
Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
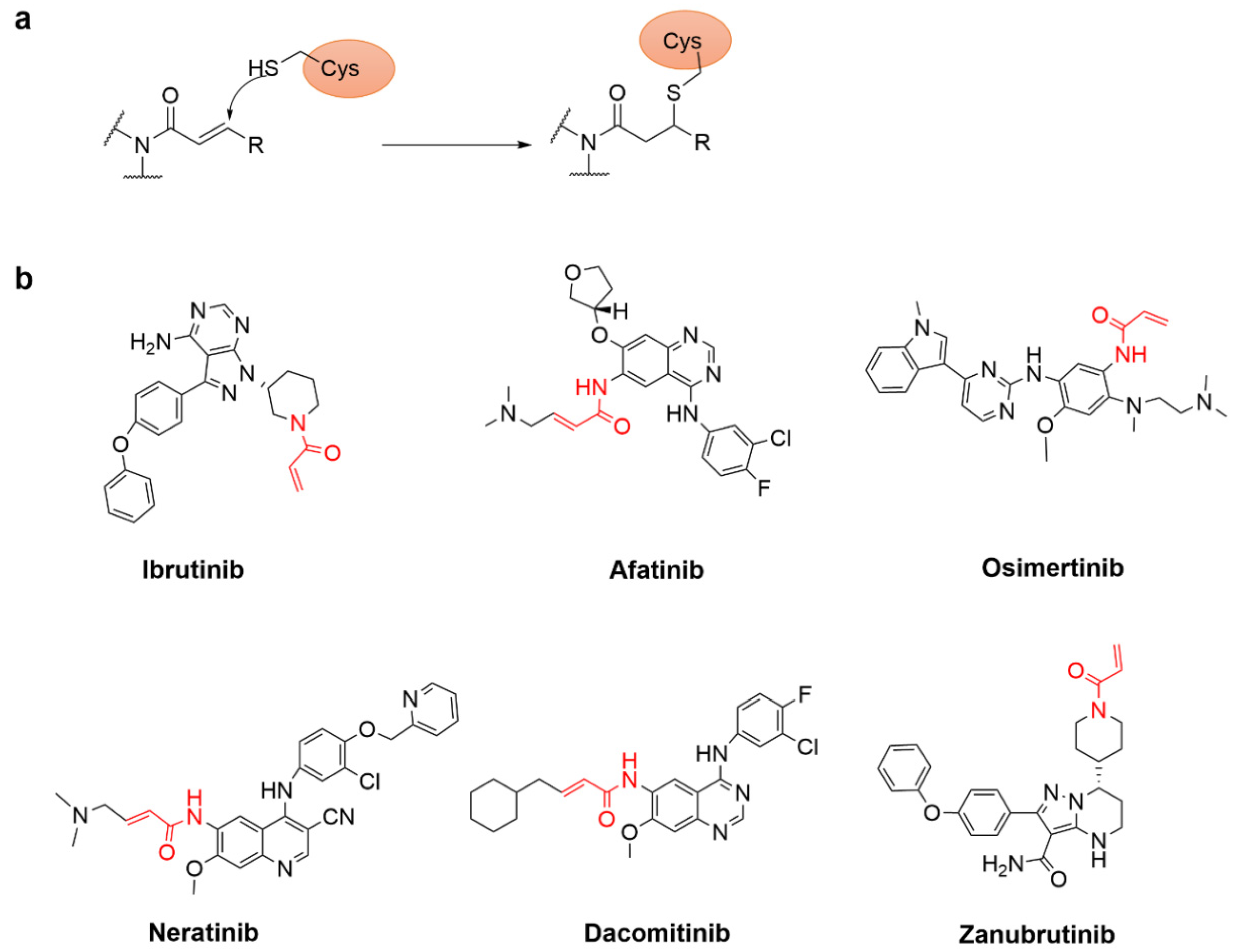{kind=link}
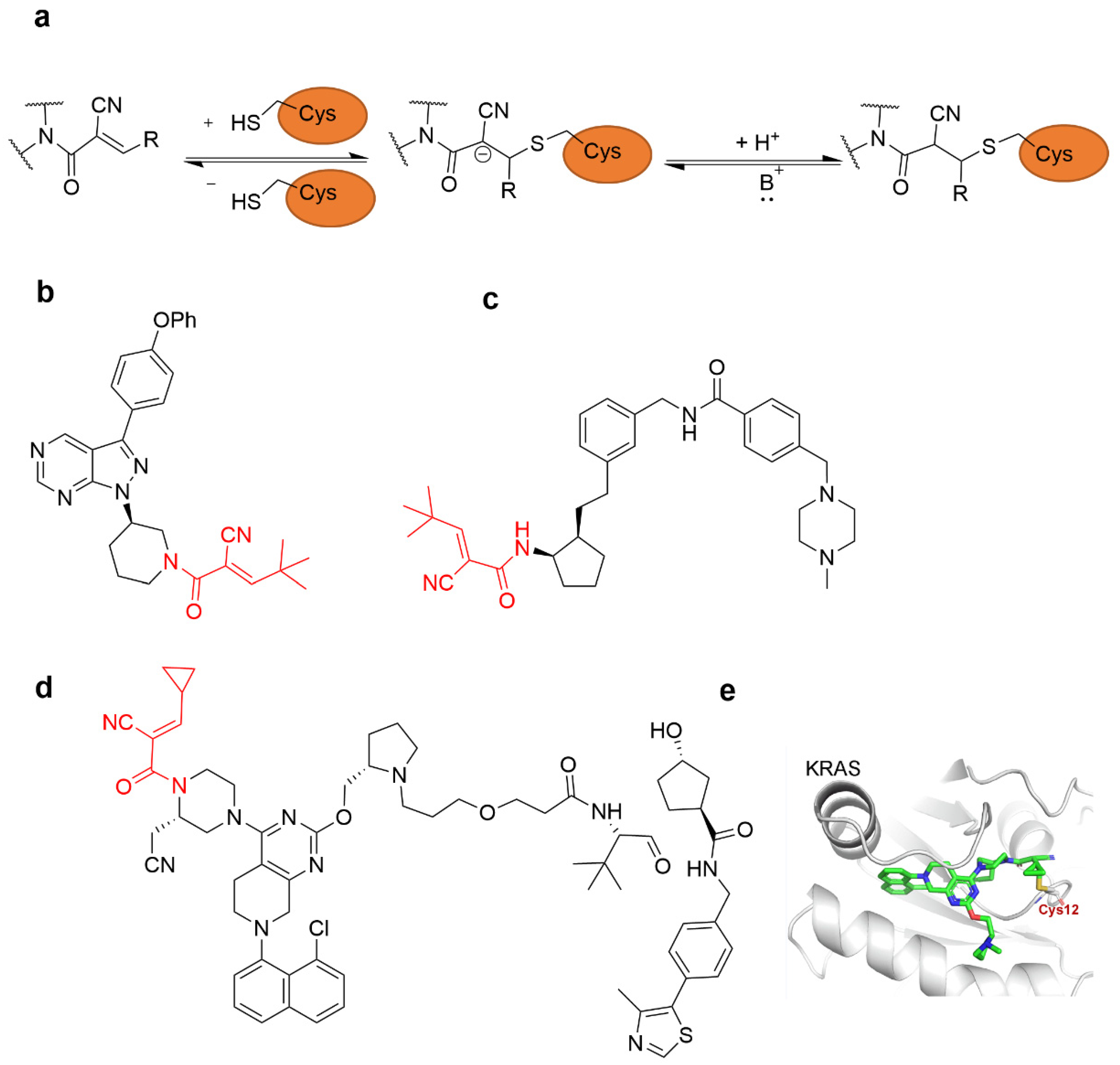{kind=link}
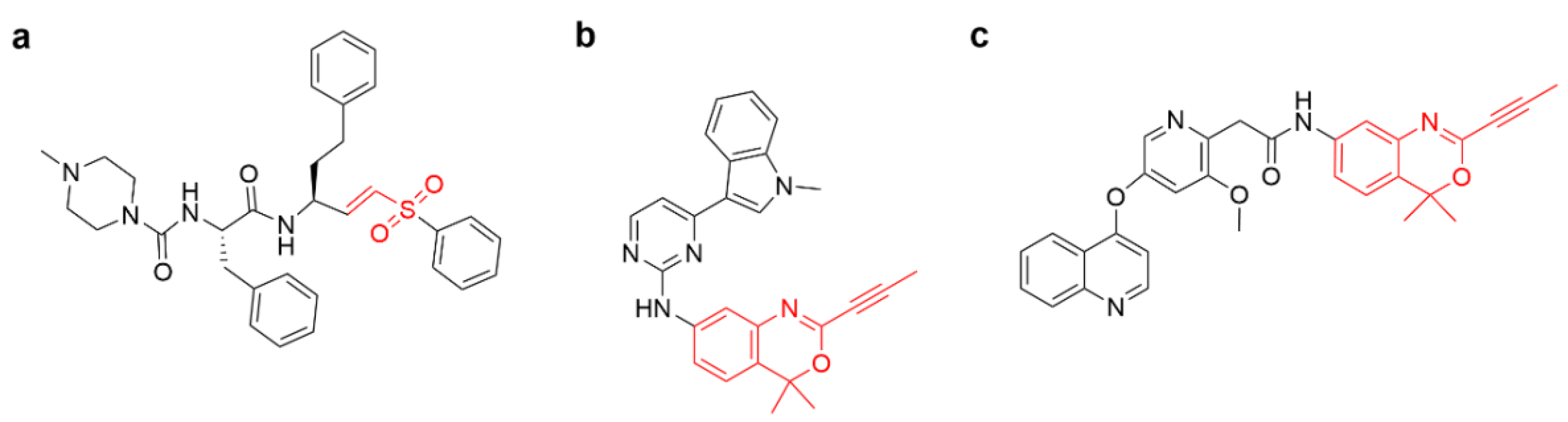{kind=link}
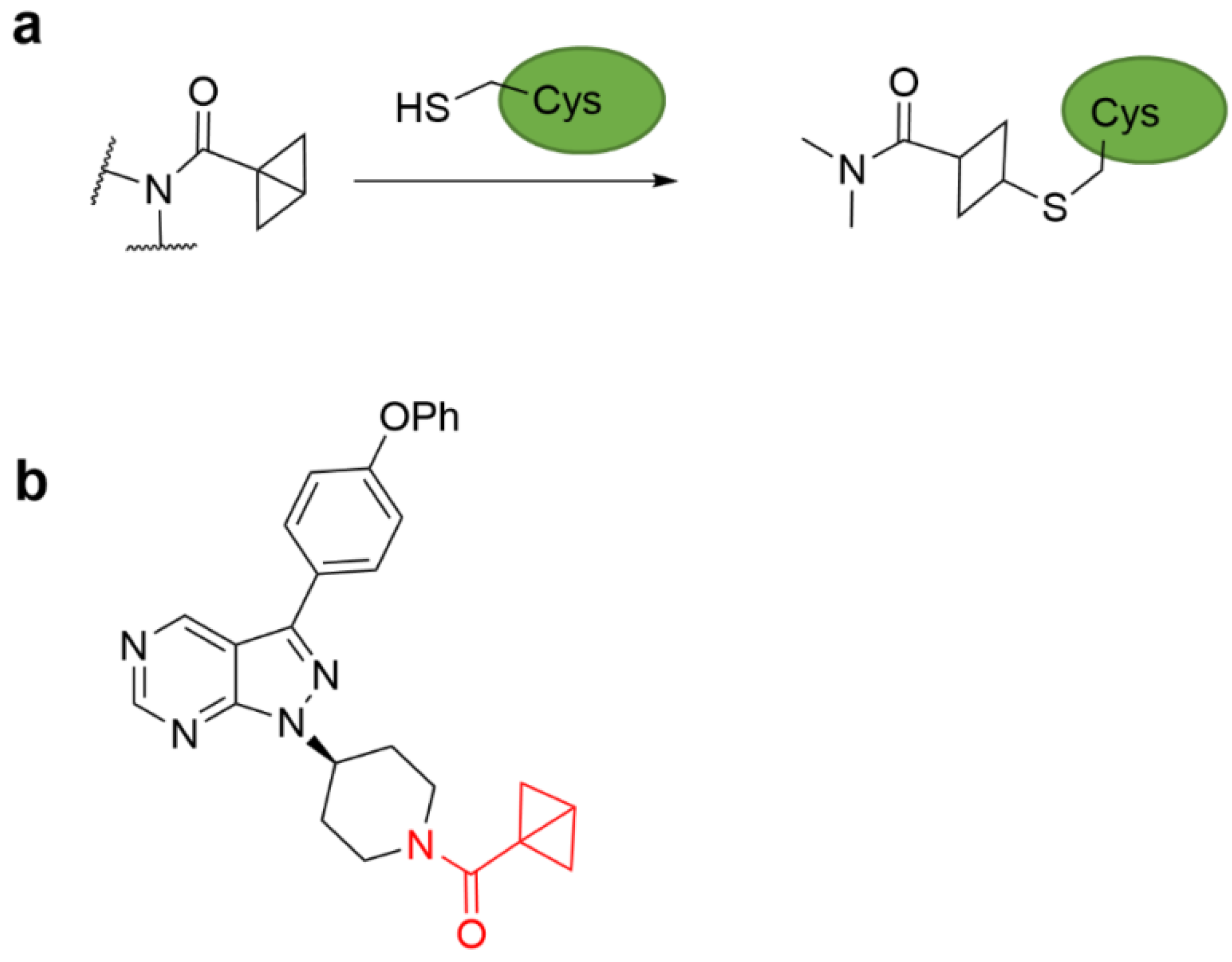{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Covalent Inhibitors
3. Covalent Inhibitors Covalently Bound to Cysteine
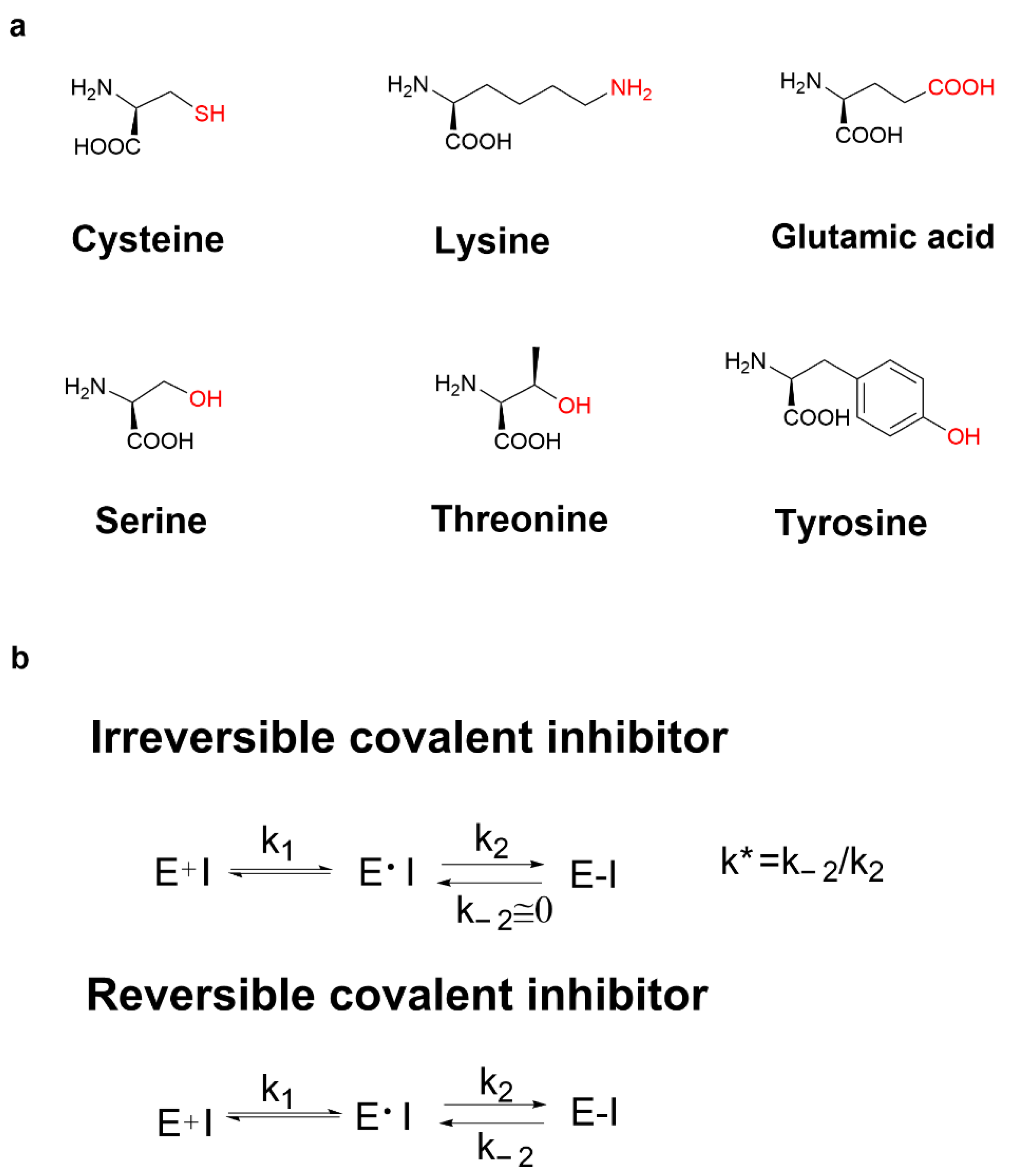
3.1. Cysteine Profile
3.2. Cysteine-Directed Covalent Drugs
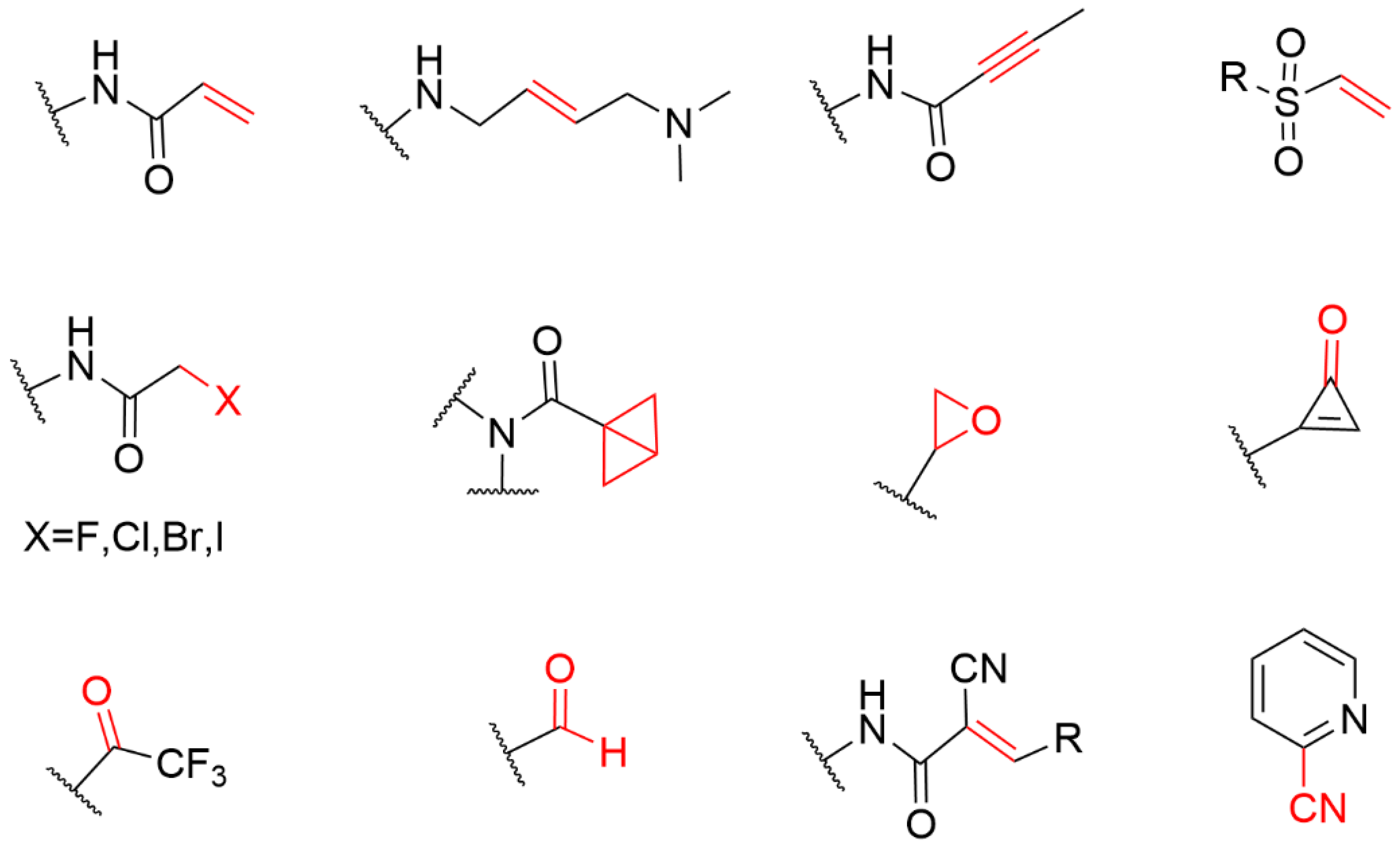
4. The Covalent Warheads Targeting Cysteine Residue
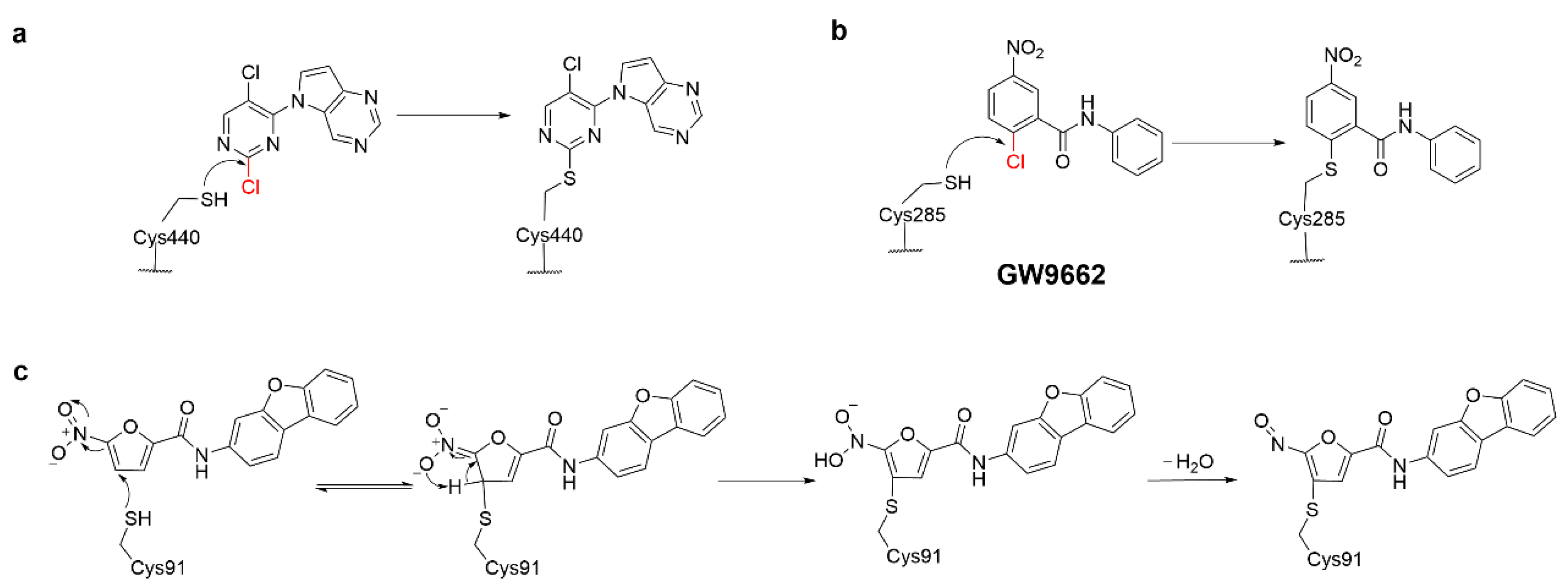
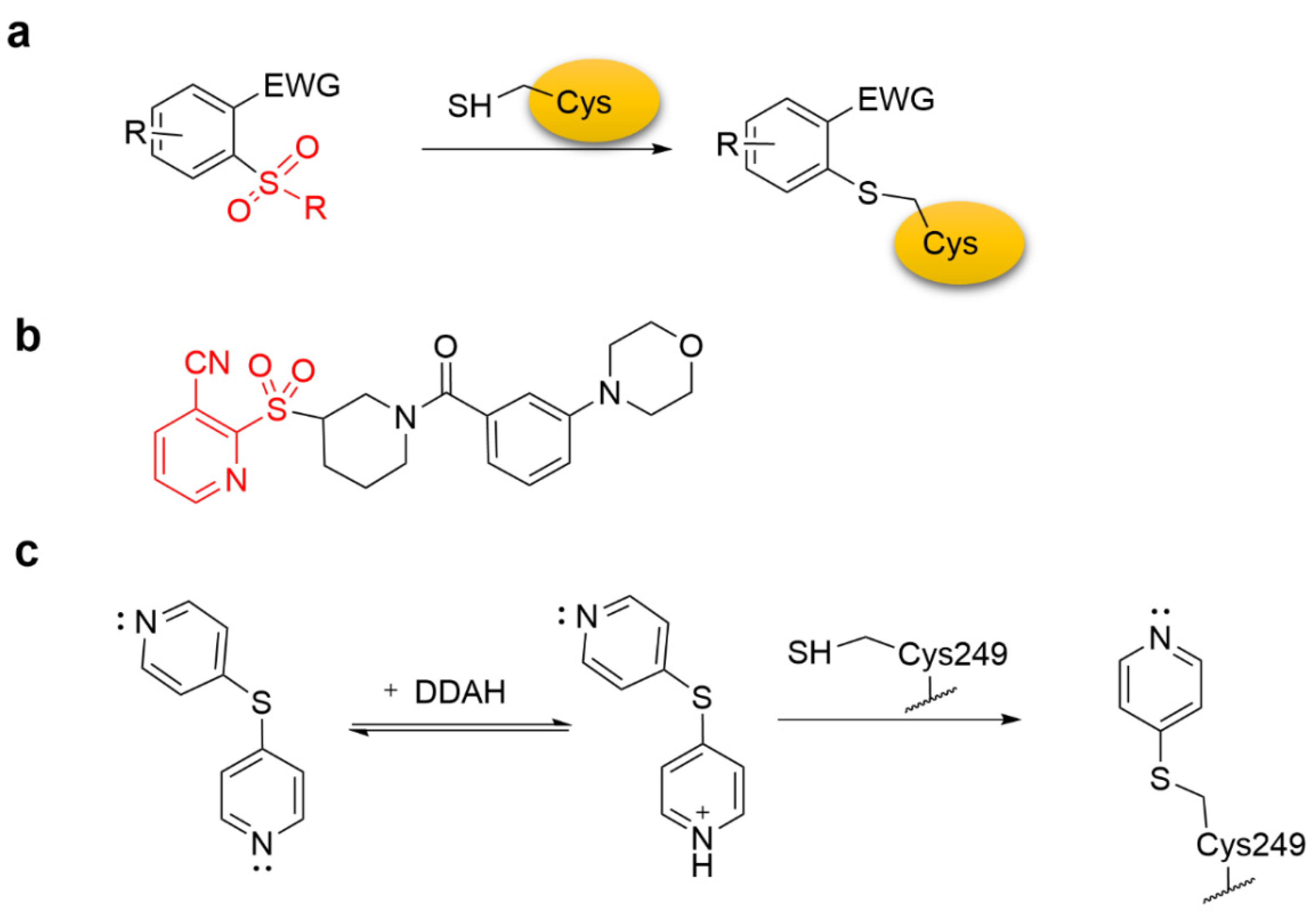
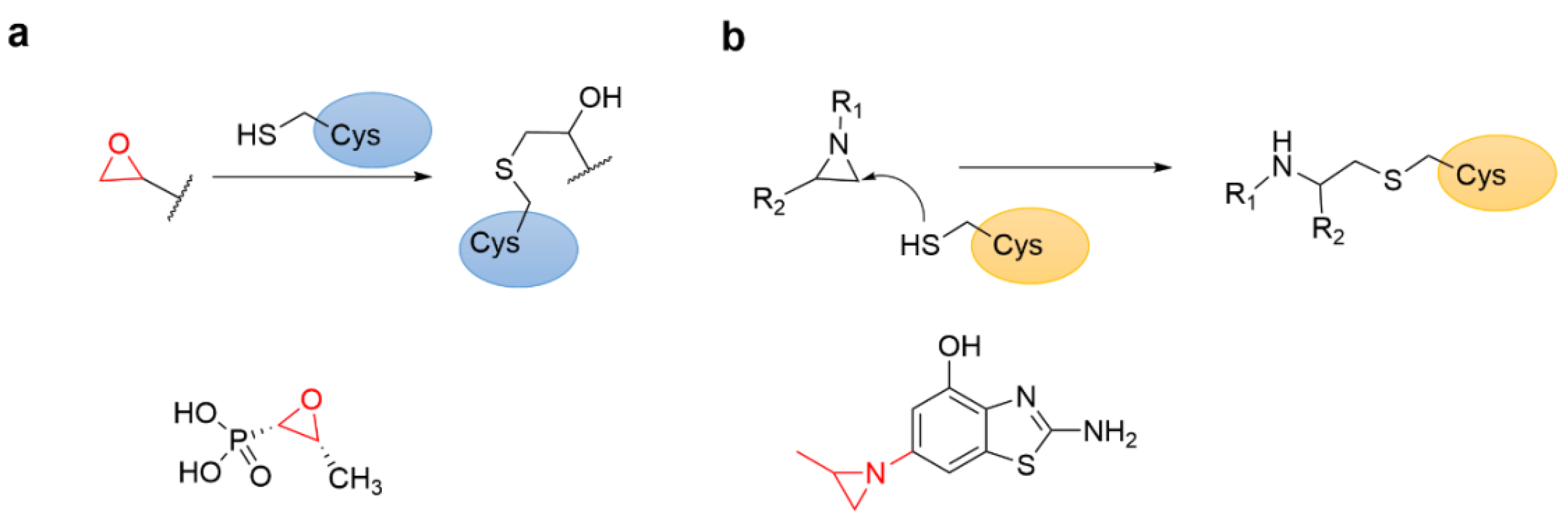
4.1. Heteroaromatic Warheads
4.2. α,β-Unsaturated Carbonyl Warhead
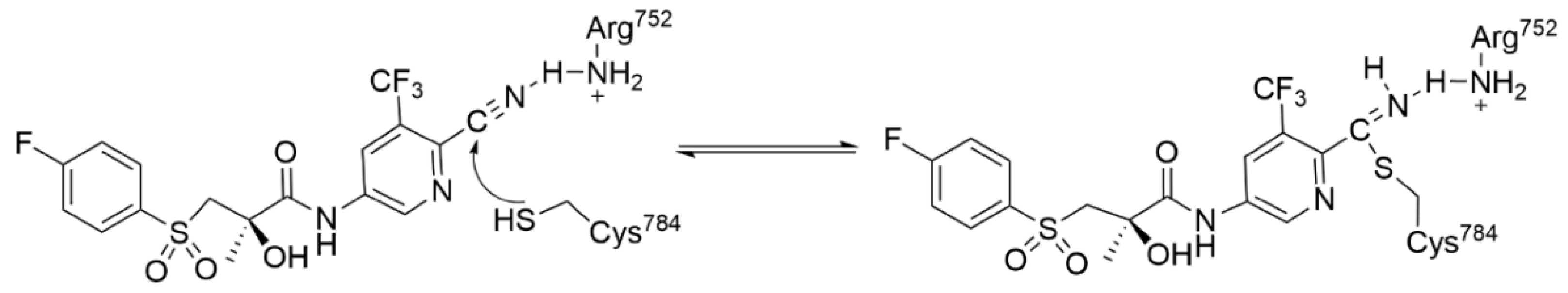
4.2.1. Acrylamide Warhead
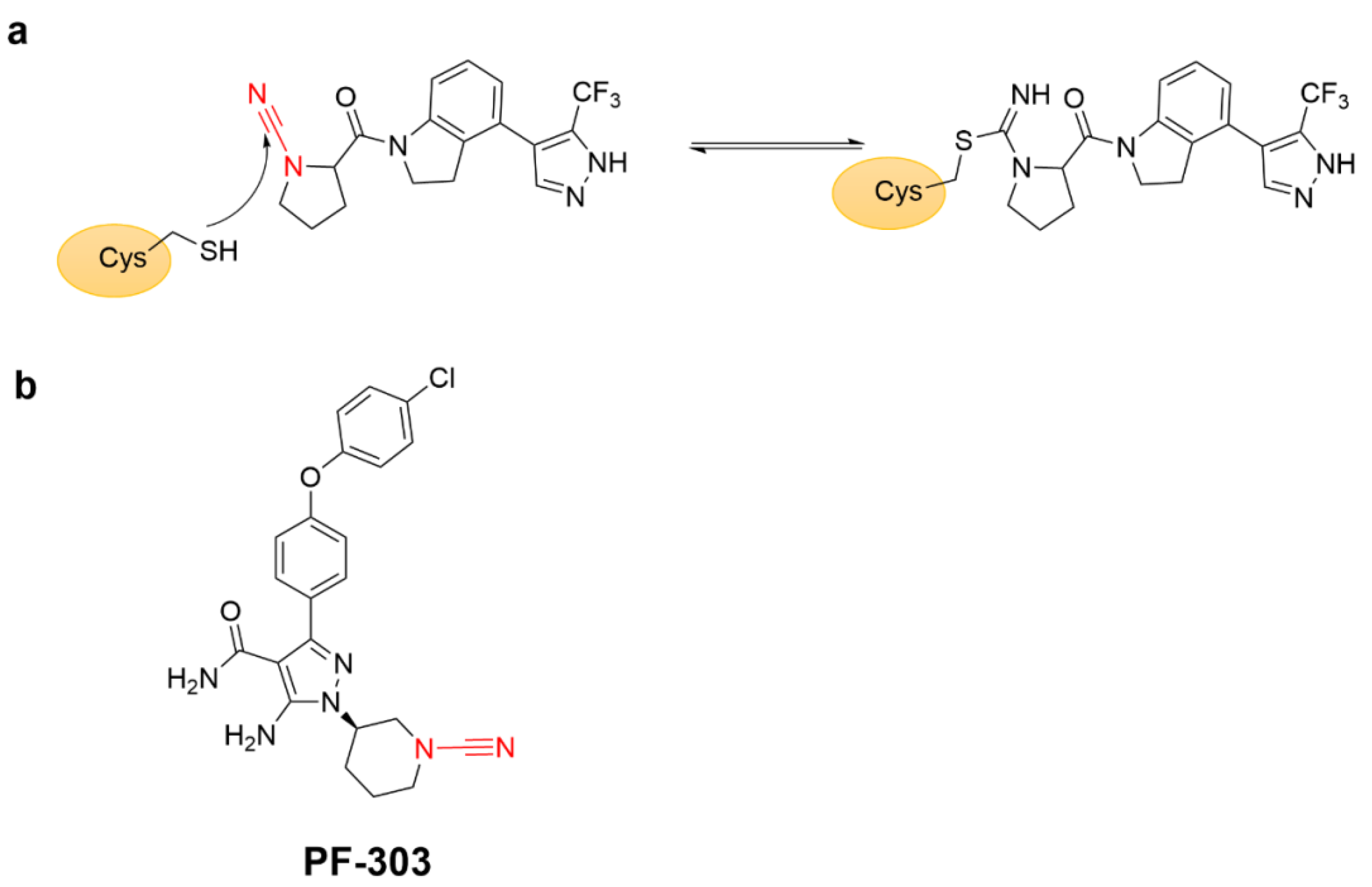
4.2.2. α-Cyan Acrylamide Warhead
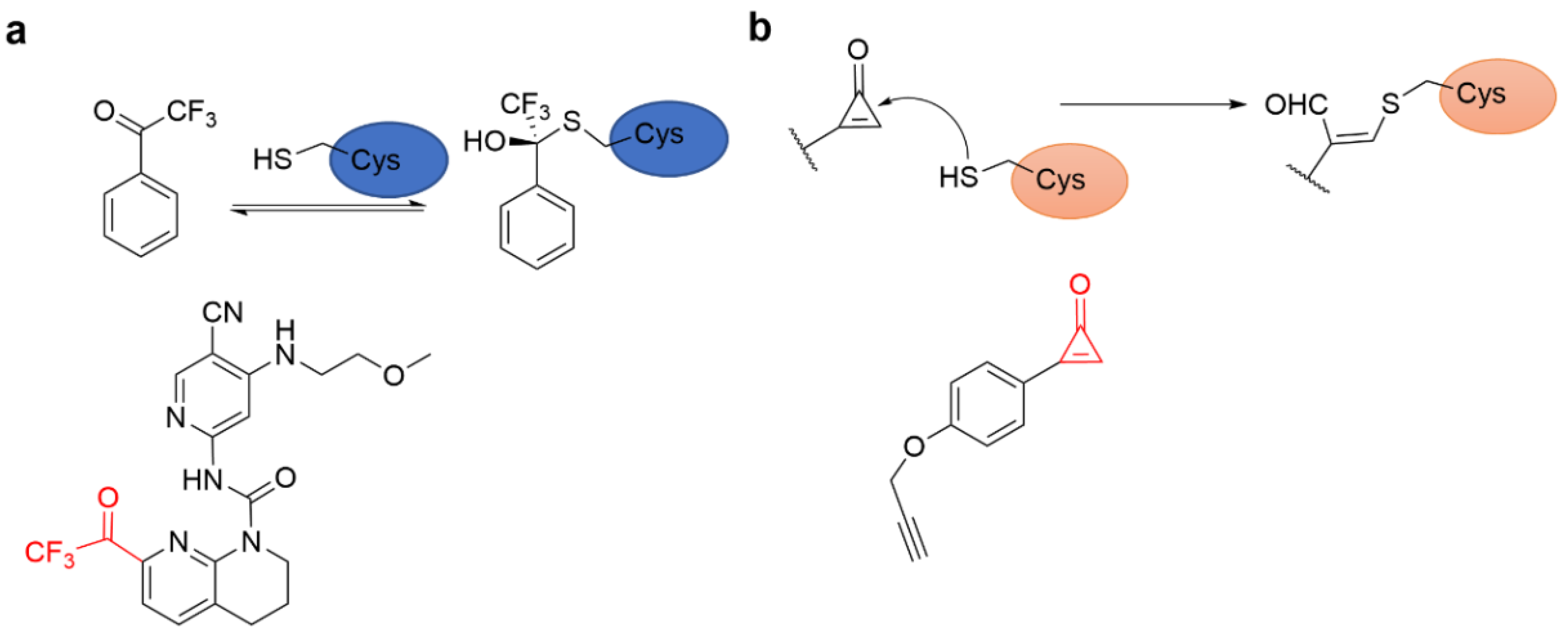
4.2.3. Other Alkenes or Alkynes
4.3. Strain Release Motif Warhead
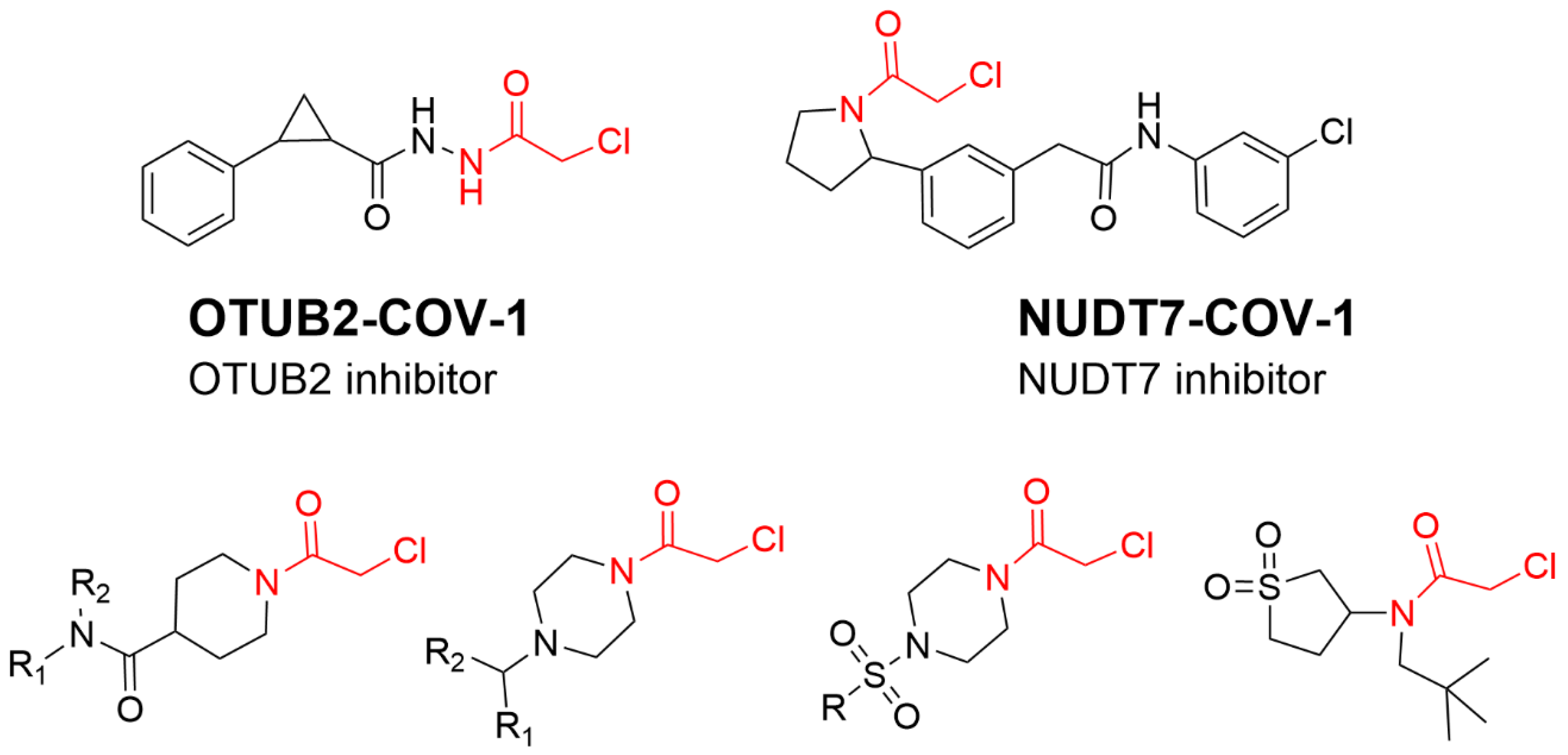
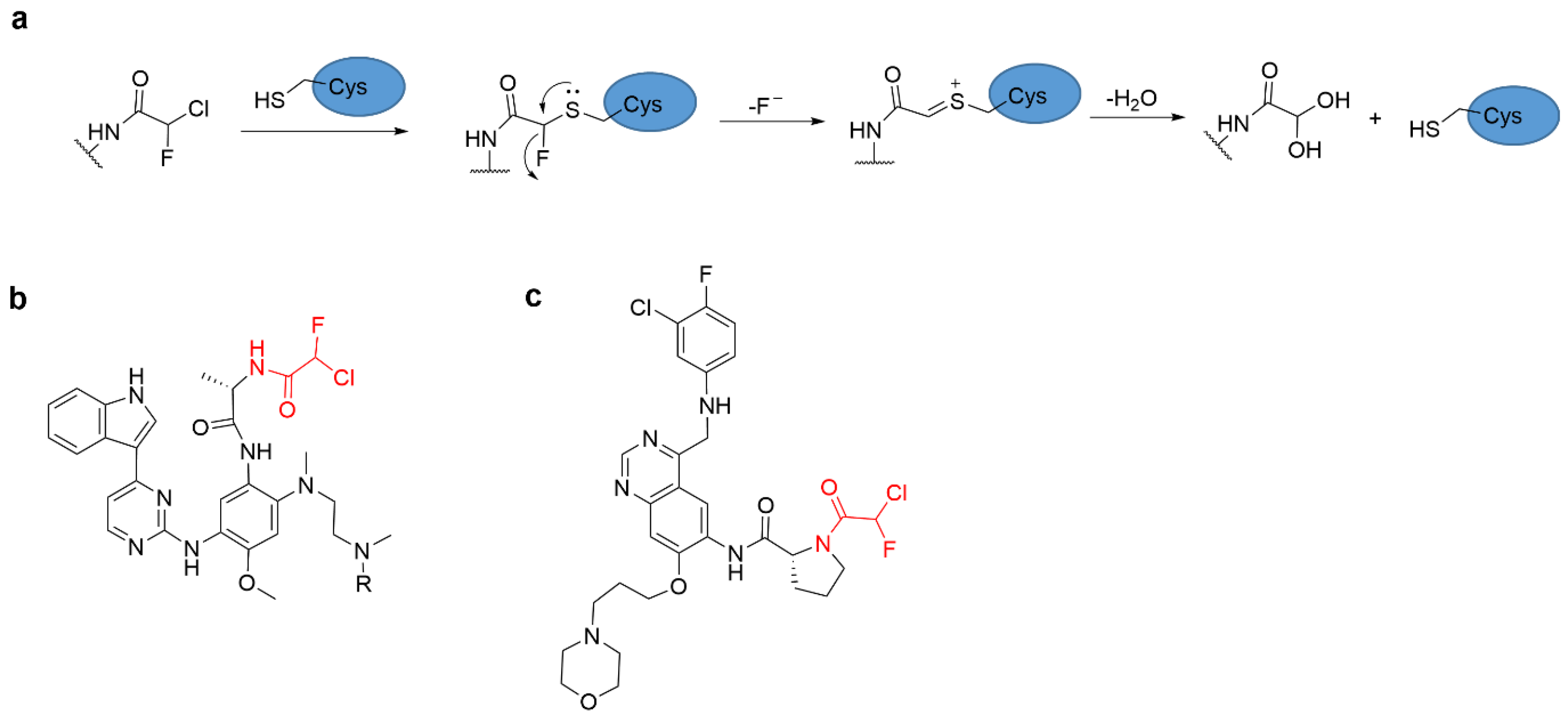
4.4. Alkyl Halide Warhead
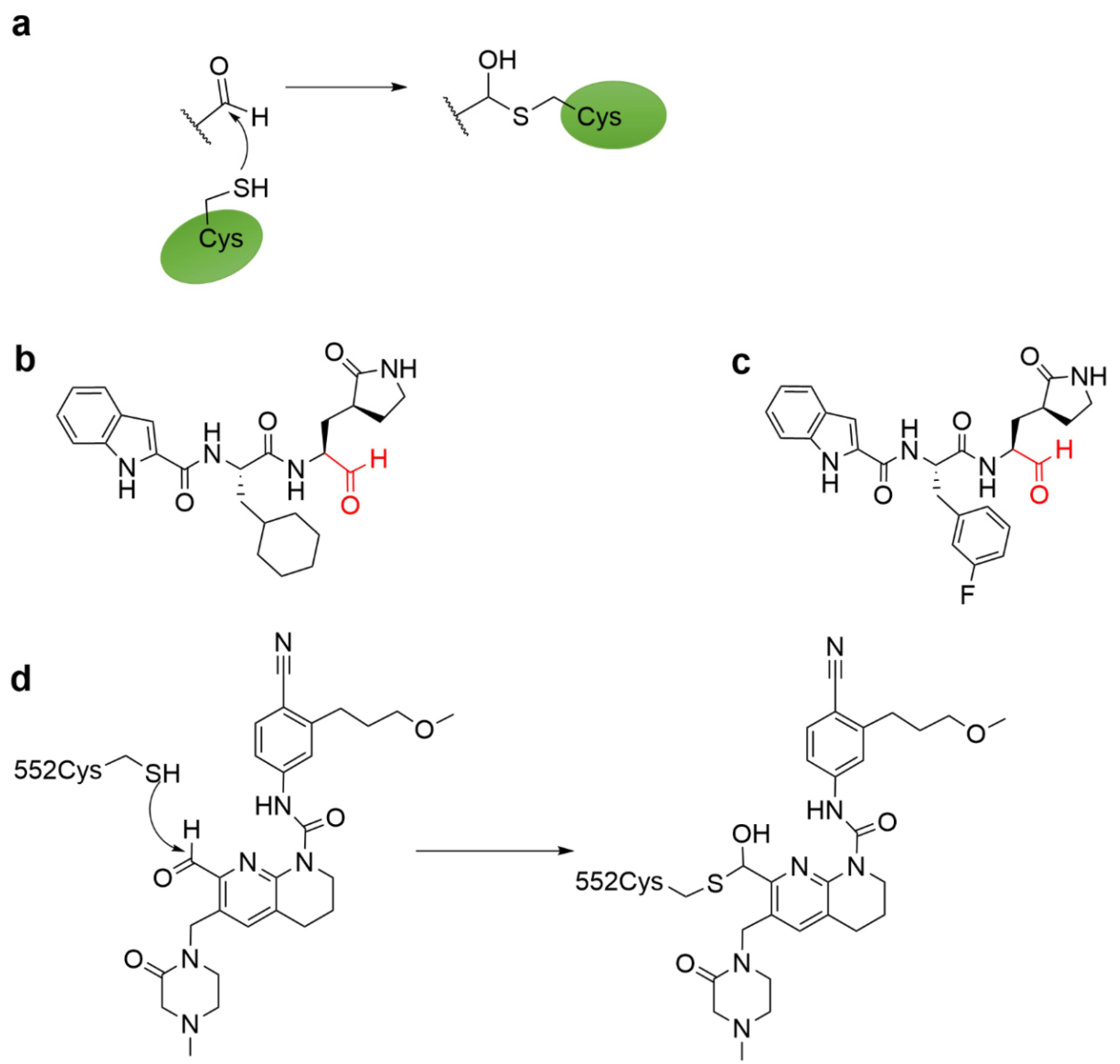
4.5. Aldehyde Ketone Warhead
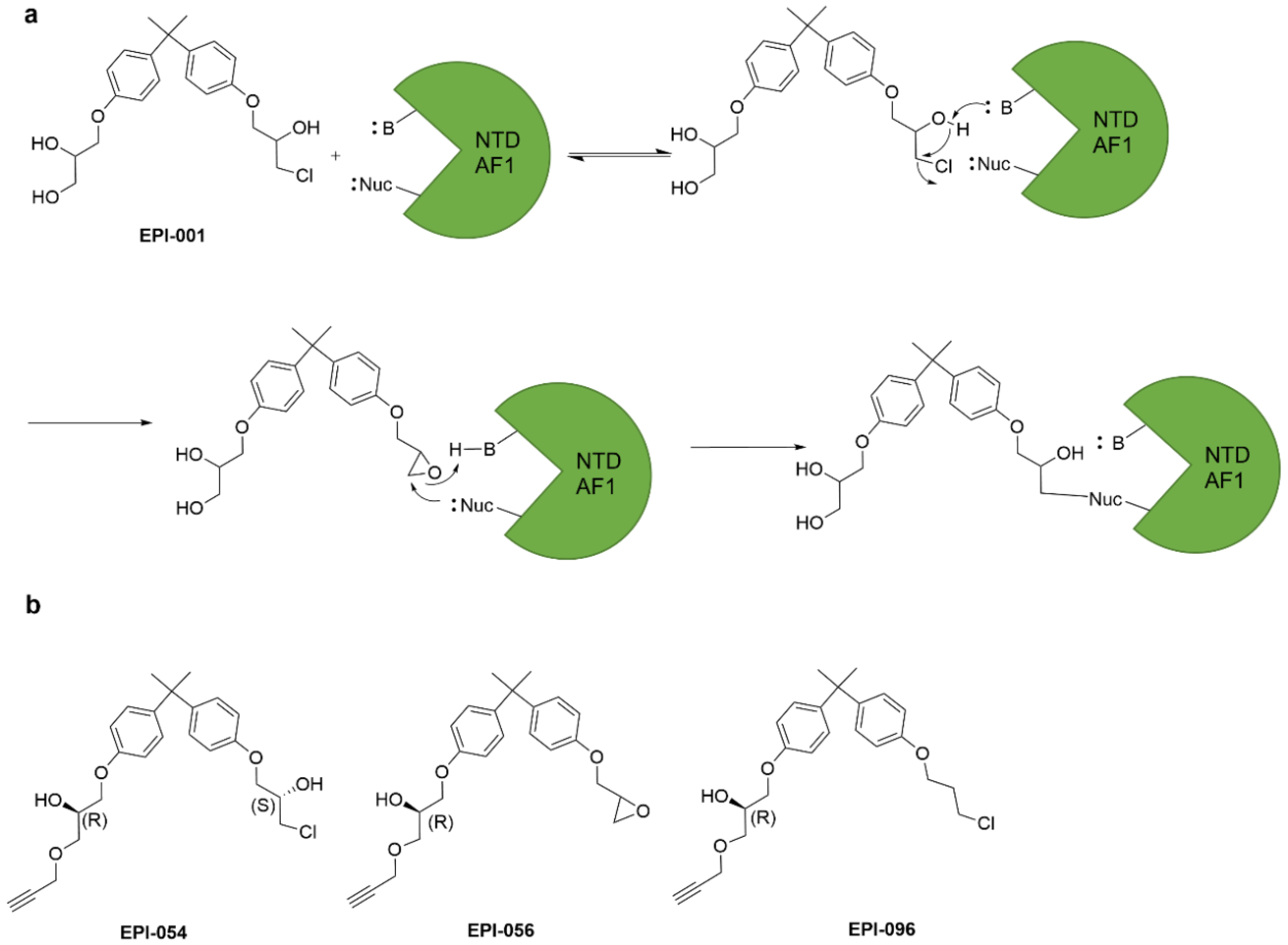
4.6. Epoxides and Other Three-Membered Rings
4.7. Covalent Inhibitors Targeting Cyan Groups
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uetrecht, J. Idiosyncratic drug reactions: Past, present, and future. Chem. Res. Toxicol. 2008, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS–MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Noda, N.; Takaya, J.; Abo, M.; Toh, K.; Tajiri, K.; Cui, C.; Zhou, L.; Sato, S.-I.; Uesugi, M. Discovery of Non-Cysteine-Targeting Covalent Inhibitors by Activity-Based Proteomic Screening with a Cysteine-Reactive Probe. ACS Chem. Biol. 2022, 17, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Tagirasa, R.; Yoo, E. Covalent Small Molecule Immunomodulators Targeting the Protease Active Site. J. Med. Chem. 2021, 64, 5291–5322. [Google Scholar] [CrossRef] [PubMed]
- Carrera, A.C.; Alexandrov, K.; Roberts, T.M. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc. Natl. Acad. Sci. USA 1993, 90, 442–446. [Google Scholar] [CrossRef]
- Quach, D.; Tang, G.; Anantharajan, J.; Baburajendran, N.; Poulsen, A.; Wee, J.L.K.; Retna, P.; Li, R.; Liu, B.; Tee, D.H.Y.; et al. Strategic Design of Catalytic Lysine-Targeting Reversible Covalent BCR-ABL Inhibitors. Angew Chem Int Ed Engl 2021, 60, 17131–17137. [Google Scholar] [CrossRef]
- Martín-Gago, P.; Fansa, E.K.; Winzker, M.; Murarka, S.; Janning, P.; Schultz-Fademrecht, C.; Baumann, M.; Wittinghofer, A.; Waldmann, H. Covalent Protein Labeling at Glutamic Acids. Cell Chem. Biol. 2017, 24, 589–597. [Google Scholar] [CrossRef]
- Powers, J.C.; Asgian, J.L.; Ekici, Ö.D.; James, K.E. Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef]
- Fadeyi, O.O.; Hoth, L.R.; Choi, C.; Feng, X.; Gopalsamy, A.; Hett, E.C.; Kyne, R.E.; Robinson, R.P.; Jones, L.H. Covalent Enzyme Inhibition through Fluorosulfate Modification of a Noncatalytic Serine Residue. ACS Chem. Biol. 2017, 12, 2015–2020. [Google Scholar] [CrossRef]
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s Tyrosine Kinase Inhibitors to Sequester Y551 and Prevent Phosphorylation Determines Potency for Inhibition of Fc Receptor but not B-Cell Receptor Signaling. Mol. Pharmacol. 2017, 91, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Shannon, D.A.; Colby, T.; Wang, Z.; Shabab, M.; Kumari, S.; Villamor, J.G.; McLaughlin, C.J.; Weerapana, E.; Kaiser, M. Chemical Proteomics with Sulfonyl Fluoride Probes Reveals Selective Labeling of Functional Tyrosines in Glutathione Transferases. Chem. Biol. 2013, 20, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef]
- Pace, N.J.; Weerapana, E. Diverse Functional Roles of Reactive Cysteines. ACS Chem. Biol. 2013, 8, 283–296. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α, β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.-M.; Winssinger, N. Cysteine Mapping in Conformationally Distinct Kinase Nucleotide Binding Sites: Application to the Design of Selective Covalent Inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [Google Scholar] [CrossRef]
- Visscher, M.; Arkin, M.R.; Dansen, T.B. Covalent targeting of acquired cysteines in cancer. Curr. Opin. Chem. Biol. 2016, 30, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, Q.; Bliven, S.; Xie, L.; Bourne, P.E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 2017, 60, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Gao, J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Tucker, D.L.; Rule, S.A. A critical appraisal of ibrutinib in the treatment of mantle cell lymphoma and chronic lymphocytic leukemia. Ther. Clin. Risk Manag. 2015, 11, 979–990. [Google Scholar] [PubMed]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Johansson, M.H. Reversible michael additions: Covalent inhibitors and prodrugs. Mini-Rev. Med. Chem. 2012, 12, 1330–1344. [Google Scholar]
- Hall, A.; Abendroth, J.; Bolejack, M.J.; Ceska, T.; Dell’Aiera, S.; Ellis, V.; Fox, D.; François, C.; Muruthi, M.M.; Prével, C.; et al. Discovery and Characterization of a Novel Series of Chloropyrimidines as Covalent Inhibitors of the Kinase MSK1. ACS Med. Chem. Lett. 2022, 13, 1099–1108. [Google Scholar] [CrossRef]
- Leesnitzer, L.M.; Parks, D.J.; Bledsoe, R.K.; Cobb, J.E.; Collins, J.L.; Consler, T.G.; Davis, R.G.; Hull-Ryde, E.A.; Lenhard, J.M.; Patel, L.; et al. Functional Consequences of Cysteine Modification in the Ligand Binding Sites of Peroxisome Proliferator Activated Receptors by GW9662. Biochemistry 2002, 41, 6640–6650. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Zambaldo, C.; Vinogradova, E.V.; Qi, X.; Iaconelli, J.; Suciu, R.M.; Koh, M.; Senkane, K.; Chadwick, S.R.; Sanchez, B.B.; Chen, J.S.; et al. 2-Sulfonylpyridines as Tunable, Cysteine-Reactive Electrophiles. J. Am. Chem. Soc. 2020, 142, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.-C.; May, V.K.; Bedford, G.C.; Tuley, A.A.; Fast, W. Discovery of 4,4′-Dipyridylsulfide Analogs as “Switchable Electrophiles” for Covalent Inhibition. ACS Chem. Biol. 2021, 16, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Chen, X.; Song, X.; Tu, Z.; Chen, Y.; Zhang, Z.; Ding, K. Characterization of an aromatic trifluoromethyl ketone as a new warhead for covalently reversible kinase inhibitor design. Bioorganic Med. Chem. 2021, 50, 116457. [Google Scholar] [CrossRef] [PubMed]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Kim, H.; Hwang, Y.S.; Kim, M.; Park, S.B. Recent advances in the development of covalent inhibitors. RSC Med. Chem. 2021, 12, 1037–1045. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. 2005, 102, 7665–7670. [Google Scholar] [CrossRef]
- Michalczyk, A.; Klüter, S.; Rode, H.B.; Simard, J.R.; Grütter, C.; Rabiller, M.; Rauh, D. Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorganic Med. Chem. 2008, 16, 3482–3488. [Google Scholar] [CrossRef]
- Deeks, E.D. Neratinib: First Global Approval. Drugs 2017, 77, 1695–1704. [Google Scholar] [CrossRef]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First Approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Miller, R.M.; Paavilainen, V.O.; Krishnan, S.; Serafimova, I.M.; Taunton, J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc. 2013, 135, 5298–5301. [Google Scholar] [CrossRef]
- Jang, J.Y.; Kim, H.; Kim, H.J.; Suh, S.W.; Park, S.B.; Han, B.W. Structural basis for the inhibitory effects of a novel reversible covalent ligand on PPARγ phosphorylation. Sci. Rep. 2019, 9, 11168. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wen, Y.; Wang, C.; Zhou, Y.; Zhou, Y.; Zhang, Z.-M.; Liu, T.; Lu, X. Efficient targeted oncogenic KRASG12C degradation via first reversible-covalent PROTAC. Eur. J. Med. Chem. 2022, 230, 114088. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, R.; Emami, S. Recent applications of vinyl sulfone motif in drug design and discovery. Eur. J. Med. Chem. 2022, 234, 114255. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. Evaluation of trypanocidal activity of combinations of anti-sleeping sickness drugs with cysteine protease inhibitors. Exp. Parasitol. 2015, 151, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Doyle Patricia, S.; Zhou Yuan, M.; Engel Juan, C.; McKerrow James, H. A Cysteine Protease Inhibitor Cures Chagas’ Disease in an Immunodeficient-Mouse Model of Infection. Antimicrob. Agents Chemother. 2007, 51, 3932–3939. [Google Scholar] [CrossRef]
- McAulay, K.; Hoyt, E.A.; Thomas, M.; Schimpl, M.; Bodnarchuk, M.S.; Lewis, H.J.; Barratt, D.; Bhavsar, D.; Robinson, D.M.; Deery, M.J.; et al. Alkynyl Benzoxazines and Dihydroquinazolines as Cysteine Targeting Covalent Warheads and Their Application in Identification of Selective Irreversible Kinase Inhibitors. J. Am. Chem. Soc. 2020, 142, 10358–10372. [Google Scholar] [CrossRef]
- Tokunaga, K.; Sato, M.; Kuwata, K.; Miura, C.; Fuchida, H.; Matsunaga, N.; Koyanagi, S.; Ohdo, S.; Shindo, N.; Ojida, A. Bicyclobutane Carboxylic Amide as a Cysteine-Directed Strained Electrophile for Selective Targeting of Proteins. J. Am. Chem. Soc. 2020, 142, 18522–18531. [Google Scholar] [CrossRef]
- Lopchuk, J.M.; Fjelbye, K.; Kawamata, Y.; Malins, L.R.; Pan, C.-M.; Gianatassio, R.; Wang, J.; Prieto, L.; Bradow, J.; Brandt, T.A.; et al. Strain-Release Heteroatom Functionalization: Development, Scope, and Stereospecificity. J. Am. Chem. Soc. 2017, 139, 3209–3226. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorg. Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kostic, M.; Zhang, T.; Che, J.; Patricelli, M.P.; Jones, L.H.; Chouchani, E.T.; Gray, N.S. Fragment-based covalent ligand discovery. RSC Chem. Biol. 2021, 2, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [PubMed]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Fontan, L.; Yang, C.; Kabaleeswaran, V.; Volpon, L.; Osborne, M.; Beltran, E.; Garcia, M.; Cerchietti, L.; Shaknovich, R.; Yang, S.; et al. MALT1 Small Molecule Inhibitors Specifically Suppress ABC-DLBCL In Vitro and In Vivo. Cancer Cell 2012, 22, 812–824. [Google Scholar] [CrossRef]
- Roberts, A.M.; Miyamoto, D.K.; Huffman, T.R.; Bateman, L.A.; Ives, A.N.; Akopian, D.; Heslin, M.J.; Contreras, C.M.; Rape, M.; Skibola, C.F.; et al. Chemoproteomic Screening of Covalent Ligands Reveals UBA5 As a Novel Pancreatic Cancer Target. ACS Chem. Biol. 2017, 12, 899–904. [Google Scholar] [CrossRef]
- Dubiella, C.; Pinch, B.J.; Koikawa, K.; Zaidman, D.; Poon, E.; Manz, T.D.; Nabet, B.; He, S.; Resnick, E.; Rogel, A.; et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 2021, 17, 954–963. [Google Scholar] [CrossRef]
- Allimuthu, D.; Adams, D.J. 2-Chloropropionamide As a Low-Reactivity Electrophile for Irreversible Small-Molecule Probe Identification. ACS Chem. Biol. 2017, 12, 2124–2131. [Google Scholar] [CrossRef]
- Shindo, N.; Fuchida, H.; Sato, M.; Watari, K.; Shibata, T.; Kuwata, K.; Miura, C.; Okamoto, K.; Hatsuyama, Y.; Tokunaga, K.; et al. Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat. Chem. Biol. 2019, 15, 250–258. [Google Scholar] [CrossRef]
- Sato, M.; Fuchida, H.; Shindo, N.; Kuwata, K.; Tokunaga, K.; Xiao-Lin, G.; Inamori, R.; Hosokawa, K.; Watari, K.; Shibata, T.; et al. Selective Covalent Targeting of Mutated EGFR(T790M) with Chlorofluoroacetamide-Pyrimidines. ACS Med. Chem. Lett. 2020, 11, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.-K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.-K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Noe, M.C.; Gilbert, A.M. Chapter Twenty-Seven—Targeted Covalent Enzyme Inhibitors. In Annual Reports in Medicinal Chemistry; Desai, M.C., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 47, pp. 413–439. [Google Scholar]
- Siklos, M.; BenAissa, M.; Thatcher, G.R.J. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.A.; Doherty, W.; Evans, P.; Malthouse, J.P.G. Quantifying tetrahedral adduct formation and stabilization in the cysteine and the serine proteases. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2015, 1854, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, R.A.; Knoepfel, T.; Buschmann, N.; Leblanc, C.; Mah, R.; Todorov, M.; Nimsgern, P.; Ripoche, S.; Niklaus, M.; Warin, N.; et al. Discovery of Roblitinib (FGF401) as a Reversible-Covalent Inhibitor of the Kinase Activity of Fibroblast Growth Factor Receptor 4. J. Med. Chem. 2020, 63, 12542–12573. [Google Scholar] [CrossRef]
- Fan, Y.; Si, H.; Zhang, Z.; Zhong, L.; Sun, H.; Zhu, C.; Yin, Z.; Li, H.; Tang, G.; Yao, S.Q.; et al. Novel Electrophilic Warhead Targeting a Triple-Negative Breast Cancer Driver in Live Cells Revealed by “Inverse Drug Discovery”. J. Med. Chem. 2021, 64, 15582–15592. [Google Scholar] [CrossRef]
- Gomes, A.R.; Varela, C.L.; Tavares-da-Silva, E.J.; Roleira, F.M.F. Epoxide containing molecules: A good or a bad drug design approach. Eur. J. Med. Chem. 2020, 201, 112327. [Google Scholar] [CrossRef]
- Singh, G.S. Synthetic Aziridines in Medicinal Chemistry: A Mini-Review. Mini-Rev. Med. Chem. 2016, 16, 892–904. [Google Scholar] [CrossRef]
- Kaur, B.; Singh, P. Epoxides: Developability as active pharmaceutical ingredients and biochemical probes. Bioorganic Chem. 2022, 125, 105862. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Zhanel, M.A.; Karlowsky, J.A. Intravenous fosfomycin: An assessment of its potential for use in the treatment of systemic infections in Canada. Can. J. Infect. Dis. Med. Microbiol. 2018, 2018, 8912039. [Google Scholar] [CrossRef] [PubMed]
- Dijkmans, A.C.; Zacarías, N.V.O.; Burggraaf, J.; Mouton, J.W.; Wilms, E.B.; Van Nieuwkoop, C.; Touw, D.J.; Stevens, J.; Kamerling, I.M.C. Fosfomycin: Pharmacological, Clinical and Future Perspectives. Antibiotics 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lees, W.J.; Kempsell, K.E.; Lane, W.S.; Duncan, K.; Walsh, C.T. Characterization of a Cys115 to Asp Substitution in the Escherichia coli Cell Wall Biosynthetic Enzyme UDP-GlcNAc Enolpyruvyl Transferase (MurA) That Confers Resistance to Inactivation by the Antibiotic Fosfomycin. Biochemistry 1996, 35, 4923–4928. [Google Scholar] [CrossRef] [PubMed]
- Eschenburg, S.; Priestman, M.; Schönbrunn, E. Evidence That the Fosfomycin Target Cys115 in UDP-N-acetylglucosamine Enolpyruvyl Transferase (MurA) Is Essential for Product Release. J. Biol. Chem. 2005, 280, 3757–3763. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Howard, C.A.; Zari, S.; Cho, H.J.; Shukla, S.; Li, H.; Ndoj, J.; González-Alonso, P.; Nikolaidis, C.; Abbott, J.; et al. Covalent inhibition of NSD1 histone methyltransferase. Nat. Chem. Biol. 2020, 16, 1403–1410. [Google Scholar] [CrossRef]
- de Jesus Cortez, F.; Nguyen, P.; Truillet, C.; Tian, B.; Kuchenbecker, K.M.; Evans, M.J.; Webb, P.; Jacobson, M.P.; Fletterick, R.J.; England, P.M. Development of 5N-Bicalutamide, a High-Affinity Reversible Covalent Antiandrogen. ACS Chem. Biol. 2017, 12, 2934–2939. [Google Scholar] [CrossRef]
- Krabill, A.D.; Chen, H.; Hussain, S.; Feng, C.; Abdullah, A.; Das, C.; Aryal, U.K.; Post, C.B.; Wendt, M.K.; Galardy, P.J.; et al. Ubiquitin C-Terminal Hydrolase L1: Biochemical and Cellular Characterization of a Covalent Cyanopyrrolidine-Based Inhibitor. ChemBioChem 2020, 21, 712–722. [Google Scholar] [CrossRef]
- Bashore, C.; Jaishankar, P.; Skelton, N.J.; Fuhrmann, J.; Hearn, B.R.; Liu, P.S.; Renslo, A.R.; Dueber, E.C. Cyanopyrrolidine Inhibitors of Ubiquitin Specific Protease 7 Mediate Desulfhydration of the Active-Site Cysteine. ACS Chem. Biol. 2020, 15, 1392–1400. [Google Scholar] [CrossRef]
- Benson, M.J.; Rodriguez, V.; von Schack, D.; Keegan, S.; Cook, T.A.; Edmonds, J.; Benoit, S.; Seth, N.; Du, S.; Messing, D.; et al. Modeling the Clinical Phenotype of BTK Inhibition in the Mature Murine Immune System. J. Immunol. 2014, 193, 185. [Google Scholar] [CrossRef]
- Baillie, T.A. Approaches to mitigate the risk of serious adverse reactions in covalent drug design. Expert Opin. Drug Discov. 2021, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27, 7728. https://doi.org/10.3390/molecules27227728
Huang F, Han X, Xiao X, Zhou J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules. 2022; 27(22):7728. https://doi.org/10.3390/molecules27227728
Chicago/Turabian StyleHuang, Fangjiao, Xiaoli Han, Xiaohui Xiao, and Jinming Zhou. 2022. "Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development" Molecules 27, no. 22: 7728. https://doi.org/10.3390/molecules27227728
APA StyleHuang, F., Han, X., Xiao, X., & Zhou, J. (2022). Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules, 27(22), 7728. https://doi.org/10.3390/molecules27227728