
Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity


 , , , , , , , ,
, , , , , , , , 

Abstract
1. Introduction
2. Results and Discussion
2.1. Synthetic Aspects and Reactivity
2.1.1. Ortho-Pyridine Carboxamides
2.1.2. Meta- and Para-Pyridine Carboxamides
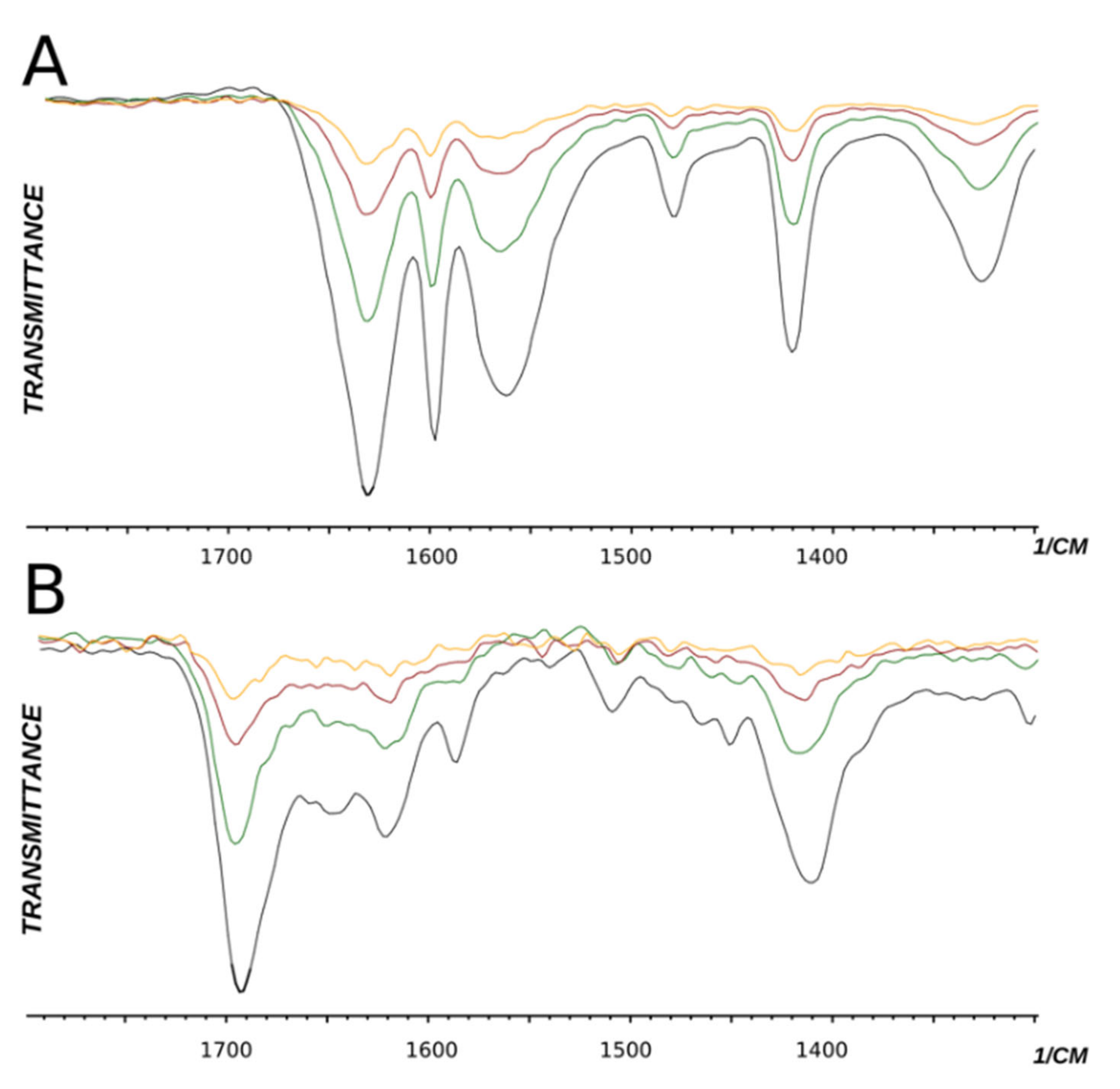
2.1.3. Investigations under High Temperature
2.2. X-ray Investigation
2.3. Quantum Chemistry
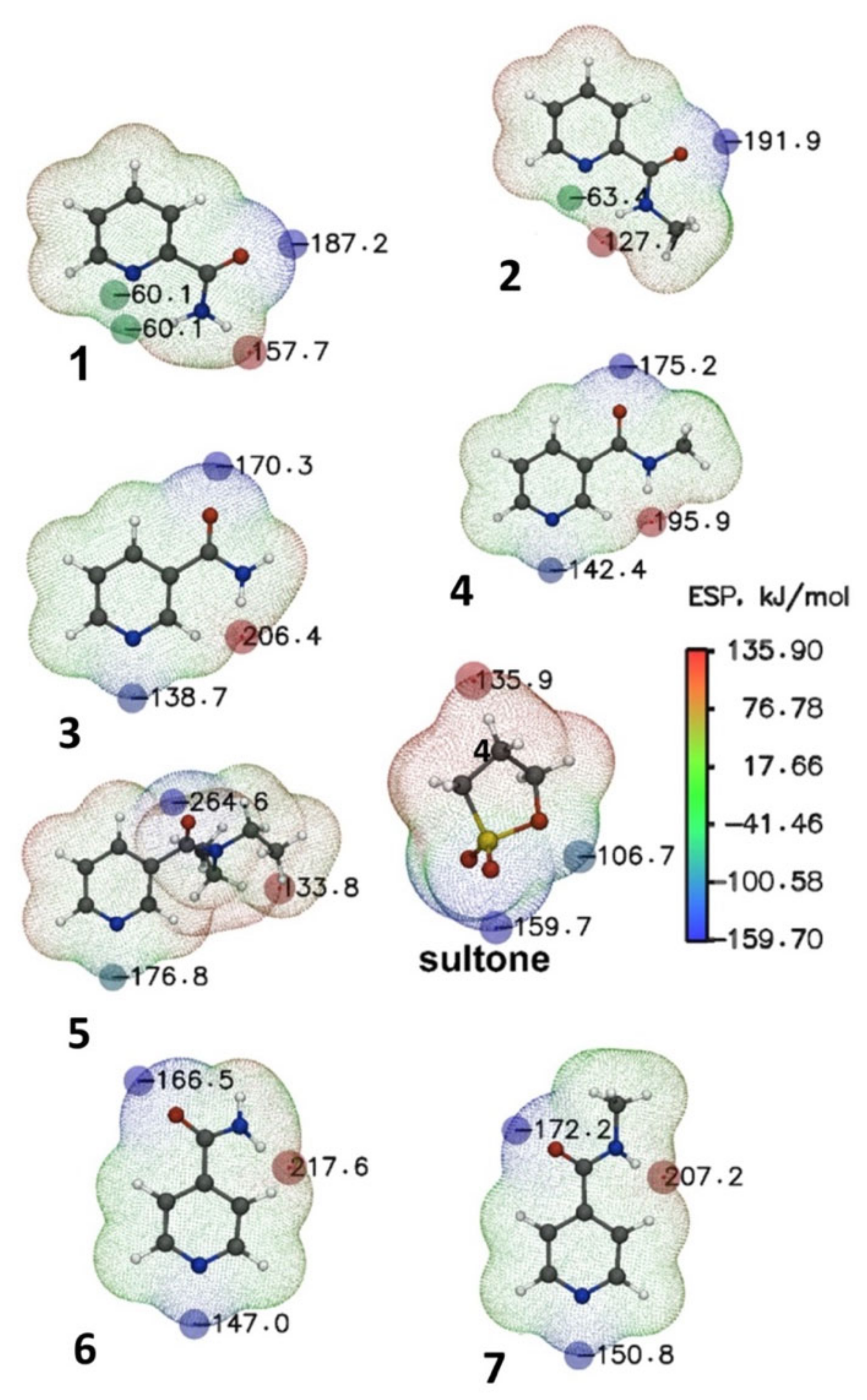
2.3.1. Electrostatic Potential Analysis
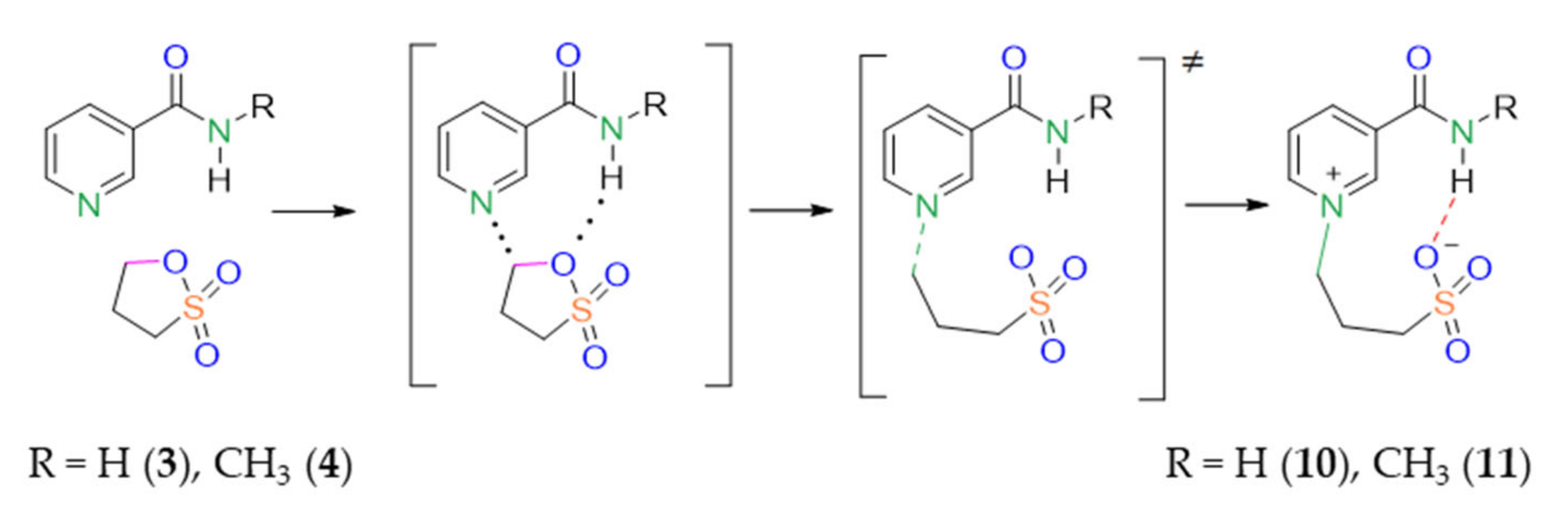
2.3.2. Transition States Search
2.3.3. Thermodynamic and Activation Parameters
2.4. Biological Activity
2.4.1. In Silico Evaluation of Possible Biological Activity
2.4.2. In Vitro Studies
3. Materials and Methods
3.1. General
3.2. Dynamic NMR
3.3. Synthesis
- 3-(2-(Carbamoylpyridinium-1-yl)propane-1-sulfonate (8). The reaction was performed in 5 mL of methanol. Obtained 0.5 g (24% yield) sulfonate, m.p. 187-188 °C. IR (solid, ν/cm−1): 1706 (C=O), 1602 (C=C pyridine), 1167, 1146, 1171 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 2.41–2.58 (m, 2H, C-CH2-C), 2.92–3.06 (m, 2H, CH2SO3), 4.76–4.91 (m, 2H, N-CH2), 8.38 (d, 2H, 3J = 6.4, H4, H6), 9.07 (d, 2H, 3J = 6.4, H3, H6); 13C-NMR (75.5 MHz, D2O, ppm): δ 26.22, 47.10, 60.36, 126.63, 148.80, 166.81; Anal. calcd. for C9H12N2O4S: C, 44.26; H, 4.92; N, 11.48; S, 13.11; Found: C, 43.98; H, 5.01; N, 11.30; S, 12.89.
- 3-(2-(Methylcarbamoyl) pyridinium-1-yl)propane-1-sulfonate (9). Reaction was performed in 5 mL of methanol. Recrystallized from ethanol-water, 60:1. Obtained 0.08 g (36% yield) sulfonate, m.p. 237–240 °C. IR (solid, ν/cm−1): 1677 (C=O), 1618 (C=C pyridine), 1210, 1150, 1036 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 2.35–2.49 (m, 2H, C-CH2-C), 2.97–3.03 (m, 2H, CH2SO3), 3.00 (s, 3H, CH3) 4.84 (t, 2H, 3J = 7.7, N-CH2), 8.66–8.70 (m, 2H, H4, H5), 8.65 (t, H, 3J = 7.5, H6), 9.01–9.04 (m, 1H, H3); 13C-NMR (75.5 MHz, D2O, ppm): δ 26.33, 26.62, 47.34, 58.11, 127.95, 129.53, 132.51, 146.78, 147.38, 162.07; Anal. calcd. for C10H14N2O4S: C, 44.93; H, 5.65; N, 10.48; S, 11.99; Found: C, 45.28; H, 5.70; N, 10.27; S, 12.38.
- 3-(3-Carbamoylpyridinium-1-yl)propane-1-sulfonate (10). (a) Reaction was performed in 5 mL of methanol. Recrystallized from ethanol-water, 2:1. Obtained 1.86 g (89% yield) sulfonate, m.p. > 300 °C (276–280 °C, ethanol-water, 2:1 [27]). IR (solid, ν/cm−1): 1685 (C=O), 1654 (C=C pyridine), 1211, 1156, 1140 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 2.36–2.46 (m, 2H, C-CH2-C), 2.93 (t, 2H, 3J = 7.1 CH2SO3), 4.77 (t, 2H, 3J = 7.3, N-CH2), 8.12 (t, 1H, 3J = 7.0, H5), 8.82 (d, 1H, 3J = 8.1, H6), 8.98 (d, 1H, 3J = 6.1, H4), 9.28 (s, 1H, H2); 13C-NMR (75.5 MHz, D2O, ppm): δ 26.08, 47.00, 60.48, 128.55, 134.11, 144.30, 144.49, 146.74, 165.79; Anal. calcd. for C9H12N2O4S: C, 44.26; H, 4.92; N, 11.48; S, 13.11; Found: C, 44.18; H, 4.96; N, 11.49; S, 13.06. (b) Reaction was performed in 5 mL of methanol. The mixture was heated under reflux for 4 h. Obtained 1.67 g (80% yield) sulfonate. The parameters of the 1H, 13C NMR, and FT-IR spectra of compounds obtained at different temperatures (a and b) have the same values.
- 3-(3-(Methylcarbamoyl)pyridinium-1-yl)propane-1-sulfonate (11). Reaction was performed in 5 mL of methanol. Recrystallized from ethanol–water, 5:1. Obtained 1.84 g (97% yield) sulfonate, m.p. 280–282 °C. IR (solid, ν/cm1): 1661, 1631 (C=O), 1593 (C=C pyridine), 1202, 1174, 1150 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 2.32–2.48 (m, 2H, C-CH2-C), 2.84–2.96 (m, 2H, CH3), 2.96 (s, 3H, CH2SO3), 4.76 (t, 2H, 3J = 7.8, N-CH2), 8.11 (t, 1H, 3J = 6.8, H5), 8.76 (d, 1H, 3J = 7.8, H6), 8.96 (d, 1H, 3J = 6.6, H4); 9.22 (s, 1H, H2); 13C-NMR (75.5 MHz, D2O, ppm): δ 26.03, 26.61, 46.97, 60.46, 128.54, 134.69, 143.87, 144.07, 146.45, 164.16; Anal. calcd. for C10H14N2O4S: C, 46.50; H, 5.46; N, 10.84; S, 12.41; Found: C, 46.09; H, 5.45; N, 10.66; S, 12.43.
- 3-(3-(Diethylcarbomoyl)pyridinium-1-yl)propane-1-sulfonate (12). The reaction was performed in 5 mL of methanol. Recrystallized from acetonitrile–ethanol, 10: 1. Obtained 1.52 g (59% yield) sulfonate, m.p. 197–199 °C. IR (solid, ν/cm−1): 1620 (C=O), 1201, 1182, 1037 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz):δ 1.07 (t, 3H, 3J = 7.2, CH2-CH3), 1.17 (t, 3H, 3J = 7.2, CH2-CH3), 2.36–2.46 (m, 2H, C-CH2-C), 2.86–2.96 (m, 2H, CH2SO3), 3.23 (q, 2H, 3J = 7.4, CH2-CH3), 3.50 (q, 2H, 3J = 7.4, CH2-CH3), 4.76 (t, 2H, 3J = 7.5, N-CH2), 8.12 (t, 1H, 3J = 7.1, H5), 8.09–8.15 (m, 1H, H6), 8.53–8.58 (m, 1H, H4), 9.04 (s, 1H, H2); 13C-NMR (75.5 MHz, D2O, ppm): δ 11.76, 13.02, 26.18, 40.72, 44.42, 47.00, 60.49, 129.06, 136.49, 142.31, 143.55, 145.61, 165.37. Anal. calcd. for C13H20N2O4S∙H2O: C, 49.04; H, 6.96; N, 8.80; S, 10.07; Found: C, 48.54; H, 6.71; N, 8.34.
- 3-(4-Carbamoylpyridinium-1-yl)propane-1-sulfonate (13). (a) Reaction was performed in 5 mL of methanol. Recrystallized from ethanol–water, 5:2. Obtained 1.20 g (58% yield) sulfonate, m.p. 283–284 °C. IR (solid, ν/cm−1): 1692 (C=O), 1626 (C=C pyridine), 1199, 1164, 1130 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 2.49–2.51 (m, 2H, C-CH2-C), 3.01 (t, 2H, 3J = 7.3, CH2SO3), 4.85 (t, 2H, 3J = 7.5, N-CH2), 8.38 (d, 2H, 3J = 6.3, H3, H5), 9.06 (d, 2H, 3J = 6.3, H2, H6); 13C-NMR (75.5 MHz, D2O, ppm): δ 26.22, 47.12, 60.34, 126, 145, 166.82, 34.99, 92.23, 111.47, 113.98, 119.10, 124.92, 139.13, 151.75, 159.51. Anal. calcd. for C9H12N2O4S: C, 44.25; H, 4.95; N, 11.47; S, 13.13; Found (%): C, 44.01; H, 4.90; N, 11.25; S, 13.04. (b) Reaction was performed in 5 mL of methanol. The mixture was heated under reflux for 4 h. Obtained 1.30 g (67% yield) sulfonate. The parameters of the 1H, 13C NMR, and FT-IR spectra of compounds obtained at different temperatures have the same values.
- 3-(4-(Methylcarbamoyl)pyridinium-1-yl)propane-1-sulfonate (14). The reaction was performed in 5 mL of methanol. Obtained 1.66 g (76% yield) sulfonate, m.p. > 300 °C. IR (solid, ν/cm−1): 1665 (C=O), 1645 (C=C pyridine), 1178, 1143, 1036 (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 2.33–2.44 (m, 2H, C-CH2-C), 2.83–2.95 (m, 2H, CH2SO3), 2.88 (s, 3H, CH3), 4.75 (t, 2H, 3J = 7.6, N-CH2), 8.24 (d, 2H, 3J = 6.1, H3, H5), 8.96 (d, 2H, 3J = 6.1, H2, H6); 13C-NMR (75.5 MHz, D2O, ppm): δ 26.10, 26.64, 46.96, 60.13, 126.22, 145.64, 149.09, 165.04. Anal. calcd. for C10H14N2O4S: C, 46.50; H, 5.46; N, 10.85; S, 12.41; Found: C, 46.15; H, 5.31; N, 10.53.
- 2-Carbamoylpyridin-1-ium 3-methoxypropane-1-sulfonate (15).(a) Reaction was performed in 5 mL of methanol. Mixture was heated under reflux for 4 h. Obtained 1.52 g (65% yield) complex (15), m.p. 153–155 °C (benzene-acetonitrile, 1:2). IR (solid, ν, cm−1): 1707 (C=O), 1602 (C=C pyridine), 1179, 1124, 1035, (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 1.88 (m, 2H, C-CH2-C), 2.81 (t, 2H, 3J = 7.1 CH2SO3), 3.46 (t, 2H, 3J = 7.3, CH3O-CH2), 3.23 (s, 3H, CH3O-CH2), 8.08 (t, 1H, 3J = 7.0, H5), 8.77 (d, 1H, 3J = 8.1, H6), 8.35 (d, 1H, 3J = 6.1, H4), 8.58 (t, 1H, 3J = 7.0, H5); 13C-NMR (75.5 MHz, D2O, ppm): δ 25.55, 26.56, 58.18, 60.36, 71.20 122.79, 127.52, 140.35, 147.68, 148.77, 163.63; Anal. calcd. for C10H16N2O5S: C, 43.47; H, 5.84; N, 10.14; S, 11.60; O, 28.95; Found: C, 42.85; H, 6.54; N, 9.09; S, 10.40; O, 31.13.(b) Reaction was performed in 5 mL of methanol-d4. Mixture was heated under reflux for 4 h. Obtained 2.21 g (93% yield) complex (15-d), m.p. 149-153 °C (benzene-acetonitrile, 1: 2). IR (solid, ν, sm−1): 1687 s (C=O), 1609 (C=C pyridine), 1184, 1128, 1035, (SO3). 1H-NMR (300.1 MHz, DMSO-d6, ppm, J/Hz): δ 1.75–1.87 (m, 2H, C-CH2-C), 2.56-2.66 (m, 2H, CH2SO3), 3.33 (t, 2H, 3J = 7.3, CD3O-CH2), 7.82 (m, 1H, H5), 8.71 (m, 1H, H6), 8.03 (m, 1H, H4), 8.29 (m, 1H, H5); 13C-NMR (75.5 MHz, DMSO-d6, ppm): δ 25.58, 48.79, 71.11, 123.43, 127.99, 141.45, 147.12, 148.02, 164.42.
- 2-(Methylcarbamoyl)pyridin-1-ium 3-methoxypropane-1-sulfonate (16). Reaction was performed in 5 mL of methanol. Mixture was heated under reflux for 4 h. Obtained 2.00 g (81% yield) oily complex, m.p. 60–64 °C. IR (solid, ν/cm−1): 1679 s (C=O), 1604 (C=C pyridine), 1212, 1108, 1032, (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 1.92 (m, 2H, C-CH2-C), 2.85 (t, 2H, 3J = 7.1 CH2SO3), 3.50 (t, 2H, 3J = 7.3, CH3O-CH2), 3.26 (s, 3H, CH3O-CH2), 2.95 (s, 3H, HNCH3), 8.12 (t, 1H, 3J = 7.0, H5), 8.81 (d, 1H, 3J = 8.1, H6), 8.34 (d, 1H, 3J = 6.1, H4), 8.63 (t, 1H, 3J = 7.0, H5); 13C-NMR (75.5 MHz, D2O, ppm): δ 24.03, 26.90, 47.78, 57.61, 70.47, 125.06, 129.57, 143.21, 146.96, 162.39. C11H18N2O5S HR-MS (ESI) calcd. for C6H6N2OCH3+: 137.16 [M + H+]+, found 137.07; calculated for C4H9O4S+: 153.17, found 153.02 [M + H+]+.
3.4. Calculation Details
3.5. X-ray Diffraction Studies
3.6. Cell Culture
3.6.1. Cell Viability Assay
3.6.2. Quantitative Real-Time PCR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Borozdenko, D.A.; Ezdoglian, A.A.; Shmigol, T.A.; Gonchar, D.I.; Lyakhmun, D.N.; Tarasenko, D.V.; Golubev, Y.V.; Cherkashova, E.A.; Namestnikova, D.D.; Gubskiy, I.L.; et al. A Novel Phenylpyrrolidine Derivative: Synthesis and Effect on Cognitive Functions in Rats with Experimental Ishemic Stroke. Molecules 2021, 26, 6124. [Google Scholar] [CrossRef] [PubMed]
- Borozdenko, D.A.; Lyakhmun, D.N.; Golubev, Y.V.; Tarasenko, D.V.; Kiseleva, N.M.; Negrebetsky, V.V. Study of the new 4-phenylpyrrolidinone-2 derivative pharmacokinetics and neuroprotective effect in the ischemic stroke animal model. Bull. RSMU 2020, 1, 49–56. [Google Scholar] [CrossRef]
- Borozdenko, D.A.; Shmigol, T.A.; Ezdoglian, A.A.; Gonchar, D.I.; Karpechenko, N.Y.; Lyakhmun, D.N.; Shagina, A.D.; Cherkashova, E.A.; Namestnikova, D.D.; Gubskiy, I.L.; et al. The Effect of a New N-hetero Cycle Derivative on Behavior and Inflammation against the Background of Ischemic Stroke. Molecules 2022, 27, 5488. [Google Scholar] [CrossRef] [PubMed]
- Shipov, A.G.; Kramarova, E.P.; Negrebetsky, V.V.; Pogozhikh, S.A.; Baukov, Y.I. Synthesis, Molecular and Crystal Structure of Phe Notropyl. Bull. RSMU 2006, 1, 56–61. [Google Scholar]
- DeBruler, C.; Hu, B.; Moss, J.; Liu, X.; Luo, J.; Sun, Y.; Liu, T.L. Designer Two-Electron Storage Viologen Anolyte Materials for Neutral Aqueous Organic Redox Flow Batteries. Chem 2017, 3, 961–978. [Google Scholar] [CrossRef]
- Ichikawa, T.; Kato, T.; Ohno, H. 3D Continuous Water Nanosheet as a Gyroid Minimal Surface Formed by Bicontinuous Cubic Liquid-Crystalline Zwitterions. J. Am. Chem. Soc. 2012, 134, 11354–11357. [Google Scholar] [CrossRef]
- Frederickson, M.; Vuillard, L.; Abell, C. Novel Selenium-Containing Non-Detergent Sulfobetaines. Tetrahedron Lett. 2003, 44, 7925–7928. [Google Scholar] [CrossRef]
- Bondock, S.; Khalifa, W.; Fadda, A.A. Synthesis and antimicrobial evaluation of some new thiazole, thiazolidinone and thiazoline derivatives starting from 1-chloro-3,4-dihydronaphthalene-2-carboxaldehyde. Eur. J. Med. Chem. 2007, 42, 948–954. [Google Scholar] [CrossRef]
- Fadda, A.A.; El-Mekawy, R.E.-D.; AbdelAal, M.T. Synthesis and antimicrobial evaluation of some new N-pyridinium, quinolinium, and isoquinolinium sulfonate derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1148–1154. [Google Scholar] [CrossRef]
- Mostafa, A.; El Bialy, S.; Bayoumi, W.; Ueda, Y.; Ikeda, M.; Kato, N.; Abdelal, A. Synthesis and In Vitro Activity of Pyrrolo[3,4-d]pyrimidine-2,5-diones as Potential Non-nucleoside HCV Inhibitors. Curr. Enzym. Inhib. 2016, 12, 170–176. [Google Scholar] [CrossRef]
- Tian, X.; Liu, T.; Fang, B.; Wang, A.; Zhang, M.; Hussain, S.; Luo, L.; Zhang, R.; Zhang, Q.; Wu, J.; et al. NeuN-Specific Fluorescent Probe Revealing Neuronal Nuclei Protein and Nuclear Acids Association in Living Neurons under STED Nanoscopy. ACS Appl. Mater. Interfaces 2018, 10, 31959–31964. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Acker, C.D.; Loew, L.M. Tethered Bichromophoric Fluorophore Quencher Voltage Sensitive Dyes. ACS Sens. 2018, 3, 2621–2628. [Google Scholar] [CrossRef]
- Khalili, A.; Nasr-Esfahani, M.; Mohammadpoor-Baltork, I.; Tangestaninejad, S.; Mirkhani, V.; Moghadam, M. Synthesis and characterization of 4-methyl-1-(3-sulfopropyl)pyridinium hydrogen sulfate as a new ionic liquid immobilized on silica nanoparticles: A recyclable nanocomposite ionic liquid for the production of various substituted phthalazine-ones. J. Mol. Liq. 2018, 253, 1–10. [Google Scholar] [CrossRef]
- Wang, P.; Meng, J.; Xu, M.; Yuan, T.; Yang, N.; Sun, T.; Zhang, Y.; Feng, X.; Cheng, B. A simple but efficient zwitterionization method towards cellulose membrane with superior antifouling property and biocompatibility. J. Membr. Sci. 2015, 492, 547–558. [Google Scholar] [CrossRef]
- An, L.; Yu, Y.H.; Chen, J.; Bae, J.H.; Youn, D.H.; Jeong, H.M.; Kim, Y.S. Synthesis and characterization of tailor-made zwitterionic lignin for resistance to protein adsorption. Ind. Crops Prod. 2021, 167, 113514. [Google Scholar] [CrossRef]
- Cai, M.; Leng, M.; Lu, A.; He, L.; Xie, X.; Huang, L.; Ma, Y.; Cao, J.; Chen, Y.; Luo, X. Synthesis of amphiphilic copolymers containing zwitterionic sulfobetaine as pH and redox responsive drug carriers. Colloids Surf. B Biointerfaces 2015, 126, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shechter, I.; Gu, P.; Jiang, G.; Onofrey, T.J.; Cann, R.O.; Castro, A.; Spencer, T.A. Sulfobetaine zwitterionic inhibitors of squalene synthase. Bioorganic Med. Chem. Lett. 1996, 6, 2585–2588. [Google Scholar] [CrossRef]
- Laschewsky, A.; Zerbe, I. Polymerizable and polymeric zwitterionic surfactants: 1. Synthesis and bulk properties. Polymer 1991, 32, 2070–2080. [Google Scholar] [CrossRef]
- Anton, P.; Köberle, P.; Laschewsky, A. Structure and properties of zwitterionic polysoaps: Functionalization by redox-switchable moieties. In Trends in Colloid and Interface Science VI; Steinkopff: Heidelberg, Germany, 1992. [Google Scholar]
- Anton, P.; Heinze, J.; Laschewsky, A. Redox-active monomeric and polymeric surfactants. Langmuir 1993, 9, 77–85. [Google Scholar] [CrossRef]
- Nakagawa, M.; Rikukawa, M.; Watanabe, M.; Sanui, K.; Ogata, N. Photochromic, Electrochemical, and Photoelectrochemical Properties of Novel Azopyridinium Derivatives. Bull. Chem. Soc. Jpn. 1997, 70, 737–744. [Google Scholar] [CrossRef]
- Suvorova, O.N.; Domrachev, G.A.; Shchupak, E.A.; Kudryavtseva, G.S.; Kirillov, A.I.; Zaitsev, A.A. Studies of porphyrin zinc compounds intercalated into V2O5 xerogel. Russ. Chem. Bull. 2009, 58, 2233–2239. [Google Scholar] [CrossRef]
- Gromov, S.P.; Vedernikov, A.I.; Kuz’mina, L.G.; Kondratuk, D.V.; Sazonov, S.K.; Strelenko, Y.A.; Alfimov, M.V.; Howard, J.A.K. Photocontrolled Molecular Assembler Based on Cucurbit[8]uril: [2+2]-Autophotocycloaddition of Styryl Dyes in the Solid State and in Water. Eur. J. Org. Chem. 2010, 2010, 2587–2599. [Google Scholar] [CrossRef]
- Mondal, S. Recent Developments in the Synthesis and Application of Sultones. Chem. Rev. 2012, 112, 5339–5355. [Google Scholar] [CrossRef]
- Mondal, S.; Debnath, S.; Das, B. Synthesis of seven-membered fused sultones by reductive Heck cyclization: An investigation for stereochemistry through DFT study. Tetrahedron 2015, 71, 476–486. [Google Scholar] [CrossRef]
- Mondal, S.; Debnath, S.; Pal, S.; Das, A. Synthesis of Uracil-, Coumarin- and Quinolone-Fused Benzosultams and Benzosultones. Synthesis 2015, 47, 3423–3433. [Google Scholar] [CrossRef]
- Willner, I.; Ford, W.E. Preparation of zwitterionic electron acceptors and donors. J. Heterocycl. Chem. 1983, 20, 1113–1114. [Google Scholar] [CrossRef]
- Enders, D.; Backes, M. Asymmetric synthesis of alpha, gamma-substituted gamma-alkoxy methyl sulfonates via diastereospecific ring-opening of sultones. ARKIVOC 2004, 2, 181–188. [Google Scholar] [CrossRef]
- Enders, D.; Iffland, D. Asymmetric Synthesis of α-Phenyl-γ-Hetero-Substituted Isopropyl Sulfonates via Diastereoselective Ring-Opening of γ-Sultones. Synthesis 2007, 2007, 1837–1840. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc.-Perkin Trans. 1 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Sjoberg, P.; Politzer, P. Use of the electrostatic potential at the molecular surface to interpret and predict nucleophilic processes. J. Phys. Chem. 1990, 94, 3959–3961. [Google Scholar] [CrossRef]
- Yokogawa, D.; Suda, K. Electrostatic Potential Fitting Method Using Constrained Spatial Electron Density Expanded with Preorthogonal Natural Atomic Orbitals. J. Phys. Chem. A 2020, 124, 9665–9673. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Gloriozova, T.A.; Lagunin, A.A.; Druzhilovskiy, D.S.; Rudik, A.V.; Stolbov, L.A.; Dmitriev, A.V.; Tarasova, O.A.; Ivanov, S.M.; et al. Computer-aided prediction of biological activity spectra for organic compounds: The possibilities and limitations. Russ. Chem. Bull. 2019, 68, 2143–2154. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Mol. Inform. 2011, 30, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Khandwekar, A.P.; Doble, M.; Patil, D.P.; Shouche, Y.S. The biocompatibility of sulfobetaine engineered poly (ethylene terephthalate) by surface entrapment technique. J. Biomater. Appl. 2010, 25, 119–143. [Google Scholar] [CrossRef]
- Liu, P.; Song, J. Sulfobetaine as a zwitterionic mediator for 3D hydroxyapatite mineralization. Biomaterials 2013, 34, 2442–2454. [Google Scholar] [CrossRef]
- Pu, Y.; Hou, Z.; Khin, M.M.; Zamudio-Vázquez, R.; Poon, K.L.; Duan, H.; Chan-Park, M.B. Synthesis and Antibacterial Study of Sulfobetaine/Quaternary Ammonium-Modified Star-Shaped Poly[2-(dimethylamino)ethyl methacrylate]-Based Copolymers with an Inorganic Core. Biomacromolecules 2017, 18, 44–55. [Google Scholar] [CrossRef]
- Racovita, S.; Trofin, M.A.; Loghin, D.F.; Zaharia, M.M.; Bucatariu, F.; Mihai, M.; Vasiliu, S. Polybetaines in Biomedical Applications. Int. J. Mol. Sci. 2021, 22, 9321. [Google Scholar] [CrossRef]
- Muhammad, W.; Zhai, Z.; Wang, S.; Gao, C. Inflammation-modulating nanoparticles for pneumonia therapy. WIREs Nanomed. Nanobiotechnol. 2022, 14, e1763. [Google Scholar] [CrossRef] [PubMed]
- Castañar, L.; Saurí, J.; Williamson, R.T.; Virgili, A.; Parella, T. Pure in-phase heteronuclear correlation NMR experiments. Angew. Chem. Int. Ed. Engl. 2014, 53, 8379–8382. [Google Scholar] [CrossRef] [PubMed]
- Rule, G.S.; Hitchens, T.K. Fundamentals of Protein Nmr Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Schafer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using atoms in molecules. Chem. Phys. Lett. 1992, 194, 1–8. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Reagent Structure | Product | Product Structure | Yield %, Average ± SD (Number of Runs). | Yield %, Best Attempt |
|---|---|---|---|---|---|
| 1 |  | 8 |  | 12 ± 11 (3) | 24 |
| 2 |  | 9 |  | 16 ± 15 (9) | 44 |
| 3 |  | 10 |  | 68 ± 16 (4) | 89 |
| 4 |  | 11 |  | 82 ± 12 (4) | 97 |
| 5 |  | 12 |  | 58 ± 3 (4) | 62 |
| 6 |  | 13 |  | 57 ± 8 (4) | 67 |
| 7 |  | 14 |  | 64 ± 16 (2) | 76 |
| Reagent | ESP (N), kJ/mol See Figure 2 | Product | Gas Phase, T = 298.15 | Methanol, T = 298.15 | |||
|---|---|---|---|---|---|---|---|
| ΔH#, kJ∙mol−1 | ΔrH°, kJ∙mol−1 | ΔH#, kJ∙mol−1 | ΔrH°, kJ∙mol−1 | ΔrG°, kJ∙mol−1 | |||
| 1 | −60.10 | 8 | 149.0 | −1.78 | 96.8 | −58.1 | −7.5 |
| 2 | −63.30 | 9 | 149.0 | −2.28 | 95.7 | −59.9 | −10.3 |
| 3 | −156.70 | 10 | 129.1 | −41.3 | 74.6 | −92.8 | −42.9 |
| 4 | −142.40 | 11 | 127.6 | −44.2 | 75.8 | −92.7 | −43.0 |
| 5 | −176.80 | 12 | 122.4 | −25.7 | 69.7 | −99.9 | −47.6 |
| 6 | −147.00 | 13 | 131.2 | −10.6 | 74.7 | −95.8 | −48.2 |
| 7 | −150.80 | 14 | 130.0 | −12.1 | 71.8 | −95.6 | −45.5 |
| No | Pa > Pi | Pa > 30% | Pa > 50% (IDs) | Pa > 70% (IDs) | Activity |
|---|---|---|---|---|---|
| 1 | 7 | 7 | 7 | 7 | Antiischemic, cerebral |
| 2 | 7 | 7 | 4 (8, 10, 11, 13) | 1 (14) | Phobic disorders treatment |
| 3 | 7 | 7 | 7 | 0 | Antianginal |
| 4 | 7 | 7 | 5 (10–14) | 0 | Antineoplastic (liver cancer) |
| 5 | 7 | 7 | 3 (8, 13, 14) | 0 | Antineoplastic (myeloid leukemia) |
| 6 | 7 | 7 | 3 (9, 11, 14) | 0 | Mucomembranous protector |
| 7 | 7 | 7 | 3 (11, 13, 14) | 0 | Kidney function stimulant |
| 8 | 7 | 7 | 1 (13) | 0 | Cytoprotectant |
| 9 | 6 | 5 | 1 (12) | 0 | Analeptic |
| ID | p.o., mg/kg (Class) | i.v., mg/kg (Class) | i.p., mg/kg (Class) | s.c., mg/kg (Class) | Cytotoxicity, Pa > 0.3 Pa Pi Tumor Cell Line, Type |
|---|---|---|---|---|---|
| 8 | 2346 (5 class) | 588 (5 class) | 768 (5 class) | 1336 (5 class) | 0.323 0.116 RKO (Colon carcinoma) |
| 9 | 809 (4 class) | 233 (4 class) | 514 (5 class) | 236 (4 class) | 0.400 0.099 Kasumi 1 (Leukemia) |
| 10 | 705 (4 class) | 619 (5 class) | 512 (5 class) | 1377 (5 class) | 0.312 0.136 RKO (Colon carcinoma) |
| 11 | 760 (4 class) | 499 (5 class) | 567 (5 class) | 660 (4 class) | 0.320 0.168 Kasumi 1 (Leukemia) |
| 12 | 1195 (4 class) | 235 (4 class) | 299 (4 class) | 447 (4 class) | - |
| 13 | 738 (4 class) | 616 (5 class) | 594 (5 class) | 858 (4 class) | 0.316 0.097 SJSA-1 (Osteosarcoma) 0.327 0.110 RKO (Colon carcinoma) |
| 14 | 946 (4 class) | 566 (5 class) | 537 (5 class) | 865 (4 class) | 0.344 0.143 Kasumi 1 (Leukemia) |
| Cell Lines | ||||
|---|---|---|---|---|
| A549 | HaCat | MCF7 | NKE | |
| Compound 8, mg/mL | 4.0 ± 2.75 | 3.8 ± 1.21 | 3.2 ± 1.80 | 4.1 ± 1.64 |
| Compound 9, mg/mL | 2.5 ± 1.02 | 3.2 ± 1.43 | 3.0 ± 2.01 | 2.4 ± 0.86 |
| Compound 10, mg/mL | 8.3 ± 1.91 | 8.5 ± 2.25 | 8.6 ± 1.60 | 9.5 ± 1.60 |
| Compound 11, mg/mL | 7.6 ± 2.33 | 8.3 ± 1.25 | 7.9 ± 2.32 | 8.3 ± 3.0 |
| Compound 12, mg/mL | 8.9 ± 2.41 | 9.3 ± 2.47 | 8.7 ± 2.15 | 8.3 ± 2.76 |
| Compound 13, mg/mL | 6.9 ± 2.01 | 7.3 ± 2.09 | 6.7 ± 2.11 | 6.2 ± 2.64 |
| Gene | Forward Primer | Reverse Rimer |
|---|---|---|
| COX2 | 5′-TTGGAGGCGAAGTGGGTTTT-3′ | 5′-TGGCTGTTTTGGTAGGCTGT-3′ |
| Il1b | 5′-CAAGGGGACATTAGGCAGCA-3′ | 5′-ATGACCAATTCATCCCCCACA-3′ |
| iNos | 5′-AGGGTCACAACTTTACAGGGAG-3′ | 5′-CAGCTCAGTCCCTTCACCAA-3′ |
| Rpl27 | 5′-AGCGATCCAAGATCAAGTCCTTT-3′ | 5′-CAACAGTCTTGTCCAAGGGGATA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramarova, E.P.; Borisevich, S.S.; Khamitov, E.M.; Korlyukov, A.A.; Dorovatovskii, P.V.; Shagina, A.D.; Mineev, K.S.; Tarasenko, D.V.; Novikov, R.A.; Lagunin, A.A.; et al. Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity. Molecules 2022, 27, 7542. https://doi.org/10.3390/molecules27217542
Kramarova EP, Borisevich SS, Khamitov EM, Korlyukov AA, Dorovatovskii PV, Shagina AD, Mineev KS, Tarasenko DV, Novikov RA, Lagunin AA, et al. Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity. Molecules. 2022; 27(21):7542. https://doi.org/10.3390/molecules27217542
Chicago/Turabian StyleKramarova, Eugene P., Sophia S. Borisevich, Edward M. Khamitov, Alexander A. Korlyukov, Pavel V. Dorovatovskii, Anastasia D. Shagina, Konstantin S. Mineev, Dmitri V. Tarasenko, Roman A. Novikov, Alexey A. Lagunin, and et al. 2022. "Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity" Molecules 27, no. 21: 7542. https://doi.org/10.3390/molecules27217542
APA StyleKramarova, E. P., Borisevich, S. S., Khamitov, E. M., Korlyukov, A. A., Dorovatovskii, P. V., Shagina, A. D., Mineev, K. S., Tarasenko, D. V., Novikov, R. A., Lagunin, A. A., Boldyrev, I., Ezdoglian, A. A., Karpechenko, N. Y., Shmigol, T. A., Baukov, Y. I., & Negrebetsky, V. V. (2022). Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity. Molecules, 27(21), 7542. https://doi.org/10.3390/molecules27217542