Abstract
Sterically shielded nitroxides, which demonstrate high resistance to bioreduction, are the spin labels of choice for structural studies inside living cells using pulsed EPR and functional MRI and EPRI in vivo. To prepare new sterically shielded nitroxides, a reaction of cyclic nitrones, including various 1-pyrroline-1-oxides, 2,5-dihydroimidazole-3-oxide and 4H-imidazole-3-oxide with alkynylmagnesium bromide wereused. The reaction gave corresponding nitroxides with an alkynyl group adjacent to the N-O moiety. The hydrogenation of resulting 2-ethynyl-substituted nitroxides with subsequent re-oxidation of the N-OH group produced the corresponding sterically shielded tetraalkylnitroxides of pyrrolidine, imidazolidine and 2,5-dihydroimidazole series. EPR studies revealed large additional couplings up to 4 G in the spectra of pyrrolidine and imidazolidine nitroxides with substituents in 3- and/or 4-positions of the ring.
1. Introduction
Cyclic nitroxides with four ethyl (or more bulky alkyl) substituents adjacent to the N-O group are known to show much higher resistance to reduction with biogenic reductants and enzymatic systems than corresponding tetramethyl analogs [1]. The higher stability of these so-called “sterically shielded” nitroxides makes them favorable for those fields of application where the decay of conventional tetramethyl nitroxides is fast. The stability of the nitroxide group is of crucial importance for structural studies of biological macromolecules in their native environment inside living cells using site-directed spin labeling (SDSL) and pulsed EPR techniques [2]. For this application, reduction-resistant spin labels were prepared from sterically shielded nitroxides of pyrroline [3,4,5], isoindoline [6] and pyrrolidine series [7,8]. Sterically shielded nitroxides of piperidine [9,10] and imidazoline [11,12] series were designed for functional MRI and EPRI in vivo.
The reaction of cyclic nitrones with organometallic reagents is a widely used method of synthesis of various nitroxides [13,14]. There are some examples of successful synthesis of sterically shielded tetraethyl nitroxides using this method. For example, the direct addition of ethylmagnesium bromide to cyclic nitrones of 4H-imidazole 3-oxide or 2H-imidazole 1-oxide series afforded 2,2,5,5-tetraethyl substituted 2,5-dihydroimidazol-1-oxyls with 40–50% yield [15,16]. However, sterically hindered 1-pyrroline 1-oxides are not prone to the addition of EtMgBr [8,17], presumably due to metalation [18]. These nitrones, nevertheless, readily react with vinylmagnesium [19], allylmagnesium [20] or ethynylmagnesium halides [8,21], which are known to show lower basicity than EtMgBr, affording corresponding nitroxides in good yields. The terminal vinyl or ethynyl groups can then be converted into ethyl ones via hydrogenation. Although the nitroxide group is reduced to hydroxylamine, it can be easily recovered via oxidation. This technique of indirect introduction of the ethyl group allowed us to prepare highly resistant to reduction nitroxides [8,19,21].
In this work, we focus on reactions of alkynylmagnesium bromides with various cyclic nitrones of pyrroline (1–3), imidazoline (4) and 4H-imidazole-3-oxide (5) series (Figure 1).
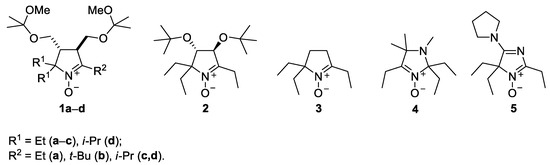
Figure 1.
The sterically hindered nitrones used in this study.
The reaction was found to give satisfactory yields of 2-ethynyl-substituted nitroxides with all these sterically hindered nitrones except for 1d. Subsequent hydrogenation of the ethynyl derivatives produced corresponding sterically shielded tetraalkylnitroxides.
Besides that, the new 2-alkynyl-substituted nitroxides themselves may be of interest as bioorthogonal spin labels capable of binding to biomolecules, modified with azide or nitrone groups via copper-catalyzed 1,3-dipolar cycloaddition reactions [22,23,24]. These alkynyl derivatives may find even broader applications because alkynes are used in the synthesis of numerous heterocyclic systems; for recent reviews, see [25,26,27].
2. Results and Discussion
2.1. Nitrones
A series of sterically hindered nitrones 1–5 have been prepared to investigate their reactions with alkynylmagnesium halides. We have previously reported on the synthesis of diastereomeric pyrrolidines 6a,b and 7a,b and corresponding nitrones 8a,b from amino acids, ketones and dimethyl fumarate according to Scheme 1 [17,19]. The pyrrolidines 6c,d and 7c,d were prepared in analogy to the published procedures. The reaction afforded the mixtures of diastereomers in a 1:1 ratio (cf. [17,19]), and the individual diastereomers were isolated using column chromatography. The mixtures were subjected to reduction of the ester groups with LiAlH4 and oxidation with tungstate–hydrogen peroxide system to give the nitrones 8c,d (racemates). We noticed earlier that treatment of 3,4-bis-(hydroxymethyl)-1-pyrroline1-oxideswith Grignard reagents mightlead to THF-insoluble precipitate formation, presumably, magnesium alcoholates [17]. To avoid extra consumption of Grignard reagent and to prevent precipitation of magnesium alcoholates from the reaction mixtures, the hydroxy groups in the nitrones 8a–d were protected via treatment with 2,2-dimethoxypropane, and the resulting nitrones 1a–d were used for nitroxides syntheses.
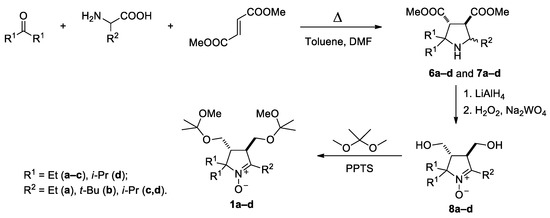
Scheme 1.
Synthesis of nitrones 1a–d.
The nitrone 2 was prepared from 9 [28,29] by successive introduction of three ethyl groups via reaction with ethylmagnesium bromide and oxidation (Scheme 2). The isomers formed in the intermediate steps were not separated, and the crude mixture was used in the second and third steps affording chiral nitrone 2 as a final product with a 25% yield.
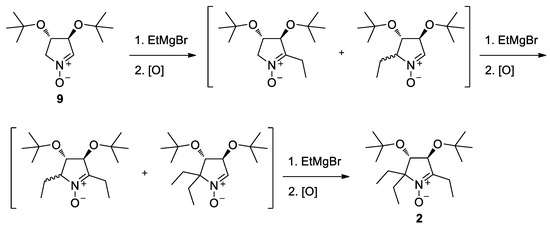
Scheme 2.
Synthesis of nitrone 2.
To prepare nitrone 4, the 2-amino-2-methylpentan-3-one oxime (10) was heated under reflux with pentane-3-one in methanol using ammonium acetate as a catalyst (cf. [30]), Scheme 3. The resulting nitrone 11 was subjected to Eschweiler-Clarke alkylation in analogy to literature protocol [31].
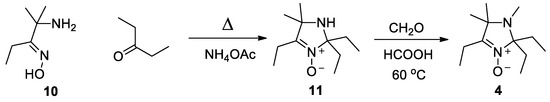
Scheme 3.
Synthesis of nitrone 4.
The nitrones 3 [20] and 5 [19] were prepared according to literature protocols.
2.2. Nitroxides
Nitrones 1–5 were treated with a 10-fold excess of ethynylmagnesium bromide in THF, then quenched with water and oxidized (Scheme 4). The reaction products, times and yields of corresponding nitroxides are given in Table 1. In the case of 1, processing of the reaction mixtures implied treatment with an aqueous acid solution to remove the protecting groups. Noteworthily, the conversion of 1b and c was incomplete under these conditions, and TLC analysis of the quenched reaction mixtures showed the presence of the starting compounds. Bulky substituents at nitrone group carbon atom of 1 strongly decreased the reaction rate but didnot influence much the yield of the nitroxide. More hindered nitrone 1d was quantitatively recovered from the reaction mixture after stirring with ethynylmagnesium bromide for a week.
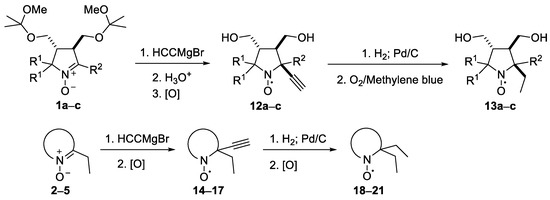
Scheme 4.
Synthesis of nitroxides.

Table 1.
Synthesis of nitroxides from nitrones: reaction times and yields (for conditions, see the Section 3).
The addition of ethynylmagnesium bromide to 1a–c proceeded with higher selectivity than that of vinylmagnesium bromide affording only one diastereomer (cf. [19]). The structures 12a–c were assigned based on X-ray analysis data (see Figures S2–S4 in the Supplementary Information in this article “X-ray diffraction data”). Selective formation of 12a–c presumably results from the coordination of the organometallic reagent with the oxygen atom of the neighboring alkoxymethyl group. Interestingly, the reaction with chiral nitrone 2 also gave a single isomer, a chiral nitroxide 14, but the ethynyl group entered from the opposite side, trans- to the neighboring tert-butoxy group (Figure S5 in the Supplementary Information in this article“X-ray diffraction data”).
To introduce larger alkynyl groups corresponding organometallic reagents were prepared from terminal acetylenes and EtMgBr. The reaction with 1a and 3 afforded nitroxides 22a–c and 23 with 54–66% yield (Scheme 5). The stereochemistry of this reaction was similar to that of ethynylmagnesium bromide addition, see the X-ray analysis data in Figures S6–S8 in the Supplementary Information in this article“X-ray diffraction data.”
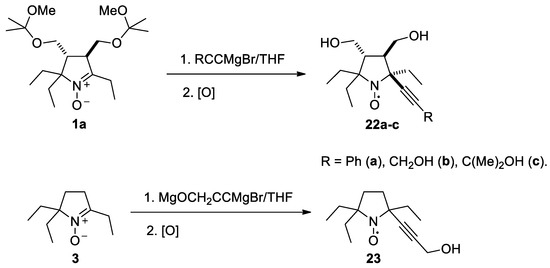
Scheme 5.
Preparation of nitroxides 22a–c and 23.
We have shown earlier that careful hydrogenation of ethynyl-substituted nitroxide in THF under atmospheric pressure allows for avoiding undesired over-reduction of the nitroxide group to an amine. The resulting ethyl-substituted hydroxylamine can then be easily oxidized to the corresponding nitroxide [8]. Using this procedure, nitroxides 12a–c, 14–17 were converted to nitroxides 13a–c, 18–21 (Scheme 4 and Table 1).
It should be noted that, in agreement with our previous observations [17,19], nitroxides 13a–c, 18–20 can’t be prepared via the direct addition of EtMgBr. The overall yield of 13a from 8a using the procedures described here is nearly the same as in the previously described procedure with vinylmagnesium bromide [19]. The reaction of 5 with EtMgBr gives a lower yield of 21 (45%) [15] than the above two-step procedure with ethynylmagnesium bromide (Table 1).
2.3. EPR Spectra
Parameters of the EPR spectra of the new nitroxides are given in Table 2. We have shown earlier that in nitroxides with a 3,4-disubstituted five-membered saturated ring, each pair of geminal ethyl groups at twoand fivepositions of the heterocycle produces one large (ca. 2 G) additional splitting due to hfi with one of the methylene hydrogens of the ethyl group [8,19,32]. Replacement of one of the geminal ethyls with another group may change the hyperfine structure of the spectrum greatly. The data on 13a–c in Table 2 and Figure 2 demonstrate the remarkable evolution of the EPR spectra upon the increase of the steric demand of the substituent. The spectrum of 13c follows the pattern described for 13a [19], with two additional large splittings on γ-hydrogens. Replacement of one of the ethyl groups with theisopropyl one resulted in an increase of both hfi constants (by ca. 50% and 12%). Interestingly, the tert-butyl group (in 13b) produces an opposite effect showing only one doublet splitting on one of the γ-hydrogens (cf. [17]).
Table 2.
Parameters of the EPR spectra of nitroxides in oxygen-free distilled water at a concentration of 0.2 mM. Modulation amplitude 0.5 G, MW power 5 mW.
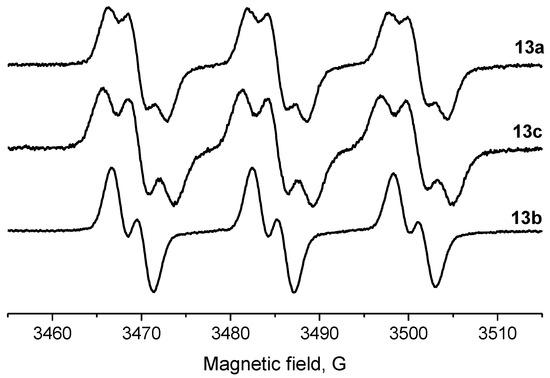
Figure 2.
EPR spectra of nitroxides 13a (top), 13c (middle) and 13b (bottom; for conditions, see the Section 3).
Another example of an unusual hyperfine structure in the EPR spectrum is demonstrated by 20. The EPR spectrum of close analog of this nitroxide, 2,2,5,5-tetraethyl-3,4-dimethylimidazolidine-1-oxyl (24; Figure 3), is known to contain two splitting constants (ca. 2 G) [32], while 20 showed a single additional splitting with aH = 3.64 G.
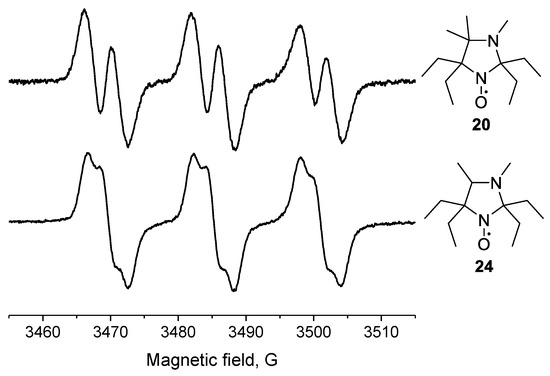
Figure 3.
EPR spectra of nitroxides 20 and 24.
The above examples demonstrate that minor changes in the stricture of 2,2,5,5-tetraethyl-substituted five-membered ring nitroxides may lead to drastic changes in their EPR spectra. To the best of our knowledge, similar effects never occur for 2,2,5,5-tetramethyl nitroxides.
3. Materials and Methods
3.1. General Information
The 2-amino-2-methylpentan-3-one oxime (10) was obtained from the Pilot Plant of N. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry SB RAS, chemprod@nioch.nsc.ru. The compounds 3 [20] and 5 [15], 8a [19], 8b [17], and 9 [29] were prepared according to the literature. Ethynyl magnesium bromide solutions (0.5–1 M) were prepared according to the protocol described in [33].
The IR spectra were recorded on a Bruker Vector 22 FT-IR spectrometer (Bruker, Billerica, MA, USA) in KBr pellets (1:150 ratio) or in neat samples (see the Supplementary Information in this article pp. 27–37) and are reported in wave numbers (cm−1). 1H NMR spectra were recorded on a Bruker AV 300 (300.132 MHz), AV 400(400.134 MHz) and DRX 500 (500.130 MHz) spectrometers (Bruker, Billerica, MA, USA). 13C NMR spectra were recorded on a Bruker AV 300 (75.467 MHz), AV 400 (100.614 MHz) and DRX 500 (125.758 MHz) spectrometers (see the Supplementary Information in this article pp. 5–26). All the NMR spectra were acquired for 5–10% solutions in CDCl3, (CD3)2SO or CDCl3-CD3OD mixtures at 300 K using the signal of the solvent as a standard. NMR spectra of nitroxides for analysis and structure assignment were recorded after reduction with Zn in CD3OD-CF3COOH at 65 °C as described in [8] or with Zn and ND4Cl in CD3OD at 5 °C. HRMS analyses were performedusinga High-Resolution Mass Spectrometer DFS (Thermo Electron, Waltham, MA, USA).
HPLC analyses were carried out using an HPLC-UV (Agilent 1100, Agilent Technologies Inc., Santa Clara, CA, USA) with a Zorbax C8 column (250 mm × 4.6 mm with 5 µm particle size; Agilent Technologies Inc., USA). The column was thermostatically controlled at 35 °C. Samples were dissolved in methanol (2 mg/mL) and 3 µL of the solutionwas injected. The mobile phase was composed of A (0.1% H3PO4 in water) and B (methanol) with the following gradient elution: 0–7 min 80% B, 7–10 min 100% B, 10–12 min 100% B, the flow rate was set to 1.0 mL/min, and peaks were detected using a wavelength of 230 nm.
Reactions were monitored by TLC on precoated TLC sheets ALUGRAM Xtra SIL G/UV254 ((Macherey-Nagel GmbH & Co. KG, Düren, Germany) using UV light 254 nm, 1% aqueous permanganate, 10% solution of phosphomolybdic acid in ethanol and Dragendorff reagent as visualizing agents. Kieselgel60 (Macherey-Nagel GmbH & Co. KG) was utilized as an adsorbent for column chromatography.
EPR experiments were performed on X-band (9.8 GHz) EPR spectrometer ER-200D (Bruker). All measurements were performed in 50 μL glass capillary. The radicals were dissolved in oxygen-free distilled water at a concentration of 0.2 mM. EPR settings: modulation amplitude 0.5 G, MW, power 5 mW time constant50 ms; total acquisition time 3 min. The water-insoluble radicals were dissolved in a water/ethanol mixture (50%/50%) at a concentration of 0.1 mM. Data simulation was performed with the free software Winsim.
CCDC 2,209,668 (8d), 2,209,669 (12a), 2,209,670 (12c), 220,9671 (12b), 2,209,672 (13b), 2,209,673 (22a), 2,209,674 (22b), 2,209,675 (22c), 2,209,676 (14) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre https://www.ccdc.cam.ac.uk/structures/, accessed on 27 September 2022.
3.2. Synthesis
3.2.1. General Method of Synthesis of 3,4-Bis(methoxycarbonyl)pyrrolidines 6c,d and 7c,d
A mixture of valine (11.7 g, 0.1 mol), dimethyl fumarate (14.4 g, 0.1 mol), ketone (1 mol), DMF (100 mL) and toluene (100 mL)was placed into Dean-Stark apparatus and stirred under reflux in a week (TLC control on silica gel, hexane–ethylacetate 4:1, visualizationwith Dragendorff’s reagent). The solvent was distilled off in a vacuum, the residue was dissolved in ethyl acetate, and the solution was extracted with 5% aqueous sulfuric acid. Acidic extracts were basified with Na2CO3 and extracted with ethyl acetate. The extract was dried with Na2CO3, and the solvent was distilled off in a vacuum to give a yellow oil, a mixture of isomers, which was used for the next step without further purification. For the analysis, the isomers were separated using column chromatography on silica gel (hexane/ethyl acetate 4/1).
2,2-Diethyl-5-isopropyl-3,4-bis(methoxycarbonyl)pyrrolidines. 6c, yield 20%, colorless oil, IR (neat) νmax: 1726 (C=O). 1H NMR (400 MHz; CDCl3,δ):0.79 (t, Jt = 7.4 Hz, 3H), 0.91 (d, Jd = 6.6 Hz, 3H), 0.94 (t, Jt = 7.4 Hz, 3H), 0.95 (d, Jd = 6.6 Hz, 3H), 1.12 (dq, Jd = 13.9 Hz, Jq = 7.2 Hz, 1H), 1.44 (dq, Jd = 13.9 Hz, Jq = 7.5 Hz, 1H), 1.51 (dsp, Jd = 9.2 Hz, Jsp = 6.6 Hz, 1H), 1.54–1.60 (br, 1H), 1.65 (dq, Jd = 15.1 Hz, Jq = 7.4 Hz, 1H), 1.68 (dq, Jd = 15.1 Hz, Jq = 7.4 Hz, 1H), 2.94 (dd, Jd1 = 9.2 Hz, Jd2 = 7.0 Hz, 1H), 3.09 (d, Jd = 4.5 Hz, 1H), 3.45 (dd, Jd1 = 7.0 Hz, Jd2 = 4.5 Hz, 1H), 3.63 (s, 3H), 3.63 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3, δ): 7.9, 8.0, 20.7, 20.9, 25.4, 29.4, 29.7, 51.0, 51.3, 51.5, 56.7, 67.3, 68.0, 173.2, 175.2. Anal. calcd for C15H27NO4: C, 63.13; H, 9.24; N, 4.91; found: C, 63.14; H, 9.54; N, 4.90. 7c, yield 20%, colorless oil, IR (neat) νmax: 1735 (C=O). 1H NMR (400 MHz; CDCl3, δ): 0.79 (t, Jt = 7.4 Hz, 3H), 0.85 (t, Jt = 7.4 Hz, 3H), 0.87 (d, Jd = 6.6 Hz, 3H), 0.91 (d, Jd = 6.6 Hz, 3H), 1.28 (q, Jq = 7.4 Hz, 2H), 1.37–1.43 (br, 1H), 1.51 (dq, Jd = 13.9 Hz, Jq = 7.2 Hz, 1H), 1.57 (dq, Jd = 13.9 Hz, Jq = 7.4 Hz, 1H), 1.71 (dsp, Jd = 5.7 Hz, Jsp = 6.6 Hz, 1H), 3.03 (dd, Jd1 = 8.8 Hz, Jd2 = 7.7 Hz, 1H), 3.08 (d, Jd = 7.7 Hz, 1H), 3.10 (dd, Jd1 = 8.8 Hz, Jd2 = 5.7 Hz, 1H), 3.62 (s, 3H), 3.62 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3, δ):7.9, 8.0, 18.8, 19.5, 28.2, 29.1, 32.0, 51.4, 51.5, 51.8, 56.4, 66.8, 67.3, 173.4, 174.7. Anal. calcd for C15H27NO4: C, 63.13; H, 9.24; N, 4.91; found: C, 63.05; H, 9.37; N, 4.82.
2,2,5-Triisopropyl-3,4-bis(methoxycarbonyl)pyrrolidines. 6d, yield 8%, colorless oil, IR (neat) νmax: 1733 (C=O). 1H NMR (300 MHz; CDCl3, δ): 0.83 (d, Jd = 7.0 Hz, 3H), 0.84 (d, Jd = 6.6 Hz, 3H), 0.88 (d, Jd = 6.6 Hz, 3H), 0.92 (d, Jd = 7.0 Hz, 3H), 0.97 (d, Jd = 6.8 Hz, 3H), 1.05 (d, Jd = 6.8 Hz, 3H), 1.50–1.59 (br, 1H), 1.67 (dsp, Jd = 7.5 Hz, Jsp = 6.6 Hz, 1H), 2.00 (sp, Jsp = 7.0 Hz, 1H), 2.16 (sp, Jsp = 6.8 Hz, 1H), 3.13 (dd, Jd1 = 9.0 Hz, Jd2 = 7.5 Hz, 1H), 3.32 (d, Jd = 8.9 Hz, 1H), 3.59 (dd, Jd1 = 9.0 Hz, Jd2 = 8.9 Hz), 3.63 (s, 6H). 13C{1H} NMR (75 MHz, CDCl3, δ): 17.9, 18.3, 18.9, 19.3, 19.5, 20.8, 29.9, 31.4, 31.6, 49.9, 51.4, 51.5, 53.3, 64.5, 71.1, 173.2, 174.7. Anal. calcd for C17H31NO4: C, 65.14; H, 9.97; N, 4.47; found: C, 65.37; H, 9.96; N, 4.50. 7d, yield 8%, colorless oil, IR (neat) νmax: 1735 (C=O). 1H NMR (300 MHz; CDCl3, δ): 0.75 (d, Jd = 6.6 Hz, 3H), 0.78 (d, Jd = 6.8 Hz, 3H), 0.80 (d, Jd = 6.9 Hz, 3H); 0.92 (d, Jd = 6.7 Hz, 3H), 0.96 (d, Jd = 6.9 Hz, 3H); 1.02 (d, Jd = 6.7 Hz, 3H), 1.14–1.21 (br, 1H), 1.71 (dsep, Jd = 4.8 Hz, Jsep = 6.9 Hz, 1H), 2.01 (sep, Jsep = 6.9 Hz, 1H), 2.12 (sep, Jsp = 6.7 Hz, 1H), 3.02 (dd, Jd1 = 10.5 Hz, Jd2 = 10.5 Hz, 1H), 3.09 (dd, Jd1 = 10.5 Hz, Jd2 = 4.8 Hz, 1H), 3.26 (d, Jd = 10.5 Hz, 1H), 3.62 (s, 3H), 3.66 (s, 3H). 13C{1H} NMR (75 MHz, CDCl3, δ): 17.2, 17.5, 18.1, 18.3, 18.5, 19.4, 31.1, 31.1, 36.8, 50.8, 51.1, 51.7, 51.7, 66.5, 69.30, 172.9, 174.8. Anal. calcd for C17H31NO4: C, 65.14; H, 9.97; N, 4.47; found: C, 64.91; H, 9.83; N, 4.63.
3.2.2. General Method of Synthesis of 3,4-Bis(hydroxymethyl)-3,4-dihydro-2H-pyrrole 1-oxides (8c,d)
A solution of crude amines (mixture of isomers 6c,d or 7c,d; 0.01 mol) in dry THF (10 mL) was added dropwise to a stirred solution of LiAlH4 (0.76 g, 0.02 mol) in dry THF (50 mL). The mixture was stirred at ambient temperature for 1 h, then the flask was placed into a cold water bath and quenched with water. The organic phase was separated via decantation, the remaining wet precipitate was washed with THF 3 × 20 mL, and the combined extracts were evaporated in a vacuum. The residue was dissolved in methanol (50 mL) and mixed with a solution of sodium tungstate dihydrate (0.33 g, 0.001 mol) and EDTA disodium salt (0.34 g, 0.001 mol) in water (25 mL) and hydrogen peroxide 30% (5 mL, 0.05 mol) was added. The solution was allowed to stand at ambient temperature for a few days (TLC control on silica gel, ethylacetate–methanol9:1, visualization with UV-254 and Dragendorff’s reagent). Then the catalytic amount of MnO2 (0.1 g, 1.1 mmol) was carefully added to quench the remaining H2O2. After oxygen evolution ceased, the solution was evaporated in a vacuum. The residue was triturated with a chloroform/ethanol 100/1 mixture, the catalyst was filtered off, and the solvent was distilled off in a vacuum. The residue was triturated with diethyl ether, and a yellowish crystalline precipitate was collected, which was used for the next step without further purification. For the analysis, the nitrone was purified using column chromatography on silica gel (ethyl acetate).
2,2-Diethyl-5-isopropyl-3,4-bis(hydroxymethyl)-3,4-dihydro-2H-pyrrole 1-oxide (8c). Yield 1.9 g (80%), colorless crystals, m.p. 87–90 °C (from ethyl acetate–diethyl ether). IR (KBr) νmax: 1592 (C=N). 1H NMR (400 MHz; CDCl3, δ):0.71 (t, Jt = 7.4 Hz, 3H), 0.84 (t, Jt = 7.4 Hz, 3H), 1.12 (d, Jd = 7.0 Hz, 3H), 1.17 (d, Jd = 7.0 Hz, 3H), 1.47 (dq, Jd = 14.4 Hz, Jq = 7.4 Hz, 1H), 1.59 (dq, Jd = 14.3 Hz, Jq = 7.4 Hz, 1H), 1.71 (dq, Jd = 14.3 Hz, Jq = 7.4 Hz, 1H), 1.82 (dq, Jd = 14.3 Hz, Jq = 7.4 Hz, 1H), 2.30 (ddd, Jd1 = 8.9 Hz, Jd2 = 8.6 Hz, Jd3 = 4.4 Hz, 1H), 2.86 (ddd, Jd1 = 8.9 Hz, Jd2 = 7.6 Hz, Jd3 = 3.2 Hz, 1H), 3.22 (sp, Jsp = 7.0 Hz, 1H), 3.35 (dd, Jd1 = 10.0 Hz, Jd2 = 8.6 Hz, 1H), 3.65 (dd, Jd1 = 10.9 Hz, Jd2 = 7.6 Hz, 1H), 3.69–3.76 (br, 1H), 4.01–4.09 (br, 1H), 5.23–5.34 (br, 1H), 5.38–5.47 (br, 1H). 13C{1H} NMR (100 MHz, CDCl3, δ): 7.4, 9.2, 17.4, 18.0, 26.2, 27.5, 30.5, 45.3, 50.4, 61.1, 63.5, 80.1, 153.4. Anal. calcd for C13H25NO3: C, 64.16; H, 10.36; N, 5.76; found: C, 64.14; H, 10.90 N, 5.50.
2,2,5-Triisopropyl-3,4-bis(hydroxymethyl)-3,4-dihydro-2H-pyrrole 1-oxide (8d). Yield 1.1 g (42%), colorless crystals, m.p. 160–161 °C (from ethyl acetate–diethyl ether). IR (KBr) νmax: 1595 (C=N). 1H NMR (500 MHz; CDCl3,δ):0.77 (d, Jd = 6.6 Hz, 3H), 0.79 (d, Jd = 6.6 Hz, 3H), 0.87 (d, Jd = 7.0, 3H), 1.07 (d, Jd = 7.0 Hz, 3H), 1.14 (d, Jd = 7.0 Hz, 3H), 1.16 (d, Jd = 7.0 Hz, 3H), 1.78 (sp, Jsp = 6.6 Hz, 1H), 2.20 (ddd, Jd1 = 10.5 Hz, Jd2 = 6.9 Hz, Jd3 = 3.7 Hz, 1H), 2.64 (sp, Jsp = 7.0 Hz, 1H), 2.83 (ddd, Jd1 = 9.4 Hz, Jd2 = 6.9 Hz, Jd3 = 3.3 Hz, 1H), 3.18 (sp, Jsp = 7.0 Hz, 1H), 3.26 (dd, Jd1 = 10.3 Hz, Jd2 = 9.4 Hz, 1H), 3.57 (dd, Jd1 = 10.5 Hz, Jd2 = 10.0 Hz, 1H), 3.69 (dd, Jd1 = 10.0 Hz, Jd2 = 3.7 Hz, 1H), 4.01 (dd, Jd1 = 10.3 Hz, Jd2 = 3.3 Hz, 1H), 5.22–5.26 (br, 1H), 5.35–5.40 (br, 1H). 13C{1H} NMR (125 MHz, CDCl3, δ): 15.4, 16.5, 17.0, 17.6, 17.8, 18.9, 25.9, 29.3, 30.1, 42.8, 51.1, 62.3 63.4, 83.5, 151.2. Anal. calcdfor C15H29NO3: C, 66.38; H, 10.77; N, 5.16; found: C, 66.45; H, 10.71; N, 5.14.
3.2.3. General Method of Protecting Hydroxyl Groups with 2,2-Dimethoxypropane
2,2-Dimethoxypropane (18 mL, 0.15 mol) was added dropwise to a mixture of nitrone (8a–d) (5 mmol), pyridinium p-toluenesulfonate (0.1 g, 0.4 mmol), molecular saves 3A and dry chloroform (50 mL). The mixture was stirred at ambient temperature for 1 day (TLC control on silica gel, ethyl acetate–methanol9:1, visualization with UV-254 and Dragendorff’s reagent). Then the molecular saves 3A were filtered off. The organic phase was washed with a 10% solution of sodium carbonate in water (50 mL) and was dried with Na2CO3, and filtered off. The solvent was evaporated in a vacuum, and the residue was crystallized from hexane.
2,2,5-Triethyl-3,4-bis(((2-methoxypropan-2-yl)oxy)methyl)-3,4-dihydro-2H-pyrrole 1-oxide (1a). Yield 1.2 g (63%), colorless crystals, m.p. 41–42 °C. IR (KBr) νmax: 2829 (OC-H), 1600 (C=N), 1153 (C-O-C). 1H NMR (400 MHz; CDCl3,δ): 0.71 (t, Jt = 7.3 Hz, 3H), 0.79 (t, Jt = 7.4 Hz, 3H), 1.03 (t, Jt = 7.4 Hz, 3H), 1.21 (s, 3H), 1.22 (s, 3H), 1.24 (s, 6H), 1.52 (dq, Jd = 14.6 Hz, Jq = 7.3 Hz, 1H), 1.57 (dq, Jd = 14.4 Hz, Jq = 7.4 Hz, 1H), 1.73 (dq, Jd = 14.6 Hz, Jq = 7.3 Hz, 1H), 1.87 (dq, Jd = 14.4 Hz, Jq = 7.4 Hz, 1H), 2.14 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 2.45 (ddd, Jd1 = 8.8 Hz, Jd2 = 7.6 Hz, Jd3 = 7.1 Hz, 1H), 2.61 (ddd, Jd1 = 8.8 Hz, Jd2 = 4.3 Hz, Jd3 = 3.8 Hz, 1H), 2.73 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 3.09 (s, 3H), 3.10 (s, 3H), 3.46 (dd, Jd1 = 7.1 Hz, Jd2 = 9.0 Hz, 1H), 3.47 (dd, Jd1 = 4.3 Hz, Jd2 = 9.4 Hz, 1H), 3.49 (dd, Jd1 = 7.6 Hz, Jd2 = 9.0 Hz, 1H), 3.51 (dd, jd1 = 9.4 Hz, Jd2 = 3.8 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3, δ): 7.6, 9.1, 9.3, 18.2, 27.4, 30.5, 24.0, 24.1, 24.1, 24.1, 38.7, 45.6, 48.4, 48.5, 59.8, 60.1, 79.6, 99.8, 99.8, 148.5. Anal. calcd for C20H39NO5: C, 64.31; H, 10.52; N, 3.75; found: C, 64.09; H, 10.48; N, 3.66.
5-tert-Butyl-2,2-diethyl-3,4-bis(((2-methoxypropan-2-yl)oxy)methyl)-3,4-dihydro-2H-pyrrole 1-oxide (1b). Yield 1.2 g (78%), colorless crystals, m.p. 55–57 °C. IR (KBr) νmax: 2829 (OC-H), 1554 (C=N), 1152 (C-O-C). 1H NMR (500 MHz; CDCl3, δ): 0.72 (t, Jt = 7.4 Hz, 3H), 0.83 (t, Jt = 7.4 Hz, 3H), 1.25 (s, 3H), 1.25 (s, 3H), 1.25 (s, 3H), 1.26 (s, 3H), 1.30 (s, 9H), 1.58 (dq, Jd = 14.7 Hz, Jq = 7.1 Hz, 1H), 1.60 (dq, Jd = 13.6 Hz, Jq = 7.4 Hz, 1H), 1.71 (dq, Jd = 14.7 Hz, Jq = 7.4 Hz, 1H), 1.85 (dq, Jd = 13.6 Hz, Jq = 7.4 Hz, 1H), 2.43 (q, Jq = 7.3 Hz, 1H), 2.66 (dt, Jd = 7.3 Hz, Jt = 3.3 Hz, 1H), 3.11 (s, 3H), 3.16 (s, 3H), 3.47 (d, Jd = 7.3 Hz, 2H), 3.60 (d, Jd = 3.3 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, δ): 7.6, 9.2, 23.9, 24.0, 24.1, 24.2, 25.7, 31.5, 34.5, 40.2, 46.8, 48.3, 48.6, 60.4, 61.8, 79.7, 99.7, 99.9, 150.4. Anal. calcd for C22H43NO5: C, 65.80; H, 10.79; N, 3.49; found: C, 65.86; H, 10.53; N, 3.66.
2,2-Diethyl-5-isopropyl-3,4-bis(((2-methoxypropan-2-yl)oxy)methyl)-3,4-dihydro-2H-pyrrole 1-oxide (1c). Yield 1.5 g (76%) of colorless crystals, m.p. 31–33 °C (dec.). IR (KBr) νmax: 2829 (OC-H), 1589 (C=N), 1153 (C-O-C). 1H NMR (500 MHz; CDCl3,δ):0.73 (t, Jt = 7.3 Hz, 3H), 0.82 (t, Jt = 7.5 Hz, 3H), 1.17 (d, Jd = 7.1 Hz, 3H), 1.19 (d, Jd = 7.1 Hz, 3H), 1.25 (s, 3H), 1.26 (s, 3H), 1.27 (s, 6H), 1.58 (dq, Jd = 14.3 Hz, Jq = 7.4 Hz, 2H), 1.75 (dq, Jd = 14.6 Hz, Jq = 7.5 Hz, 1H), 1.89 (dq, Jd = 14.0 Hz, Jq = 7.3 Hz, 1H), 2.46 (ddd, Jd1 = 8.5 Hz, Jd2 = 7.4 Hz, Jd3 = 7.4 Hz, 1H), 2.62 (ddd, jd1 = 8.5 Hz, Jd2 = 4.5 Hz, Jd3 = 3.7 Hz, 1H), 3.09 (sp, Jsp = 7.1 Hz, 1H), 3.12 (s, 3H), 3.15 (s, 2H), 3.48 (dd, Jd1 = 9.6 Hz, Jd2 = 7.4 Hz, 1H), 3.49 (dd, Jd1 = 9.2 Hz, Jd2 = 4.5 Hz, 1H), 3.54 (dd, Jd1 = 9.2 Hz, Jd2 = 7.4 Hz, 1H), 3.58 (dd, Jd1 = 9.6 Hz, Jd2 = 3.7 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3, δ): 7.5, 9.2, 17.3, 17.4, 23.9,24.0,24.1,24.1, 26.2, 27.5, 30.6, 39.4, 45.9, 48.3, 48.5, 60.2, 60.5, 79.5, 99.8, 99.9, 150.1. Anal. calcd for C21H41NO5: C, 65.08; H, 10.66; N, 3.61.; found: C, 64.95; H, 10.75; N, 3.66.
2,2,5-Triisopropyl-3,4-bis(((2-methoxypropan-2-yl)oxy)methyl)-3,4-dihydro-2H-pyrrole 1-oxide (1d). Yield 1.2 g (58%), colorless crystals, m.p. 59–63 °C. IR (KBr) νmax: 2829 (OC-H), 1594 (C=N), 1153 (C-O-C). 1H NMR (400 MHz; CDCl3,δ): 0.82 (d, Jd = 6.6 Hz, 3H), 0.83 (d, Jd = 6.6 Hz, 3H), 0.86 (d, Jd = 7.1 Hz, 3H), 1.17 (d, Jd = 7.1 Hz, 3H), 1.19 (d, Jd = 7.1 Hz, 3H), 1.20 (d, Jd = 6.6 Hz, 3H), 1.28 (s, 3H), 1.28 (s, 9H), 1.86 (sp, Jsp = 6.6 Hz, 1H), 2.43 (ddd, Jd1 = 9.0 Hz, Jd2 = 7.8 Hz, Jd3 = 5.7 Hz, 1H), 2.63 (ddd, Jd1 = 7.8 Hz, Jd2 = 3.8 Hz, Jd3 = 3.7 Hz, 1H), 2.70 (sp, Jd = 6.8 Hz, 1H), 3.07 (sp, Jsp = 7.1 Hz, 1H), 3.14 (s, 3H), 3.19 (s, 3H), 3.43 (dd, Jd1 = 9.1 Hz, Jd2 = 9.0 Hz, 1H), 3.49 (dd, Jd1 = 9.1 Hz, Jd2 = 5.7 Hz, 1H), 3.57 (dd, Jd1 = 9.5 Hz, Jd2 = 3.8 Hz, 1H), 3.65 (dd, Jd1 = 9.5 Hz, Jd2 = 3.7 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3, δ): 16.1, 16.9, 17.0, 17.1, 18.1, 19.2, 24.0, 24.1, 24.2, 26.1, 36.3, 28.0, 48.4, 48.6, 60.7, 61.8, 82.2, 99.8, 99.9, 149.5. Anal. calcd for C23H45NO5: C, 66.47; H, 10.91; N, 3.37; found: C, 66.31; H, 10.87; N, 3.58.
(3S,4S)-3,4-Di-tert-butoxy-2,2,5-triethyl-3,4-dihydro-2H-pyrrole 1-oxide (2). A solution of ethyl magnesium bromide was prepared from ethyl bromide (1.4 g, 12.87 mmol) and Mg chips (0.36 g, 14.85 mmol) in dry Et2O. A solution of (3S,4S)-3,4-di-tert-butoxy-3,4-dihydro-2H-pyrrole 1-oxide (2.27 g, 9.9 mmol) in dry Et2O (15 mL) was added dropwise. The reaction mixture was stirred for 2 h, quenched with water (0.21 mL, 11.85 mmol), the organic phase was separated, and dry air was bubbled into the solution for 5 h (TLC control, silica gel, eluent EtOAc). The resulting solution was treated with another portion of ethylmagnesium bromide and processed as described above. This procedure was repeated once again, the organic layer was concentrated in a vacuum, and the residue was purified using column chromatography (silica gel, EtOAc) to give 2 as a colorless waxy solid. Yield 0.76 g (25%). IR (KBr) νmax: 2973 (C-H). UV (EtOH): 239 (3,94). [α]26D = + 12.7 (c 0.85, CHCl3). 1H NMR (500 MHz, CDCl3, δ): 0.77 (t, Jt = 7.4 Hz, 3H), 0.78 (t, Jt = 7.4 Hz, 3H), 1.07 (t, Jt = 7.4 Hz, 3H), 1.17 (s, 9H), 1.21 (s, 9H), 1.43 (dq, Jd = 14.7 Hz, Jq = 7.4 Hz, 1H), 1.64 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 1.67 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 2.17 (dq, Jd = 14.7 Hz, Jq = 7.4 Hz, 1H), 2.37 (dq, Jd = 13.5 Hz, Jq = 7.4 Hz, 1H), 2.50 (dq, Jd = 13.5 Hz, Jq = 7.4 Hz, 1H), 3.94 (d, Jd = 5.7 Hz, 1H), 4.40 (d, Jd = 5.7 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3, δ): 7.5, 8.7, 9.0, 17.6, 26.8, 27.8, 29.0, 29.1, 74.4, 74.7, 76.7, 77.8, 78.9, 148.4. Anal. calcd for C18H35NO3: C, 68.97; H, 11.25; N, 4.47; found: C, 69.11; H, 11.17; N, 4.55.
2,2,4-Triethyl-5,5-dimethyl-2,5-dihydroimidazole 3-oxide (11). A solution of 2-amino-2-methylpentan-3-one oxime (10) (6.8 g, 52 mmol), pentane-3-one (14 g, 162 mmol) and ammonium acetate (5 g, 64 mmol) in methanol (30 mL) was heated under reflux for 3.5 h. The solution was concentrated in a vacuum, diluted with brine (100 mL) and extracted with EtOAc. The extract was dried with Na2CO3 and evaporated in a vacuum, with a bath temperature of 50 °C. The viscous residue was left overnight at 20 °C and solidified. The resulting crude 11 was used in the next step without purification. Yield 10 g (97%), colorless crystals, m.p. 40–44 °C (from hexane). IR(KBr) νmax: 3275 (NH), 1606 (C=N). 1H NMR (500 MHz; CDCl3, δ): 0.82 (t, Jt = 7.4 Hz, 6H), 1.13 (t, Jt = 7.5 Hz, 3H), 1.28 (s, 6H), 1.67 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 2H), 1.83 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 2H), 2.32 (q, Jq = 7.5 Hz, 2H). 13C{1H} NMR (125 MHz, CDCl3, δ): 7.5, 9.2, 17.3, 28.3, 30.1, 61.4, 92.7, 149.2. Anal. calcd for C11H22N2O: C, 66.62; H, 11.18; N, 14.13; found: C, 66.41; H, 10.97; N, 13.98.
2,2,4-Triethyl-1,5,5-trimethyl-2,5-dihydroimidazole 3-oxide (4). A solution of 11 (9.5 g, 48 mmol) in 37% aqueous formaldehyde (20 mL, 270 mmol) and 90% formic acid (20 mL, 470 mmol) was stirred at 60 °C overnight. The mixture was diluted with water (200 mL) and extracted with chloroform. The extract was washed with a saturated aqueous solution of Na2CO3 and dried with Na2CO3. The solvent was distilled off, affording 12 as a colorless oil, 7.8 g (77%). The compound was used without further purification. IR (neat) νmax: 2806 (CH, NMe), 1603 (C=N). 1H NMR (500 MHz; CDCl3, δ): 0.71 (t, Jt = 7.4 Hz, 6H), 1.17 (t, Jt = 7.4 Hz, 3H), 1.23 (s, 6H), 1.50 (dq, Jd = 14.5 Hz, Jq = 7.4 Hz, 2H), 1.92 (dq, Jd = 14.5 Hz, Jq = 7.4 Hz, 2H), 2.32 (s, 3H), 2.36 (q, Jq = 7.5 Hz, 2H). 13C{1H} NMR (125.77 MHz, CDCl3, δ): 8.1, 9.2, 17.4, 24.4, 25.9, 27.9, 61.2, 94.2, 150.3. Anal. calcd for C12H24N2O: C, 67.88; H, 11.39; N, 13.19; found: C, 67.49; H, 11.17; N, 13.28.
3.2.4. General Method of Reaction of Nitrones 12 with Ethynyl Magnesium Bromide
A 0.5–1 M solution of ethynylmagnesium bromide in THF (20–10 mL, 10 mmol) was added to nitrone (1 mmol) and kept at ambient temperature for 1–8 weeks (TLC control on silica gel, ethylacetate, visualization with UV-254 and Dragendorff’s reagent). Then the mixture was quenched with water, the organic phase was separated via decantation, and the remaining wet precipitate was washed with THF 5 × 10 mL. The THF was distilled off in a vacuum, and the residue was dissolved in methanol (20 mL). A 1 M solution of sodium hydroxide in water (6 mL) was added to the mixture. Then, the methylene blue (3 mg, 0.01 mmol) was added to the mixture, and the air was bubbled until the solution turned dark blue. Then the mixture was diluted with water (20 mL) and a 1M solution of sulphuric acid (4 mL). Methanol was evaporated in a vacuum. The mixture was extracted with ethyl acetate (3 × 10 mL). The organic phase was evaporated in a vacuum, and the residue was purified using column chromatography on silica gel (hexane–ethyl acetate 1:1).
2,2,5-Triethyl-5-ethynyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (12a). Yield 0.173 g (68%), yellow crystals, m.p. 107–113 °C (diethyl ether). IR (KBr) νmax: 3220 (≡C-H), 2102 (C≡C). 1H NMR (300 MHz; CD3OD, Zn/CF3COOH,δ): 0.85 (t, Jt = 7.4 Hz, 3H), 0.87 (t, Jt = 7.4 Hz, 3H), 1.07 (t, Jt = 7.4 Hz, 3H), 1.72 (q, Jq = 7.4 Hz, 2H), 2.21–2.35 (m, 4H), 2.53–2.61 (m, 2H), 2.90 (s, 1H), 3.57 (dd, Jd1 = 10.9 Hz, Jd2 = 5.3 Hz, 1H), 3.67 (dd, Jd1 = 10.9 Hz, Jd2 = 3.0 Hz, 1H), 3.68 (dd, Jd1 = 11.4 Hz, Jd2 = 5.9 Hz, 1H), 3.80 (dd, Jd1 = 11.4 Hz, Jd2 = 3.5 Hz,1H). Anal. calcd for C14H24NO3: C, 66.11; H, 9.51; N, 5.51; found: C, 66.18; H, 9.65; N, 5.50.HRMS (EI/DFS) m/z [M]+calcd for C14H24NO3: 254.1751; found: 254.1753.
5-tert-Butyl-2,2-diethyl-5-ethynyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (12b). Yield 0.189 g (67%), yellow crystals, m.p. 113–114 °C (diethyl ether). IR (KBr) νmax: 3305 (≡C-H). 1H NMR (300 MHz; CD3OD, Zn/CF3COOH, δ): 1.01 (t, Jt = 7.4 Hz, 3H), 1.05 (t, Jt = 7.4 Hz, 3H), 1.27 (s, 9H), 2.00 (q, Jq = 7.4, 2H), 2.08 (dq, Jd = 14.0 Hz, Jq = 7.4, 1H), 2.31 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), dt, Jd = 6.2 Hz, Jt = 4.7 Hz, 1H), 2.70 (ddd, Jd1 = 8.5 Hz, Jd2 = 6.2 Hz, Jd3 = 4.4 Hz, 1H), 3.49 (s, 1H), 3.73 (d, Jd = 4.7 Hz, 2H), 3.76 (dd, Jd1 = 11.2 Hz, Jd2 = 8.5 Hz, 1H), 3.95 (dd, Jd1 = 11.2 Hz, Jd2 = 4.4 Hz, 1H). Anal. Calcd for C16H28NO3: C, 68.05; H, 9.99; N, 4.96.; found: C, 67.99; H, 10.00; N, 4.87. HRMS (EI/DFS) m/z: [M]+calcd for C16H28NO3 282.2064, found 282.2060.
2,2-Diethyl-5-isopropyl-5-ethynyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (12c). Yielding 0.137 g (51%) of yellow crystals. m.p. 132–133 °C (diethyl ether). IR (KBr) νmax: 3219 (≡C-H), 2100 (C≡C). 1H NMR (300 MHz; CD3OD, Zn/CF3COOH, δ): 0.98 (t, Jt = 7.4 Hz, 3H), 1.02 (t, Jt = 7.4 Hz, 3H), 1.14 (d, Jd = 6.5 Hz, 3H), 1.17 (d, Jd = 6.5 Hz, 3H), 1.93 (dq, Jd = 15.6 Hz, Jq = 7.4 Hz, 1H), 2.01–2.12 (m, 2H), 2.21 (dq, Jd = 13.7 Hz, Jq = 7.4 Hz, 1H), 2.38 (sp, Jsp = 6.5 Hz, 1H), 2.42 (ddd, Jd1 = 3.8 Hz, Jd2 = 3.8 Hz, Jd3 = 3.1 Hz, 1H), 2.56 (ddd, Jd1 = 10.0 Hz, Jd2 = 3.8 Hz, Jd3 = 4.9 Hz, 1H), 3.41 (s, 1H), 3.60 (dd, Jd1 = 10.5 Hz, Jd2 = 3.8 Hz, 1H), 3.74 (dd, Jd1 = 11.1 Hz, Jd2 = 10.0 Hz, 1H), 3.82 (dd, Jd1 = 10.5 Hz, Jd2 = 3.1 Hz, 1H), 3.92 (dd, Jd1 = 11.1 Hz, Jd2 = 4.9 Hz, 1H). Anal. calcd for C15H26NO3: C, 67.13; H, 9.76; N, 5.22.; found: C, 66.73; H, 9.79; N, 5.21.HRMS (EI/DFS) m/z [M]+calcd for C15H26NO3: 268.1907; found: 268.1909.
(3S,4S,5S)-3,4-Di-tert-butoxy-2,2,5-triethyl-5-ethynylpyrrolidine 1-oxyl (14). To the solution of 2 (0.7 g, 2.23 mmol) in dry THF (5 mL), the solution of ethynylmagnesium bromide in THF (22.3 mL of a 0.5M solution) was added in one portion. The mixture was allowed to stand at r.t. for one week. The reaction mixture was quenched with H2O (2 mL), and the inorganic precipitate was filtered off. The dry air was bubbled into the solution until the oxidation to radical was complete (TLC control, silica gel, EtOAc). The organic layer was concentrated in a vacuum, and the residue was purified using column chromatography (silica gel, EtOAc) to give 14 as an orange crystal, m.p. 91–93 °C (from hexane). Yield: 490 mg (65%), UV (EtOH): 244 (3,21). [α]26D = + 106.1 (c 0.85, CHCl3). IR (KBr) νmax: 3271 (≡C-H). 1H NMR (500 MHz; CD3OD, Zn/CF3COOH, δ): 0.99 (t, Jt = 7.4 Hz, 3H), 1.03 (t, Jt = 7.4 Hz, 3H), 1.16 (t, Jt = 7.4 Hz, 3H), 1.31 (s, 9H), 1.33 (s, 9H), 1.90–2.01 (m, 4H), 2.05 (dq, Jd = 15.5 Hz, Jq = 7.4 Hz, 1H), 2.18 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 3.27 (s, 1H), 4.02 (s, 1H), 4.20 (s, 1H). Anal. calcd for C20H36NO3: C, 70.96; H, 10.72; N, 4.14; found: C, 70.85; H, 10.65; N, 4.10.
2,2,5-Triethyl-5-ethynylpyrrolidin-1-oxyl (15). A solution of 3 (1.0 g, 5.9 mmol) in anhydrous THF (10 mL) was added to a 0.5–1 M solution of ethynyl-magnesium bromide in THF (50 mL) upon stirring. The mixture was allowed to stand at room temperature for 24 h (TLC control, hexane-ethyl acetate 4:1, UV detection), then quenched with NaCl saturated solution (10 mL). The organic layer was separated, and the residue was washed with ethyl acetate (2 × 20 mL). The combined organic layers were dried with anhydrous Na2SO4. The solvent was evaporated in a vacuum, and the crude residue was dissolved in methanol (15 mL) and basified with sodium hydroxide solution (1 M, 5 mL). Methylene blue (6 mg, 0.02 mmol) was added to the mixture, and the air was bubbled until the solution turned dark blue. The methanol was distilled off in a vacuum, and the remaining aqueous solution was extracted with ether (3 × 20 mL). The combined organic solution was washed with water (3 × 20 mL). The organic phase was dried with Na2SO4, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography on silica gel, eluent hexane–ethyl acetate 4:1, to give 15, with a yield of 800 mg (70%), as a yellow liquid. IR (KBr) νmax: 3309, 3244 (≡C-H), 2107 (C≡C). 1H NMR (300 MHz; CD3OD, Zn/CF3COOH, δ): 0.99 (t, Jt = 7.4 Hz, 3H), 1.01 (t, Jt = 7.4 Hz, 3H), 1.18 (t, Jt = 7.4 Hz, 3H), 1.76–1.86 (m, 2H), 1.94–2.17 (m, 6H), 2.23–2.24 (m, 2H), 3.39 (s, 1H). Anal. calcd for C12H20NO: C, 74.16; H, 10.38; N, 7.21; found: C, 73.95; H, 10.65; N, 7.10.
2,2,5-Triethyl-5-ethynyl-3,4,4-trimethylimidazolidin-1-oxyl (16). A solution of 5 (850 mg, 4 mmol) in THF (5 mL) was added to a 0.9 M solution of ethynylmagnesium bromide (45 mL, 40 mmol) in THF, and the flask was sealed and left at 20 °C for 21 days. The reaction mixture was quenched with brine, the organic phase was separated, and the aqueous phase was washed with diethyl ether. Combined organic phases were dried with Na2CO3, then PbO2 (10 g, 42 mmol) was added, and the reaction mixture was stirred for 24 h. The led oxides were filtered off, and the solution was concentrated in a vacuum and separated using column chromatography of silica gel, and the eluent hexane–diethyl ether 3:1 to give 16 as an orange oil, with a yield of 620 mg (65%). IR (KBr) νmax: 3309, 3251 (≡C-H), 2808 (C-H, NMe), 2112 (C≡C). For NMR investigation, the sample (10 mg) was stirred with Zn powder (100 mg) and ND4Cl (30 mg) in CD3OD (0.5 mL) at 5 °C for 10 min and filtered into an NMR tube. 1H NMR (300 MHz; CD3OD, δ): 0.97 (t, Jt = 7.4 Hz, 3H), 0.99 (t, Jt = 7.4 Hz, 3H), 1.06 (s, 3H), 1.12 (t, Jt = 7.4 Hz, 3H), 1.27 (s, 3H), 1.48 (dq, Jd = 13.6 Hz, Jq = 7.4 Hz, 1H), 1.67 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 1.78 (dq, Jd = 13.6 Hz, Jq = 7.4 Hz, 1H), 1.87 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 1.95 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 2.13 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 1H), 2.31 (s, 3H), 2.72 (s, 1H). HRMS (EI/DFS) m/z [M]+calcdforC14H25N2O: 237.1961; found: 237.1960.
2,5,5-Triethyl-2-ethynyl-4-pyrrolidino-2,5-dihydroimidazol-1-oxyl (17) was prepared using the above procedure. The reaction was completed in 10 h, with a yield of 70%, producing a yellow crystalline solid, m.p. 61–62 °C. IR (KBr) νmax: 3290 (≡C-H), 2104 (C≡C), 1583 (C=N). 1H NMR (400 MHz; CD3OD, Zn/ND4Cl, δ): 0.88 (t, Jt = 7.4 Hz, 3H), 1.00 (t, Jt = 7.4 Hz, 3H), 1.06 (t, Jt = 7.4 Hz, 3H), 1.73 (q, Jq = 7.4 Hz, 2H), 1.81 (dq, Jd = 13.6 Hz, Jq = 7.4 Hz, 1H), 1.84–2.00 (m, 6H) 2.06 (dq, Jd = 15.2 Hz, Jq = 7.4 Hz, 1H), 2.84 (s, 1H), 3.34–3.42 (m, 2H), 3.44–3.52 (m, 2H). Anal. calcd for C15H24N3O: C, 68.67; H, 9.22; N, 16.02; found: C, 68.34; H, 9.11; N, 15.89.
3.2.5. General Method of Hydrogenation
A solution of ethynyl-substituted nitroxide (10 mmol) in THF (100 mL) was placed in the reaction vessel equipped with a magnetic stirrer and a connection line to a gasometer filled with hydrogen. The catalyst (Pd/C, 4%, 200 mg) was added, and the system was purged with hydrogen and closed. The mixture was vigorously stirred until hydrogen absorption ceased (ca. 5 h, 0.6 L of hydrogen absorbed), after which the catalyst was filtered off and washed with THF. The THF was distilled off in a vacuum, and the residue was dissolved in methanol (20 mL). A 1 M solution of sodium hydroxide in water (6 mL) and methylene blue (3 mg, 0.01 mmol) was added to the mixture, and the air was bubbled until the solution turned dark blue. Then the mixture was diluted with water (20 mL) and acidified with a 1 M solution of sulfuric acid (4 mL). Methanol was evaporated in a vacuum. The mixture was extracted with ethyl acetate (3 × 10 mL). The organic phase was evaporated in a vacuum, and the residue was purified using column chromatography on silica gel (hexane–ethyl acetate 1:1).
2,2,5-Triethyl-5-tert-butyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (13b). Yield 2.35 g (82%), yellow crystals, m.p. 109–111 °C (from hexane). IR(KBr) νmax: 3373, 3294 (O-H). 1H NMR (300 MHz; CD3OD, Zn/CF3COOH, δ): 1.08 (t, Jt = 7.4 Hz, 3H), 1.12 (t, Jt = 7.4 Hz, 3H), 1.15 (t, Jt = 7.4 Hz, 3H), 1.19 (s, 9H), 1.77–2.20 (m, 6H), 2.42 (ddd, Jd1 = 12.3 Hz, Jd2 = 6.2 Hz, Jd3 = 5.5 Hz, 1H), 2.54 (ddd, Jd1 = 12.3 Hz, Jd2 = 5.3 Hz, Jd3 = 1.5 Hz, 1H), 3.79 (dd, Jd1 = 11.5 Hz, Jd2 = 5.3 Hz, 1H), 3.82 (dd, Jd1 = 11.1 Hz, Jd2 = 6.2 Hz, 1H), 3.85 (dd, Jd1 = 11.1 Hz, Jd2 = 5.5 Hz, 1H), 3.94 (dd, Jd1 = 11.5 Hz, Jd2 = 1.5 Hz, 1H). Anal. calcd for C16H32NO3: C, 67.09;H, 11.26; N, 4.89; found: C, 67.44; H, 11.51; N, 4.84. HRMS (EI/DFS) m/z [M]+calcd for C16H32NO3: 286.2377; found: 286.2380.
2,2,5-Triethyl-5-isopropyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (13c). Yield 1.39 g (51%), yellow oil. IR(neat) νmax: 2966, 2941, 2861 (C-H). 1H NMR (400 MHz; CD3OD, Zn/CF3COOH, δ): 1.03 (t, Jt = 7.4 Hz, 3H), 1.04 (t, Jt = 7.4 Hz, 3H), 1.06 (t, Jt = 7.4 Hz, 3H), 1.10 (d, Jd = 6.9 Hz, 3H), 1.11 (d, Jd = 6.9 Hz, 3H), 1.78 (dq, Jd = 14.7 Hz, Jq = 7.4 Hz, 1H), 1.81 (dq, Jd = 15.1 Hz, Jq = 7.4 Hz, 1H), 1.91 (dq, Jd = 15.1 Hz, Jq = 7.4 Hz, 1H), 1.92 (q, Jq = 7.4 Hz, 2H), 1.99 (dq, Jd = 14.7 Hz, Jq = 7.4 Hz, 1H), 2.26 (sep, Jsep = 6.9 Hz, 1H), 2.36–2.51 (m, 2H), 3.67–3.85 (m, 4H). Anal.calcd for C15H30NO3: C, 66.14; H, 11.10; N, 5.14; found: C, 66.35; H, 10.98; N, 5.07. HRMS (EI/DFS) m/z [M]+calcd for C15H30NO3: 272.2220; found: 272.2217.
(3S,4S)-3,4-Di-tert-butoxy-2,2,5,5-tetraethylpyrrolidine 1-oxyl (18). A solution of 14 (0.5 g, 1.48 mmol) in THF (10 mL) was placed in the reaction vessel equipped with a magnetic stirrer and a connection line to a gasometer filled with hydrogen. The catalyst (Pd/C, 4%, 100 mg) was added, and the system was purged with hydrogen and closed. The mixture was vigorously stirred until hydrogen absorption ceased (ca. 5 h, 66 mL of hydrogen absorbed). The catalyst was filtered off, the organic layer was concentrated in a vacuum, and the residue was purified using column chromatography (silica gel, EtOAc–hexane 1:1) to give 18 as yellow crystals, m.p. 117–119 °C (from hexane). Yield: 405 mg (80%).UV (EtOH): 241 (3,22). [α]26D = +128.1 (c 0.85, CHCl3). IR (KBr): 2966, 2937, 2879 (C-H). 1H NMR (500 MHz, CD3OD, Zn/CF3COOH, δ): 0.86 (t, Jt = 7.4 Hz, 6H), 0.90 (t, Jt = 7.4 Hz, 6H), 1.20 (s, 18H), 1.63 (dq, Jd = 15.7 Hz, Jq = 7.4 Hz, 2H), 1.84 (dq, Jd = 15.7 Hz, Jq = 7.4 Hz, 2H), 1.85 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 2H), 2.24 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 2H), 3.93 (s, 2H). 13C{1H} NMR (125 MHz, CD3OD, Zn/CF3COOH,δ): 8.0, 9.1, 25.8, 26.6, 29.0, 76.6, 76.7, 80.9. Anal. calcd for C20H40NO3: C, 70.13; H, 11.77; N, 4.09; found: C, 70.25; H, 11.65; N, 4.21.
2,2,5,5-Tetraethylpyrrolidin-1-oxyl (19). A solution of 15 (0.8 g, 4.1 mmol) in dry THF (10 mL) was placed in the reaction vessel equipped with a magnetic stirrer and a connection line to a gasometer filled with hydrogen. The catalyst (Pd/C, 4%, 30 mg) was added, and the system was purged with hydrogen and closed. The mixture was vigorously stirred until hydrogen absorption ceased (ca. 5 h, 0.25 L of hydrogen absorbed), then the catalyst was filtered off and washed with THF. The filtrate was evaporated in a vacuum, and the crude residue was dissolved in methanol (15 mL) and basified with sodium hydroxide solution (1 M, 5 mL). Methylene blue (6 mg, 0.02 mmol) was added to the mixture, and the air was bubbled until the solution turned dark blue. The methanol was distilled off in a vacuum, and the remaining aqueous solution was extracted with ether (3 × 20 mL). The organic phase was dried with Na2SO4, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (silica gel, eluent hexane–ethyl acetate 4:1) to give 19, with a yield of 800 mg (70%), yellow liquid. IR (neat, cm−): 2966, 2937, 2881 (C-H). 1H NMR (300 MHz; CD3OD, Zn/CF3COOH, δ): = 0.97 (t, Jt = 7.4 Hz, 12H), 1.75 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 4H), 1.84 (dq, Jd = 14.0 Hz, Jq = 7.4 Hz, 4H), 1.98 (s, 4H). Anal. calcd for C12H24NO: C, 72.20; H, 12.20; N, 7.06; found: C, 72.25; H, 12.35; N, 7.21. HRMS (EI/DFS) m/z [M]+calcd for C12H24NO: 198.1852, found: 198.1851.
2,2,5,5-Tetraethyl-3,4,4-trimethylimidazolidin-1-oxyl (20). A solution of 16 (355 mg, 1.5 mmol) in THF (2 mL) was placed in the reaction vessel equipped with a magnetic stirrer and a connection line to a gasometer filled with hydrogen. The catalyst (Pd/C, 4%, 50 mg) was added, and the system was purged with hydrogen and closed. The mixture was vigorously stirred until hydrogen absorption ceased (ca. 5 h, 100 mL of hydrogen absorbed), then the catalyst was filtered off and washed with THF. The solution was bubbled with air overnight, THF was distilled off in a vacuum, and the residue was separated using column chromatography on silica gel, eluent hexane–diethyl ether 3:1 to give 20 as a yellow oil, with a yield of 620 mg (65%). IR (KBr) νmax: 2972, 2943, 2881 (C-H). For the NMR investigation, the sample (10 mg) was stirred with Zn powder (100 mg) and ND4Cl (30 mg) in CD3OD (0.5 mL) at 5 °C for 10 min and filtered into an NMR tube. 1H NMR (300 MHz; CD3OD, Zn/ND4Cl,δ): = 0.94 (m, 12H), 1.04 (s, 6H), 1.55–1.80 (m, 6H), 1.82–1.96 (m, 2H), 2.31 (s, 3H). HRMS (EI/DFS) m/z [M]+calcd. forC14H25N2O: 237.1961; found: 237.1960.
2,2,5,5-Tetraethyl-4-pyrrolidino-2,5-dihydroimidazol-1-oxyl (21) was prepared using the above procedure, with a yield of 94%.IR(KBr) νmax: 2968, 2939, 2877 (C-H), 1593 (C=N). 1H NMR (400 MHz; CD3OD, Zn/ND4Cl, δ): 0.96 (t, Jt = 7.4 Hz, 6H), 0.97 (t, Jt = 7.4 Hz, 6H), 1.70 (dq, Jd = 14.2 Hz, Jq = 7.4 Hz, 2H), 1.81 (dq, Jd = 14.6 Hz, Jq = 7.4 Hz, 2H), 1.82 (dq, Jd = 14.2 Hz, Jq = 7.4 Hz, 2H), 1.92 (dq, Jd = 14.6 Hz, Jq = 7.4 Hz, 2H), 1.94–1.99 (m, 4H), 3.46–3.51 (m, 4H).
3.2.6. General Method of Reaction of Nitrones with Alkynyl Magnesium Bromides
The terminal alkyne (0.107 mol) was added dropwise to a 2 M solution of ethylmagnesium bromide in THF (50 mL, 0.100 mol). The mixture was stirred at ambient temperature for 1 h. Then a solution of 1a (3.7 g, 0.01 mol) in dry THF (10 mL) was added to the mixture and kept at ambient temperature for 2 days (TLC control on silica gel, eluent ethyl acetate, visualization with UV-254 and Dragendorff’s reagent). Then the mixture was quenched with water, the organic phase was separated via decantation, and the remaining wet precipitate was washed with THF 5 × 10 mL. The THF was distilled off in a vacuum, and the residue was dissolved in methanol (50 mL). A 1 M solution of sodium hydroxide in water (10 mL) was added to the mixture. Then, the methylene blue (3 mg, 0.01 mmol) was added to the mixture, and the air was bubbled until the solution turned dark blue. Then the mixture was diluted with water (50 mL) and a 1M solution of sulfuric acid (6 mL). Methanol was distilled off in a vacuum, and the mixture was extracted with ethyl acetate (3 × 10 mL). The organic phase was concentrated in a vacuum, and the resulting crude nitroxide 22 was purified as described below.
2,2,5-Triethyl-5-phenylethynyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (22a) was purified using column chromatography on silica gel (hexane–ethyl acetate 1:1). Yield 1.93 g (54%), yellow crystals, m.p. 100–103 °C (dec.) (from diethyl ether). IR (KBr) νmax: 3384 (O-H), 3311 (O-H), 1055 (C-OH). 1H NMR (400 MHz, CD3OD, Zn/CF3COOH, δ): 0.98 (t, Jt = 7.5 Hz, 3H), 1.01 (t, Jt = 7.5 Hz, 3H), 1.252 (t, Jt = 7.5 Hz, 3H), 1.86 (dq, Jd = 14.9 Hz, Jq = 7.5 Hz, 1H), 1.88 (dq, Jd = 14.9 Hz, Jq = 7.5 Hz, 1H), 2.10 (dq, Jd = 14.1 Hz, Jq = 7.2 Hz, 1H), 2.15 (dq, Jd = 14.5 Hz, Jq = 7.3 Hz, 1H), 2.17 (dq, Jd = 14.1 Hz, Jq = 7.9 Hz, 1H), 2.25 (dq, Jd = 14.5 Hz, Jq = 7.7 Hz, 1H), 2.39 (ddd, Jd1 = 10.2 Hz, Jd2 = 6.6 Hz, Jd3 = 4.5 Hz, 1H), 2.41 (ddd, Jd1 = 10.2 Hz, Jd2 = 5.2 Hz, Jd3 = 3.5 Hz, 1H), 3.71 (dd, Jd1 = 11.5 Hz, Jd2 = 5.2 Hz, 1H), 3.78 (dd, Jd1 = 11.5 Hz, Jd2 = 3.5 Hz, 1H), 3.86 (dd, Jd1 = 11.6 Hz, Jd2 = 6.6 Hz, 1H), 3.97 (dd, Jd1 = 11.6 Hz, Jd2 = 4.5 Hz, 1H), 7.38 (dddd, Jd1 = 7.8 Hz, Jd2 = 7.6 Hz, Jd3 = 1.3 Hz, Jd4 = 0.6 Hz, 2H), 7.42 (dddd, Jd1 = 7.6 Hz, Jd2 = 7.6 Hz, Jd3 = 1.3 Hz, Jd4 = 1.3 Hz, 1H), 7.46 (dddd, Jd1 = 7.8 Hz, Jd2 = 1.8 Hz, Jd3 = 1.3 Hz, Jd4 = 0.6 Hz, 2H). Anal. calcd for C20H28NO3: C, 72.69; H, 8.54; N, 4.24.; found: C, 72.75; H, 8.54; N, 4.46. HRMS (EI/DFS) m/z [M]+calcd for C20H28NO3: 330.2064; found: 330.2062.
2,2,5-Triethyl-5-(3-hydroxyprop-1-yn-1-yl)-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (22b) was crystallized from diethyl ether. Yield 1.7 g (60%), yellow crystals, m.p. 104–105 °C. IR (KBr) νmax: 3493, 3412, 3365 (O-H), 1045 (C-OH). 1H NMR (300 MHz, CD3OD, Zn/CF3COOH, δ): 0.94 (t, Jt = 7.5 Hz, 3H), 1.14 (t, Jt = 7.4 Hz, 3H), 1.55 (t, Jt = 7.2 Hz, 3H), 1.77 (dq, Jd = 15.5 Hz, Jq = 7.4 Hz, 1H), 1.80 (dq, Jd = 15.5 Hz, Jq = 7.4 Hz, 1H), 1.98 (dq, Jd = 13.8 Hz, Jq = 7.5 Hz, 1H), 2.04 (dq, Jd = 14.4 Hz, Jq = 7.2 Hz, 1H), 2.07 (dq, Jd = 13.8 Hz, Jq = 7.5 Hz, 1H), 2.08 (dq, Jd = 14.4 Hz, Jq = 7.2 Hz, 1H), 2.28 (ddd, Jd1 = 9.8 Hz, Jd2 = 6.5 Hz, Jd3 = 5.1 Hz, 1H), 2.30 (ddd, Jd1 = 9.8 Hz, Jd2 = 5.9 Hz, Jd3 = 3.7 Hz, 1H), 3.64 (dd, Jd1 = 11.3 Hz, Jd2 = 5.9 Hz, 1H), 3.74 (dd, Jd1 = 11.3 Hz, Jd2 = 3.7 Hz, 1H), 3.76 (dd, Jd1 = 11.4 Hz, Jd2 = 6.5 Hz, 1H), 3.85 (dd, Jd1 = 11.4 Hz, Jd1 = 5.1 Hz, 1H). Anal. calcd for C15H26NO4: C, 63.35; H, 9.22; N, 4.93.; found: C, 62.85; H, 9.25; N, 4.86. HRMS (EI/DFS) m/z: [M]+calcdfor C15H26NO4: 284.1856; found 284.1851.
2,2,5-Triethyl-5-(3-hydroxy-3-methylbut-1-yn-1-yl)-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (22c) was crystallized from diethyl ether to give 22c. Yield 2.0 g (66%) of yellow crystals, m.p. 111–114 °C. IR (KBr) νmax: 3340, 3182 (O-H). 1H NMR (300 MHz, CD3OD, Zn/CF3COOH, δ): 0.96 (t, Jt = 7.4 Hz, 3H), 0.97 (t, Jt = 7.6 Hz, 3H), 1.16 (t, Jt = 7.3 Hz, 3H), 1.50 (s, 6H), 1.80 (dq, Jd = 14.1 Hz, Jq = 7.4 Hz, 1H), 1.87 (dq, Jd = 14.1 Hz, Jq = 7.2 Hz, 1H), 1.98 (dq, Jd = 14.3 Hz, Jq = 7.7 Hz, 1H), 2.06 (dq, Jd = 15.2 Hz, Jq = 7.3 Hz, 1H), 2.11 (dq, Jd = 14.1 Hz, Jq = 7.2 Hz, 1H), 2.13 (dq, Jd = 14.3 Hz, Jq = 7.5 HZ, 1H), 2.29 (dd, Jd1 = 4.4 Hz, Jd2 = 3.1, 1H), 2.29 (dd, Jd1 = 5.7, Jd2 = 4.4, 1H) 3.66 (dd, Jd1 = 11.7 Hz, Jd2 = 4.4 Hz, 1H), 3.74 (dd, Jd1 = 11.7 Hz, Jd2 = 3.1 Hz, 1H), 3.76 (dd, Jd1 = 11.6 Hz, Jd2 = 5.7 Hz, 1H), 3.86 (dd, Jd1 = 11.6 Hz, Jd2 = 4.4 Hz, 1H). Anal. calcd for C17H30NO4: C, 65.35; H, 9.68; N, 4.48.; found: C, 65.82; H, 9.65; N, 4.52. HRMS (EI/DFS) m/z [M]+calcdfor C17H30NO4: 312.2169; found 312.2164.
2,2,5-Triethyl-5-(3-hydroxyprop-1-yn-1-yl)pyrrolidin-1-oxyl (23). A solution of 2-propyn-1-ol (1.75 mL, 29.6 mmol) in dry THF (5 mL)was slowly added to the ethylmagnesium bromide solution in THF (2.0 M, 30 mL) at 0 °C under argon atmosphere. The resulting grey mixture was stirred for 30 min at room temperature. After that, nitrone3 solution (0.5 g, 3.0 mmol) in 5 mL of anhydrous THF was added there. The final mixture was stirred for 20 h at room temperature under an argon atmosphere and then poured into a mixture of ice (10 g) and NaCl (10 g). The organic layer was separated, and the residue was washed with ethyl acetate (3 × 30 mL). The combined organic phase was dried with Na2SO4, and the solvent was evaporated in a vacuum. The crude residue was dissolved in methanol (10 mL) and basified with sodium hydroxide solution (1 M, 2 mL). Methylene blue (3 mg, 0.01 mmol) was added to the mixture, and the air was bubbled until the solution turned dark blue. The methanol was distilled off in a vacuum, and the remaining aqueous solution was extracted with ether (3 × 20 mL). The organic phase was dried with Na2SO4, and the solvent was evaporated in a vacuum. The residue was purified by column chromatography (silica gel, eluent hexane–ethyl acetate 1:1) to give 23, with a yield of 400 mg (65%), yellow liquid. IR (neat) νmax: 3417 (O-H), 2970, 2939, 2879 (C-H). 1H NMR (400 MHz; CD3OD, Zn/CF3COOH, δ): 0.99 (t, Jt = 7.4 Hz, 3H), 1.01 (t, Jt = 7.4 Hz, 3H), 1.17 (t, Jt = 7.4 Hz, 3H), 1.76–1.83 (m, 2H), 1.90–2.13 (m, 6H), 2.23–2.37 (m, 2H), 4.29 (s, 2H). HRMS (EI/DFS) m/z [M]+calcd for C13H22NO2: 224.1645; found: 224.1642
4. Conclusions
The synthesis of nitroxides via reaction of 1-pyrroline 1-oxides with alkynylmagnesium halides was earlier investigated by K. Hideg et al. [34,35,36]. Recently, we suggested using this reaction for the preparation of sterically shielded 2,2,5,5-tetraalkylpyrrolidine nitroxides [8,21]. In this paper, we showed how these reagents could be applied for the preparation of various highly strained nitroxides of pyrrolidine, imidazolidine and 2,5-dihydroimidazole series. The described two-step addition & hydrogenation protocol was suitable for the introduction of an ethyl group to the carbon atom of cyclic α-ethyl-, α-isopropyl- and α-tert-butyl nitrones and may find broader application. Some of the new nitroxides can hardly be prepared in any other way.
The new data on the feasibility of the addition of alkynylmagnesium halides to highly hindered alkylnitrones may have another consequence. In this work, we did not utilize the synthetic potential of alkynyl groups. However, taking into account the broad application of alkynes in organic synthesis, the use of 2-alkynyl nitroxides for biorthogonal spin labeling and for the synthesis of bioactive nitroxide derivatives certainly deserves attention.
The new set of nitroxides gives impressive examples of the variability of EPR spectra of five-membered nitroxides with bulky alkyl substituents adjacent to the nitroxide group. The high additional coupling constants are highly dependent on nitroxide structure. The high sensitivity of spectral parameters to minor structural changes is a promising basis for the molecular design of functional spin probes of a new generation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27217626/s1, IR and NMR spectra of all new compounds, X-ray diffraction data for 8d, 12a–c, 14, 22a–c, EPR spectra of all new nitroxides, HPLC chromatogram of 14.
Author Contributions
Conceptualization, S.A.D. and I.A.K.; validation, S.A.D., M.M.G., D.A.M., I.F.Z., A.I.T. and I.A.K.; formal analysis, Y.S.S., Y.I.G. and Y.V.G.; investigation, S.A.D., M.M.G., D.A.M., I.F.Z., Y.I.G., Y.V.G., A.I.T. and I.A.K.; writing—original draft preparation, S.A.D., M.M.G., D.A.M. and I.A.K.; writing—review and editing, S.A.D. and I.A.K.; visualization, I.A.K., Y.S.S., Y.I.G. and Y.V.G.; supervision, I.A.K.; project administration, S.A.D.; funding acquisition, S.A.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Scientific Foundation (grant number 22-73-00098).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are in the text and the Supplementary Information in this article.
Acknowledgments
We thank the personnel of the Multi-Access Center of SB RAS for recording theIR, NMR and HRMS spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Rad. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef]
- Bonucci, A.; Ouari, O.; Guigliarelli, B.; Belle, V.; Mileo, E. In-cell EPR: Progress towards structural studies inside cells. ChemBioChem 2020, 21, 451–460. [Google Scholar] [CrossRef]
- Wang, Y.; Paletta, J.T.; Berg, K.; Reinhart, E.; Rajca, S.; Rajca, A. Synthesis of unnatural amino acids functionalized with sterically shielded pyrroline nitroxides. Org. Lett. 2014, 16, 5298–5300. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. Bioresistant nitroxide spin label for in-cell EPR spectroscopy: Invitro and in oocytes protein structural dynamics studies. Angew. Chem. Int. Ed. Engl. 2018, 57, 1366–1370. [Google Scholar] [CrossRef]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. Gem-diethyl pyrroline nitroxide spin labels: Synthesis, EPR characterization, rotamer libraries and biocompatibility. ChemistryOpen 2019, 8, 1057–1065. [Google Scholar] [CrossRef]
- Saha, S.; Jagtap, A.P.; Sigurdsson, S.T. Site-directed spin labeling of 20-amino groups in RNA with isoindoline nitroxides that are resistant to reduction. Chem. Commun. 2015, 51, 13142–13145. [Google Scholar] [CrossRef]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and reduction kinetics of sterically shielded pyrrolidine nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A simple method of synthesis of 3-carboxy-2,2,5,5-tetraethylpyrrolidine-1-oxyl and preparation of reduction-resistant spin labels and probes of pyrrolidine series. Molecules 2021, 26, 5761. [Google Scholar] [CrossRef]
- Wanga, X.; Emoto, M.; Sugimoto, A.; Miyake, Y.; Itto, K.; Amasaka, M.; Xu, S.; Hirata, H.; Fujii, H.; Arimoto, H. Synthesis of 15N-labeled 4-oxo-2,2,6,6-tetraethylpiperidine nitroxide for EPR brain imaging. Tetrahedron Lett. 2014, 55, 2146–2149. [Google Scholar] [CrossRef]
- Soikkeli, M.; Kettunen, M.I.; Nivajärvi, R.; Olsson, V.; Rönkkö, S.; Laakkonen, J.P.; Lehto, V.P.; Kavakka, J.; Heikkinen, S. Assessment of the relaxation-enhancing properties of a nitroxide-based contrast agent TEEPO-Glc with in vivo magnetic resonance imaging. Contrast Media Mol. Imaging 2019, 2019, 5629597. [Google Scholar] [CrossRef]
- Komarov, D.A.; Ichikawa, Y.; Yamamoto, K.; Stewart, N.J.; Matsumoto, S.; Yasui, H.; Kirilyuk, I.A.; Khramtsov, V.V.; Inanami, O.; Hirata, H. In vivo extracellular pH mapping of tumors using electron paramagnetic resonance. Anal. Chem. 2018, 90, 13938–13945. [Google Scholar] [CrossRef]
- Samouilov, A.; Efimova, O.V.; Bobko, A.A.; Sun, Z.; Petryakov, S.; Eubank, T.D.; Trofimov, D.G.; Kirilyuk, I.A.; Grigor’ev, I.A.; Takahashi, W.; et al. In vivoproton–electron double-resonance imaging of extracellular tumor pH using an advanced nitroxide probe. Anal. Chem. 2014, 86, 1045–1052. [Google Scholar] [CrossRef]
- Keana, J.F.W. New aspects of nitroxide chemistry. In Spin Labelling II: Theory and Application; Berliner, L.J., Ed.; Academic Press: New York, NY, USA, 1979; pp. 115–172. [Google Scholar]
- Hideg, K.; Kálai, T.; Sár, C.P. Recent results in chemistry and biology of nitroxides. J. Heterocycl. Chem. 2005, 42, 437. [Google Scholar] [CrossRef]
- Kirilyuk, I.A.; Bobko, A.A.; Grigor’ev, I.A.; Khramtsov, V.V. Synthesis of the tetraethyl substituted pH-sensitive nitroxides of imidazoline series with enhanced stability towards reduction. Org. Biomol. Chem. 2004, 2, 1025–1030. [Google Scholar] [CrossRef]
- Zubenko, D.; Kirilyuk, I.; Roshchupkina, G.; Zhurko, I.; Reznikov, V.; Marque, S.R.A.; Bagryanskaya, E. Imidazoline-based nitroxides as prospective mediators in living free radical polymerization. Helv. Chim. Acta 2006, 89, 2341–2353. [Google Scholar] [CrossRef]
- Zhurko, I.F.; Dobrynin, S.; Gorodetskii, A.A.; Glazachev, Y.I.; Rybalova, T.V.; Chernyak, E.I.; Asanbaeva, N.; Bagryanskaya, E.G.; Kirilyuk, I.A. 2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and properties. Molecules 2020, 25, 845. [Google Scholar] [CrossRef]
- Nazarski, R.B.; Skowronski, R. Sterically crowded five-membered heterocyclic systems. Part 3. Unexpected formation of stable flexible pyrrolodinoxyl byradicals via nitrone aldol dimers: A spec-troscopic and mechanistic study. J. Chem. Soc. Perkin. Tr. I 1989, 1, 1603–1610. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-bis(hydroxymethyl)-2,2,5,5-tetraethylpyrrolidin-1-oxyl via 1,3-dipolar cycloaddition of azomethine ylide to activated alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef]
- Lampp, L.; Morgenstern, U.; Merzweiler, K.; Imming, P.; Seidel, R.W. Synthesis and characterization of sterically and electrostatically shielded pyrrolidine nitroxide radicals. J. Mol. Struct. 2019, 1182, 87–94. [Google Scholar] [CrossRef]
- Taratayko, A.I.; Glazachev, Y.I.; Eltsov, I.V.; Chernyak, E.I.; Kirilyuk, I.A. 3,4-Unsubstituted 2-tert-Butyl-pyrrolidine-1-oxyls with Hydrophilic Functional Groups in the Side Chains. Molecules 2022, 27, 1922. [Google Scholar] [CrossRef]
- Kálai, T.; Hubbell, W.L.; Hideg, K. Click reactions with nitroxides. Synthesis 2009, 2009, 1336–1340. [Google Scholar] [CrossRef]
- Kucher, S.; Korneev, S.; Tyagi, S.; Apfelbaum, R.; Grohmann, D.; Lemke, E.A.; Klare, J.P.; Steinhoff, H.-J.; Klose, D. Orthogonal spin labeling using click chemistry for in vitro and in vivo applications. J. Magn. Res. 2016, 275, 38–45. [Google Scholar] [CrossRef]
- Bilodeau, D.A.; Margison, K.D.; Serhan, M.; Pezacki, J.P. Bioorthogonal reactions utilizing nitrones as versatile dipoles in cycloaddition reactions. Chem. Rev. 2021, 121, 6699–6717. [Google Scholar] [CrossRef]
- Godoi, B.; Schumacher, R.F.; Zeni, G. Synthesis of heterocycles via electrophilic cyclization of alkynes containing heteroatom. Chem. Rev. 2011, 111, 2937–2980. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Zeni, G. Ten years of progress in the synthesis of six-membered N-heterocycles from alkynes and nitrogen sources. Tetrahedron 2020, 76, 130876. [Google Scholar] [CrossRef]
- Yamamoto, Y. Synthesis of heterocycles via transition-metalcatalyzed hydroarylation of alkynes. Chem. Soc. Rev. 2014, 43, 1575–1600. [Google Scholar] [CrossRef]
- Cicchi, S.; Hőld, I.; Brandi, A. New synthesis of five-membered cyclic nitrones from tartaric acid. J. Org. Chem. 1993, 58, 5274–5275. [Google Scholar] [CrossRef]
- Morozov, D.A.; Kirilyuk, I.A.; Komarov, D.A.; Goti, A.; Bagryanskaya, I.Y.; Kuratieva, N.V.; Grigor’ev I., A. Synthesis of a chiral C2-symmetric sterically hindered pyrrolidine nitroxide radical via combined iterative nucleophilic additions and intramolecular 1,3-dipolar cycloadditions to cyclic nitrones. J. Org. Chem. 2012, 77, 10688–10698. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Volodarsky, L.B. Ammounium acetate as a catalyst of the condensation of sterically hindered functionalized hydroxylamines with ketones. Russ. Chem. Bull. 1997, 46, 1577–1581. [Google Scholar] [CrossRef]
- Martin, V.V.; Volodarskii, L.B. Synthesis and some reactions of sterically hindered 3-imidazoline 3-oxides. Chem. Heterocycl. Compd. 1979, 15, 92–98. [Google Scholar] [CrossRef]
- Bobko, A.A.; Kirilyuk, I.A.; Gritsan, N.P.; Polovyanenko, D.N.; Grigor’ev, I.A.; Khramtsov, V.V.; Bagryanskaya, E.G. EPR and quantum chemical studies of the pH-sensitive imidazoline and imidazolidine nitroxides with bulky substituents. Appl. Magn. Reson. 2010, 39, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Skattebol, L.; Jones, E.R.H.; Whiting, M.C. 1-Phenyl-1-penten-4-yn-3-ol. Org. Synth. Coll. 1963, 4, 792–795. [Google Scholar] [CrossRef]
- Baracz, N.M.; Hankovsky, O.H.; Sár, S.P.; Jerkovich, G.; Hideg, K. Synthesis of alkynyl-substituted pyrrolidin-1-yloxyl radicals from 1-pyrroline N-oxide nitrones and alkynylmagnesium bromides. Synthesis 1996, 1996, 204–208. [Google Scholar] [CrossRef]
- Sár, S.P.; Jekő, J.; Fajer, P.; Hideg, K. Synthesis and reactions of new alkynyl substituted nitroxide radicals. Synthesis 1999, 1999, 1039–1045. [Google Scholar] [CrossRef]
- Sár, S.P.; Osz, E.; Jekő, J.; Hideg, K. Synthesis of spiro[pyrolidine-2,2-adamantane] nitrones alnd nitroxides. Synthesis 2005, 2005, 255–259. [Google Scholar] [CrossRef]























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).