
Erythrosin B as a New Photoswitchable Spin Label for Light-Induced Pulsed EPR Dipolar Spectroscopy
 , , and
, , and
Abstract
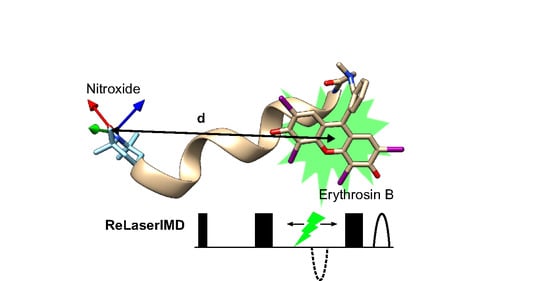
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Sample Preparation
3.2. Spectroscopy
3.3. DFT Calculations
3.4. Orientation-Dependent Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schiemann, O.; Prisner, T.F. Long-Range Distance Determinations in Biomacromolecules by EPR Spectroscopy. Q. Rev. Biophys. 2007, 40, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef]
- Borbat, P.P.; Freed, J.H. Pulse Dipolar Electron Spin Resoance: Distance Measurements. In Structural Information from Spin-Labels and Indtrinsic Paramagnetic Centres in the Biosciences; Timmel, C.R., Harmer, J.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–82. [Google Scholar]
- Jeschke, G. The Contribution of Modern EPR to Structural Biology. Emerg. Top. Life Sci. 2018, 2, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, D. Pulse EPR in Biological Systems—Beyond the Expert’s Courtyard. J. Magn. Reson. 2019, 306, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Denysenkov, V.P.; Prisner, T.F.; Stubbe, J.; Bennati, M. High-Field Pulsed Electron-Electron Double Resonance Spectroscopy to Determine the Orientation of the Tyrosyl Radicals in Ribonucleotide Reductase. Proc. Natl. Acad. Sci. USA 2006, 103, 13386–13390. [Google Scholar] [CrossRef]
- Bode, B.E.; Plackmeyer, J.; Prisner, T.F.; Schiemann, O. PELDOR Measurements on a Nitroxide-Labeled Cu(II) Porphyrin: Orientation Selection, Spin-Density Distribution, and Conformational Flexibility. J. Phys. Chem. A 2008, 112, 5064–5073. [Google Scholar] [CrossRef]
- Lovett, J.E.; Bowen, A.M.; Timmel, C.R.; Jones, M.W.; Dilworth, J.R.; Caprotti, D.; Bell, S.G.; Wong, L.L.; Harmer, J. Structural Information from Orientationally Selective DEER Spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 6840–6848. [Google Scholar] [CrossRef]
- Bowen, A.M.; Tait, C.E.; Timmel, C.R.; Harmer, J.R. Orientation-Selective DEER Using Rigid Spin Labels, Cofactors, Metals, and Clusters. In Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences; Timmel, C.R., Harmer, J.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 283–328. [Google Scholar]
- Gamble Jarvi, A.; Ranguelova, K.; Ghosh, S.; Weber, R.T.; Saxena, S. On the Use of Q-Band Double Electron-Electron Resonance to Resolve the Relative Orientations of Two Double Histidine-Bound Cu2+ Ions in a Protein. J. Phys. Chem. B 2018, 122, 10669–10677. [Google Scholar] [CrossRef]
- Banham, J.E.; Baker, C.M.; Ceola, S.; Day, I.J.; Grant, G.H.; Groenen, E.J.J.; Rodgers, C.T.; Jeschke, G.; Timmel, C.R. Distance Measurements in the Borderline Region of Applicability of CW EPR and DEER: A Model Study on a Homologous Series of Spin-Labelled Peptides. J. Magn. Reson. 2008, 191, 202–218. [Google Scholar] [CrossRef]
- Schmidt, T.; Wälti, M.A.; Baber, J.L.; Hustedt, E.J.; Clore, G.M. Long Distance Measurements up to 160 Å in the GroEL Tetradecamer Using Q-Band DEER EPR Spectroscopy. Angew. Chemie Int. Ed. 2016, 55, 15905–15909. [Google Scholar] [CrossRef]
- Joseph, B.; Tormyshev, V.M.; Rogozhnikova, O.Y.; Akhmetzyanov, D.; Bagryanskaya, E.G.; Prisner, T.F. Selective High-Resolution Detection of Membrane Protein–Ligand Interaction in Native Membranes Using Trityl-Nitroxide PELDOR. Angew. Chemie Int. Ed. 2016, 55, 11538–11542. [Google Scholar] [CrossRef]
- Jassoy, J.J.; Berndhäuser, A.; Duthie, F.; Kühn, S.P.; Hagelueken, G.; Schiemann, O. Versatile Trityl Spin Labels for Nanometer Distance Measurements on Biomolecules In Vitro and within Cells. Angew. Chemie Int. Ed. 2017, 56, 177–181. [Google Scholar] [CrossRef]
- Goldfarb, D. Gd3+ Spin Labeling for Distance Measurements by Pulse EPR Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 9685–9699. [Google Scholar] [CrossRef]
- Banerjee, D.; Yagi, H.; Huber, T.; Otting, G.; Goldfarb, D. Nanometer-Range Distance Measurement in a Protein Using Mn2+ Tags. J. Phys. Chem. Lett. 2012, 3, 157–160. [Google Scholar] [CrossRef]
- Yang, Z.; Ji, M.; Cunningham, T.F.; Saxena, S. Cu2 + as an ESR Probe of Protein Structure and Function. Methods Enzymol. 2015, 563, 459–481. [Google Scholar] [CrossRef]
- Di Valentin, M.; Albertini, M.; Zurlo, E.; Gobbo, M.; Carbonera, D. Porphyrin Triplet State as a Potential Spin Label for Nanometer Distance Measurements by Peldor Spectroscopy. J. Am. Chem. Soc. 2014, 136, 6582–6585. [Google Scholar] [CrossRef] [PubMed]
- Dal Farra, M.G.; Richert, S.; Martin, C.; Larminie, C.; Gobbo, M.; Bergantino, E.; Timmel, C.R.; Bowen, A.M.; Di Valentin, M. Light-Induced Pulsed EPR Dipolar Spectroscopy on a Paradigmatic Hemeprotein. ChemPhysChem 2019, 20, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Krumkacheva, O.A.; Timofeev, I.O.; Politanskaya, L.V.; Polienko, Y.F.; Tretyakov, E.V.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Bagryanskaya, E.G.; et al. Triplet Fullerenes as Prospective Spin Labels for Nanoscale Distance Measurements by Pulsed Dipolar EPR Spectroscopy. Angew. Chemie 2019, 131, 13405–13409. [Google Scholar] [CrossRef]
- Sannikova, N.E.; Timofeev, I.O.; Chubarov, A.S.; Lebedeva, N.S.; Semeikin, A.S.; Kirilyuk, I.A.; Tsentalovich, Y.P.; Fedin, M.V.; Bagryanskaya, E.G.; Krumkacheva, O.A. Application of EPR to Porphyrin-Protein Agents for Photodynamic Therapy. J. Photochem. Photobiol. B Biol. 2020, 211, 112008. [Google Scholar] [CrossRef]
- Williams, L.; Tischlik, S.; Scherer, A.; Fischer, J.W.A.; Drescher, M. Site-Directed Attachment of Photoexcitable Spin Labels for Light-Induced Pulsed Dipolar Spectroscopy. Chem. Commun. 2020, 56, 14669–14672. [Google Scholar] [CrossRef]
- Timofeev, I.O.; Politanskaya, L.V.; Tretyakov, E.V.; Polienko, Y.F.; Tormyshev, V.M.; Bagryanskaya, E.; Krumkacheva, O.A.; Fedin, M.V. Fullerene-Based Triplet Spin Labels: Methodology Aspects for Pulsed Dipolar EPR Spectroscopy. Phys. Chem. Chem. Phys. 2022, 24, 4475–4484. [Google Scholar] [CrossRef]
- Kay, C.W.M.; Di Valentin, M.; Möbius, K. A Time-Resolved Electron Nuclear Double Resonance (ENDOR) Study of the Photoexcited Triplet State of Free-Base Tetraphenylporphyrin. Sol. Energy Mater. Sol. Cells 1995, 38, 111–118. [Google Scholar] [CrossRef]
- Lubitz, W.; Lendzian, F.; Bittl, R. Radicals, Radical Pairs and Triplet States in Photosynthesis. Acc. Chem. Res. 2002, 35, 313–320. [Google Scholar] [CrossRef]
- Di Valentin, M.; Albertini, M.; Dal Farra, M.G.; Zurlo, E.; Orian, L.; Polimeno, A.; Gobbo, M.; Carbonera, D. Light-Induced Porphyrin-Based Spectroscopic Ruler for Nanometer Distance Measurements. Chem. A Eur. J. 2016, 22, 17204–17214. [Google Scholar] [CrossRef]
- Hintze, C.; Bücker, D.; Domingo Köhler, S.; Jeschke, G.; Drescher, M. Laser-Induced Magnetic Dipole Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 2204–2209. [Google Scholar] [CrossRef]
- Bieber, A.; Bücker, D.; Drescher, M. Light-Induced Dipolar Spectroscopy—A Quantitative Comparison between LiDEER and LaserIMD. J. Magn. Reson. 2018, 296, 29–35. [Google Scholar] [CrossRef]
- Bertran, A.; Henbest, K.B.; De Zotti, M.; Gobbo, M.; Timmel, C.R.; Di Valentin, M.; Bowen, A.M. Light-Induced Triplet−Triplet Electron Resonance Spectroscopy. J. Phys. Chem. Lett. 2021, 12, 80–85. [Google Scholar] [CrossRef]
- Bowen, A.M.; Bertran, A.; Henbest, K.B.; Gobbo, M.; Timmel, C.R.; Valentin, M. Di Orientation-Selective and Frequency-Correlated Light-Induced Pulsed Dipolar Spectroscopy. J. Phys. Chem. Lett. 2021, 12, 3819–3826. [Google Scholar] [CrossRef]
- Dal Farra, M.G.; Ciuti, S.; Gobbo, M.; Carbonera, D.; Di Valentin, M. Triplet-State Spin Labels for Highly Sensitive Pulsed Dipolar Spectroscopy. Mol. Phys. 2019, 117, 2673–2687. [Google Scholar] [CrossRef]
- Di Valentin, M.; Dal Farra, M.G.; Galazzo, L.; Albertini, M.; Schulte, T.; Hofmann, E.; Carbonera, D. Distance Measurements in Peridinin-Chlorophyll a-Protein by Light-Induced PELDOR Spectroscopy. Analysis of Triplet State Localization. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1909–1916. [Google Scholar] [CrossRef]
- Bertran, A.; Barbon, A.; Bowen, A.M.; Di Valentin, M. Light-Induced Pulsed Dipolar EPR Spectroscopy for Distance and Orientation Analysis. In Methods in Enzymology; Elsevier Inc.: Amsterdam, The Netherlands, 2022; Volume 666, pp. 171–231. ISBN 9780323999748. [Google Scholar]
- Serrer, K.; Matt, C.; Sokolov, M.; Kacprzak, S.; Schleicher, E.; Weber, S. Application of Commercially Available Fluorophores as Triplet Spin Probes in EPR Spectroscopy. Mol. Phys. 2019, 117, 2688–2699. [Google Scholar] [CrossRef]
- Marko, A.; Prisner, T.F. An Algorithm to Analyze PELDOR Data of Rigid Spin Label Pairs. Phys. Chem. Chem. Phys. 2013, 15, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis2006—A Comprehensive Software Package for Analyzing Pulsed ELDOR Data. Appl. Magn. Reson. 2006, 30, 473–498. [Google Scholar] [CrossRef]
- Meyer, V.; Swanson, M.A.; Clouston, L.J.; Boratyński, P.J.; Stein, R.A.; McHaourab, H.S.; Rajca, A.; Eaton, S.S.; Eaton, G.R. Room-Temperature Distance Measurements of Immobilized Spin-Labeled Protein by DEER/PELDOR. Biophys. J. 2015, 108, 1213–1219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Savitsky, A.; Plato, M.; Möbius, K. The Temperature Dependence of Nitroxide Spin-Label Interaction Parameters: A High-Field EPR Study of Intramolecular Motional Contributions. Appl. Magn. Reson. 2010, 37, 415–434. [Google Scholar] [CrossRef]
- Afanasyeva, E.F.; Syryamina, V.N.; De Zotti, M.; Formaggio, F.; Toniolo, C.; Dzuba, S.A. Peptide Antibiotic Trichogin in Model Membranes: Self-Association and Capture of Fatty Acids. Biochim. Biophys. Acta Biomembr. 2019, 1861, 524–531. [Google Scholar] [CrossRef]
- De Zotti, M.; Sella, L.; Bolzonello, A.; Gabbatore, L.; Peggion, C.; Bortolotto, A.; Elmaghraby, I.; Tundo, S.; Favaron, F. Targeted Amino Acid Substitutions in a Trichoderma Peptaibol Confer Activity against Fungal Plant Pathogens and Protect Host Tissues from Botrytis Cinerea Infection. Int. J. Mol. Sci. 2020, 21, 7521. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertran, A.; Morbiato, L.; Aquilia, S.; Gabbatore, L.; De Zotti, M.; Timmel, C.R.; Di Valentin, M.; Bowen, A.M. Erythrosin B as a New Photoswitchable Spin Label for Light-Induced Pulsed EPR Dipolar Spectroscopy. Molecules 2022, 27, 7526. https://doi.org/10.3390/molecules27217526
Bertran A, Morbiato L, Aquilia S, Gabbatore L, De Zotti M, Timmel CR, Di Valentin M, Bowen AM. Erythrosin B as a New Photoswitchable Spin Label for Light-Induced Pulsed EPR Dipolar Spectroscopy. Molecules. 2022; 27(21):7526. https://doi.org/10.3390/molecules27217526
Chicago/Turabian StyleBertran, Arnau, Laura Morbiato, Sara Aquilia, Laura Gabbatore, Marta De Zotti, Christiane R. Timmel, Marilena Di Valentin, and Alice M. Bowen. 2022. "Erythrosin B as a New Photoswitchable Spin Label for Light-Induced Pulsed EPR Dipolar Spectroscopy" Molecules 27, no. 21: 7526. https://doi.org/10.3390/molecules27217526
APA StyleBertran, A., Morbiato, L., Aquilia, S., Gabbatore, L., De Zotti, M., Timmel, C. R., Di Valentin, M., & Bowen, A. M. (2022). Erythrosin B as a New Photoswitchable Spin Label for Light-Induced Pulsed EPR Dipolar Spectroscopy. Molecules, 27(21), 7526. https://doi.org/10.3390/molecules27217526