 Hz, H-5′), 3.82 (2H, dd, J = 11.9, 2.6 Hz, H-6b, H-6′a), 3.63 (1H, dd, J = 12.9, 5.7 Hz, H-6′b), 3.55 (1H, td, J = 12.2, 4.4 Hz, H-5a), 3.28 (partially merged together with the solvent peak, 2H, J = 9.5 Hz, H-2′, H-4′), 3.17 (1H, dd, J = 12.5, 2.3 Hz, H-5b). 13C NMR (CD3OD, 100 MHz) δ (ppm): 168.5 (C-3), 99.7 (C-1′), 77.9, 77.6, 74.5, 71.5 (C-2′–C-5′), 63.2 (C-6′), 59.9 (C-6), 41.9 (C-5). HRMS (positive mode, m/z): 272.0739 (calculated value for C9H15NO7Na: 272.0741).
3.28. (1′S)-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-Morpholin-3-One (35)
Prepared according to General method VII. from 14 (44 mg, 0.105 mmol). Reaction time: 2 h. Column chromatography (CHCl3/MeOH 4:1 +0.5% Et3N) gave 8 mg (31%) colorless powder. Rf = 0.24 (CHCl3/MeOH 3:1), [α]D = +52 (c = 0.14, MeOH). Melting point: 142–145 °C. 1H NMR (CD3OD, 400 MHz) δ (ppm): 4.36 (1H, dd, J = 9.9, 3.4 Hz, H-3′), 4.29 (1H, td, J = 5.8, 1.1 Hz, H-6a), 4.26 (1H, td, J = 11.6, 3.2 Hz, H-6b), 3.89 (1H, dd, J = 3.3, 1.1 Hz, H-4′), 3.82 (1H, ddd, J = 11.7, 3.5, 0.9 Hz, H-5′), 3.72–3.66 (3H, m, H-2′, H-6′a, H-6′b), 3.55 (1H, td, J = 12.2, 4.4 Hz, H-5a), 3.16 (1H, ddd, J = 12.5, 2.2, 0.9 Hz, H-5b). 13C NMR (CD3OD, 100 MHz) δ (ppm): 168.7 (C-3), 100.2 (C-1′), 77.0, 74.4, 72.5, 70.3 (C-2′–C-5′), 63.0, 59.8 (C-6, C-6′), 42.0 (C-5). HRMS (positive mode, m/z): 272.0740 (calculated value for C9H15NO7Na: 272.0741).
3.29. (1′S)-1′,5′-Anhydro-D-Glucitol-Spiro-[1′,2]-5-Hydroxymorpholin-3-One (36)
Prepared according to General method VII. from 22 (75 mg, 0.11 mmol). Reaction time: 2 h. Column chromatography (CHCl3/MeOH 4:1 +0.5% Et3N) gave 20 mg (68%) colorless powder. Rf = 0.35 (CHCl3/MeOH 1:1). (As not all signals of the minor diastereomer were separately visible in the NMR spectra, we are only listing the peaks of the major product). 1H NMR (CD3OD, 400 MHz) δ (ppm): 4.37 (1H, dd, J = 12.3, 1.9 Hz, H-3′), 4.26 (1H, t, J = 8.9 Hz, H-5), 3.96 (1H, ddd, J = 9.6, 5.6, 1.9 Hz, H-5′), 3.82 (1H, dd, J = 11.9, 1.9 Hz, H-4′), 3.74 (1H, d, J = 12.2 Hz, H-2′), 3.62 (1H, dd, J = 12.0, 5.8 Hz, H-6′a), 3.38 (1H, d, J = 9.5 Hz, H-6a), 3.35–3.29 (2H, m, H-6b, H-6′b). 13C NMR (CD3OD, 100 MHz) δ (ppm): 167.5 (C-3), 99.2 (C-1′), 77.7, 77.5, 75.4, 74.9, 71.4 (C-5, C-2′–C-5′), 65.3, 62.9 (C-6, C-6′). HRMS (negative mode, m/z): 264.0725, (calculated value for C9H14NO8: 264.0725).
3.30. (1′S)-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-5-Hydroxymorpholin-3-One (37)
Prepared according to General method VII. from 23 (70 mg, 0.16 mmol). Reaction time: 2 h. Column chromatography (CHCl3/MeOH 4:1 +0.5% Et3N) gave 24 mg (55%) colorless powder. Rf = 0.16 (CHCl3/MeOH 3:1). (As not all signals of the minor diastereomer were separately visible in the NMR spectra, we are only listing the peaks of the major product) 1H NMR (CD3OD, 360 MHz) δ (ppm): 4.40 (2H, dd, J = 12.7, 2.9 Hz, H-6a, H-6b), 4.23 (1H, t, J = 6.0 Hz, H-3′), 3.91–3.90 (2H, m, H-5, H-5′), 3.78–3.71 (4H, m, H-2′, H-4′, H-6′a, H-6′b). 13C NMR (CD3OD, 90 MHz) δ (ppm): 166.2 (C-3), 98.4 (C-1′), 75.5, 73.2, 73.1, 70.9, 68.8 (C-5, C-2′–C-5′), 63.8, 61.9 (C-6, C-6′). HRMS (positive mode, m/z): 288.0690 (calculated value for C9H15NO8Na: 288.0690).
3.31. (1′S)-1′,5′-Anhydro-D-Glucitol-Spiro-[1′,2]-Morpholine-3,5-Dione (38)
Prepared according to General method VII. from 27 (200 mg, 0.29 mmol). Reaction time: 4 h. Column chromatography (CHCl3/MeOH 6:1 +0.5% Et3N) gave 40 mg (52%) colorless powder. Rf = 0.25 (CHCl3/MeOH 3:1), [α]D = +45 (c = 0.13, MeOH). Melting point: 186–188 °C. 1H NMR (CD3OD, 400 MHz) δ (ppm): 4.98 (1H, d, J = 16.7 Hz, H-6a), 4.53 (1H, d, J = 16.7 Hz, H-6b), 4.49 (1H, t, J = 9.3 Hz, H-3′), 4.07 (1H, d, J = 9.8 Hz, H-2′), 3.88–3.82 (2H, m, H-4′, H-6′a), 3.66 (1H, d, J = 9.6 Hz, H-6′b), 3.53 (1H, ddd, J = 9.5, 5.2, 1.6 Hz, H-5′). 13C NMR (CD3OD, 100 MHz) δ (ppm): 171.7, 167.9 (C-3, C-5), 97.6 (C-1′), 77.7, 76.8, 76.3, 71.3 (C-2′–C-5′), 62.7, 62.6 (C-6, C-6′). HRMS (positive mode, m/z): 286.0535 (calculated value for C9H13NO8Na: 286.0535).
3.32. (1′S)-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-Morpholine-3,5-Dione (39)
Prepared according to General method VII. from 28 (134 mg, 0.31 mmol). Reaction time: 4 h. Column chromatography (CHCl3/MeOH 4:1 +0.5% Et3N) gave 69 mg (84%) colorless powder. Rf = 0.27 (CHCl3/MeOH 4:1). 1H NMR (CD3OD, 400 MHz) δ (ppm): 4.97 (1H, d, J = 16.7 Hz, H-6a), 4.61 (1H, dd, J = 9.7, 3.5 Hz, H-3′), 4.52 (1H, d, J = 16.7 Hz, H-6b), 4.14–4.08 (2H, m, H-4′, H-5′), 4.03–3.89 (3H, m, H-2′, H-6′a, H-6′b). 13C NMR (CD3OD, 100 MHz) δ (ppm): 171.8, 168.1 (C-3, C-5), 98.1 (C-1′), 76.9, 73.6, 72.1, 70.1 (C-2′–C-5′), 62.9, 62.5 (C-6, C-6′). HRMS (positive mode, m/z): 286.0533 (calculated value for C9H13NO8Na: 286.0533).
3.33. (1′S)-1′,5′-Anhydro-D-Glucitol-Spiro-[1′,2]-(2H-1,4-Oxazin-3[4H]-One) (40)
Prepared according to General method VII. from 29 (112 mg, 0.17 mmol). Reaction time: 3 h. Column chromatography (CHCl3/MeOH 6:1 +0.5% Et3N) gave 81 mg (72%) colorless powder. Rf = 0.32 (CHCl3/MeOH 4:1), [α]D = +26 (c = 0.30, MeOH). Melting point: 176–177 °C 1H NMR (CD3OD, 400 MHz) δ (ppm): 6.16 (1H, d, J = 4.3 Hz, H-6), 5.78 (1H, d, J = 4.3 Hz, H-5), 4.31 (1H, t, J = 9.2 Hz, H-3′), 3.74 (1H, dd, J = 12.0, 2.2 Hz, H-6′a), 3.62 (1H, dd, J = 12.0, 4.9 Hz, H-6′b), 3.56 (1H, ddd, J = 9.8, 4.9, 2.2 Hz, H-5′), 3.41 (1H, d, J = 9.6 Hz, H-2′), 3.36 (1H, t, J = 9.3 Hz, H-4′). 13C NMR (CD3OD, 100 MHz) δ (ppm): 162.1 (C-3), 127.6 (C-6), 106.8 (C-5), 100.1 (C-1′), 78.2, 77.1, 75.9, 71.2 (C-2′–C-5′), 62.4 (C-6′). HRMS (positive mode, m/z): 270.0581 (calculated value for C9H13NO7Na: 270.0584).
3.34. (1′S)-1′,5′-Anhydro-D-Galactitol-Spiro-[1′,2]-(2H-1,4-Oxazin-3[4H]-One) (41)
Prepared according to General method VII. from 30 (76 mg, 0.18 mmol). Reaction time: 2 hours. Column chromatography (CHCl3/MeOH 3:1 +0.5% Et3N) gave 32 mg (71%) colorless powder. Rf = 0.20 (CHCl3/MeOH 3:1), [α]D = +62 (c = 0.21, MeOH). Melting point: 132–136 °C. 1H NMR (CD3OD, 400 MHz) δ (ppm): 6.21 (1H, d, J = 4.3 Hz, H-6), 5.81 (1H, d, J = 4.3 Hz, H-5), 4.88 (1H, dd, J = 9.9, 3.6 Hz, H-3′), 3.95 (1H, dd, J = 3.8, 1.0. Hz, H-4′), 3.86 (1H, td, J = 6.0, 1.0 Hz, H-5′), 3.83 (1H, d, J = 10.0 Hz, H-2′), 3.75–3.66 (2H, m, H-6′a, H-6′b). 13C NMR (CD3OD, 100 MHz) δ (ppm): 162.2 (C-3), 127.9 (C-6), 106.7 (C-5), 100.6 (C-1′), 77.4, 74.2, 72.8, 70.0 (C-2′–C-5′), 62.4 (C-6′). HRMS (positive mode, m/z): 270.0584 (calculated value for C9H13NO7Na: 270.0584).
3.35. (1′S)-1′,5′-Anhydro-D-Glucitol-Spiro-[1′,2]-(5-Methyl-2H-1,4-Oxazin-3[4H]-One) (42)
Prepared according to General method VII. from 33 (85 mg, 0.125 mmol). Reaction time: 3 hours. Column chromatography (CHCl3/MeOH 6:1 +0.5% Et3N) gave 30 mg (77%) colorless powder. Rf = 0.25 (CHCl3/MeOH 6:1), [α]D = +31 (c = 0.26, MeOH). Melting point: 162–164 °C. 1H NMR (CD3OD, 400 MHz) δ (ppm): 5.97 (1H, d, J = 1.4 Hz, H-6), 4.37 (1H, t, J = 9.2 Hz, H-3′), 3.79 (1H, dd, J = 11.9, 2.0 Hz, H-6′a), 3.67 (1H, dd, J = 11.9, 5.0 Hz, H-6′b), 3.61 (1H, ddd, J = 9.9, 5.0, 2.2 Hz, H-5′), 3.44 (1H, d, J = 9.6 Hz, H-2′), 3.40 (1H, t, J = 8.5 Hz, H-4′), 1.75 (3H, d, 4JH-2, Me =1.32 Hz, CH3). 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 161.1 (C-3), 121.7 (C-6), 114.4 (C-5), 98.2 (C-1′), 77.6, 75.9, 74.5, 70.1 (C-2′–C-5′), 61.3 (C-6′), 13.5 (-CH3). HRMS (positive mode, m/z): 284.0738 (calculated value for C10H15NO7Na: 284.0741).





{kind=link}
{kind=link}
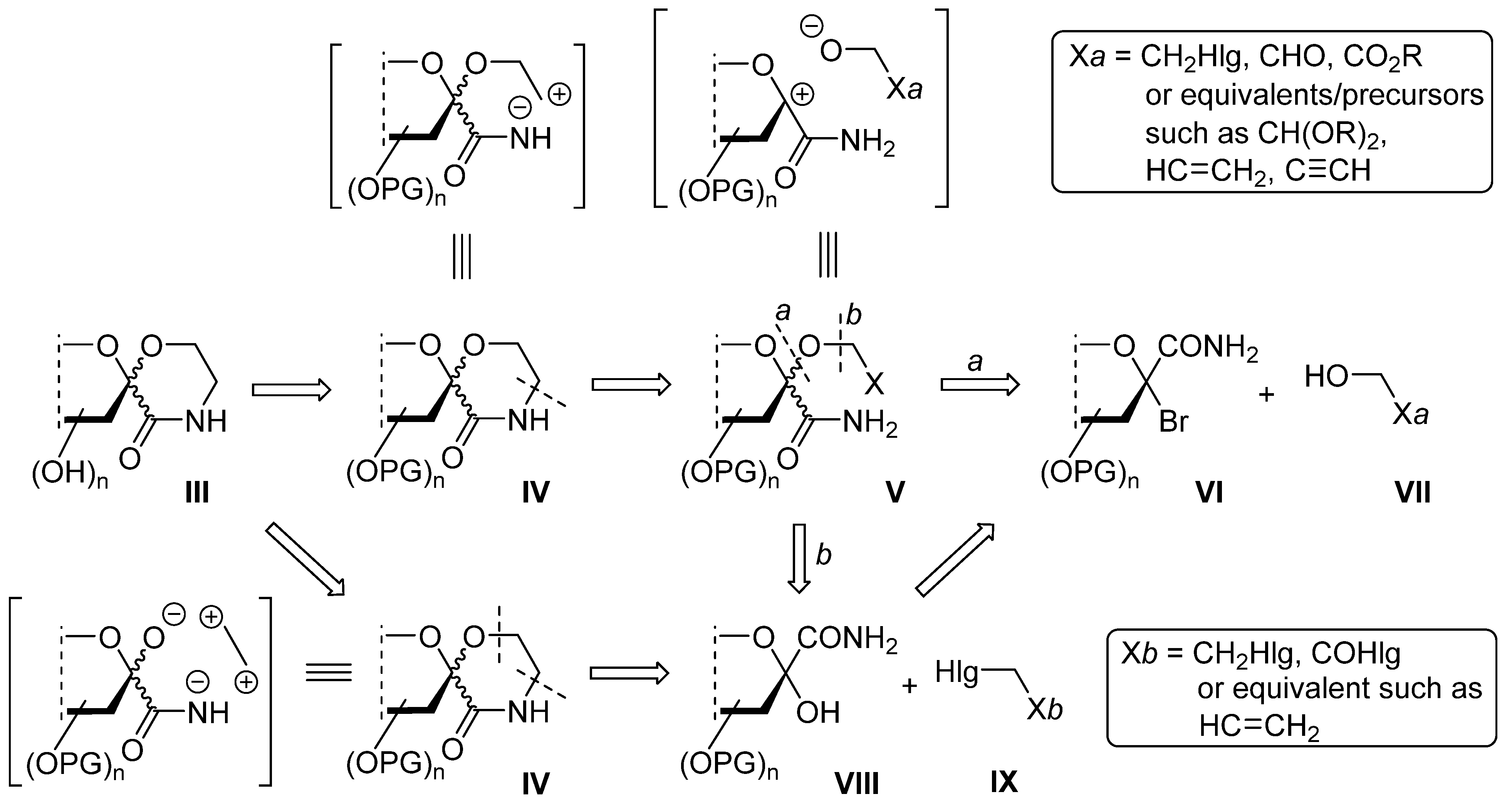{kind=link}
{kind=link}
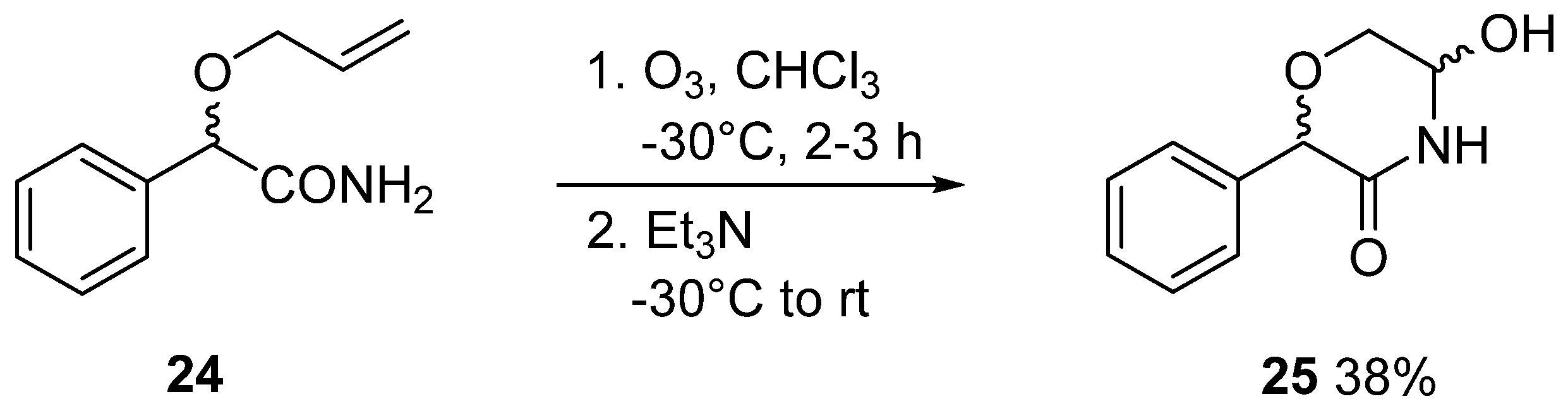{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







