


Copper-Catalyzed Homocoupling of Boronic Acids: A Focus on B-to-Cu and Cu-to-Cu Transmetalations
 ,
,
Abstract
1. Introduction
2. Results and Discussion
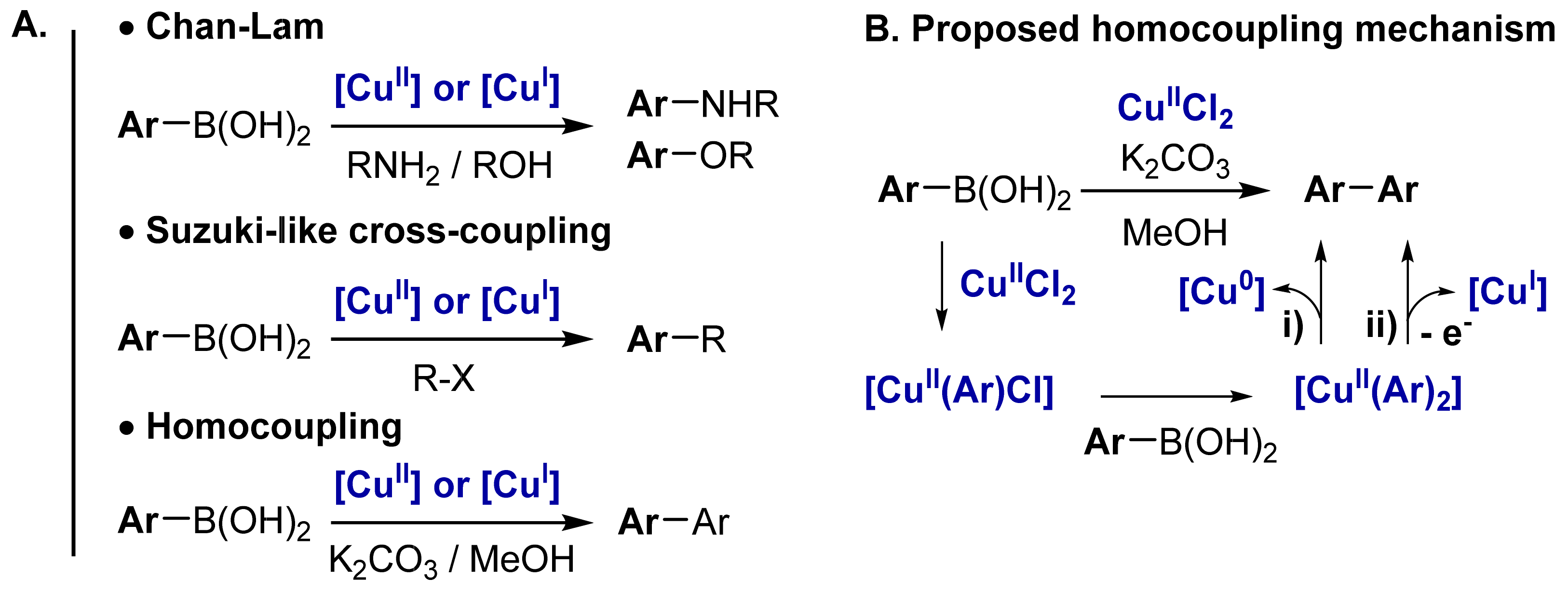
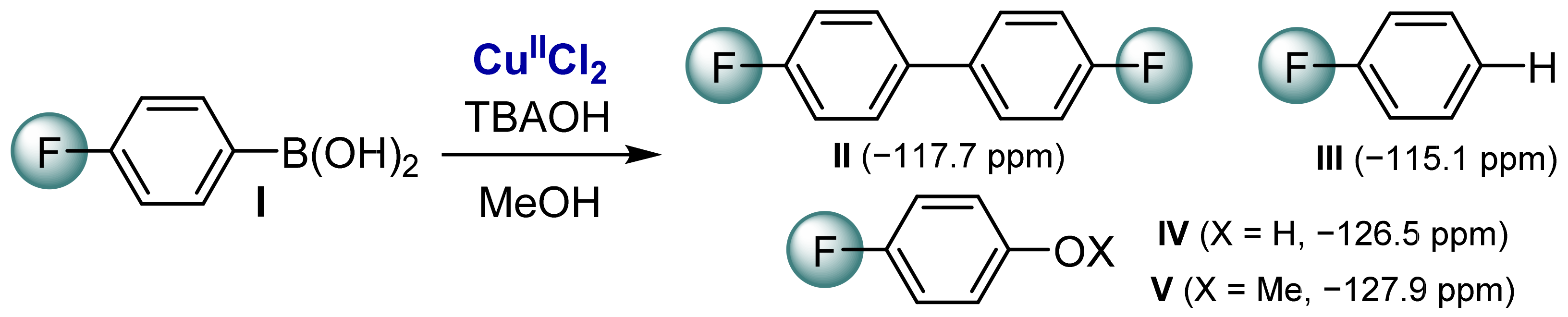
2.1. Cu-Catalyzed Homocoupling Reaction: Effect of Cu on Selectivity
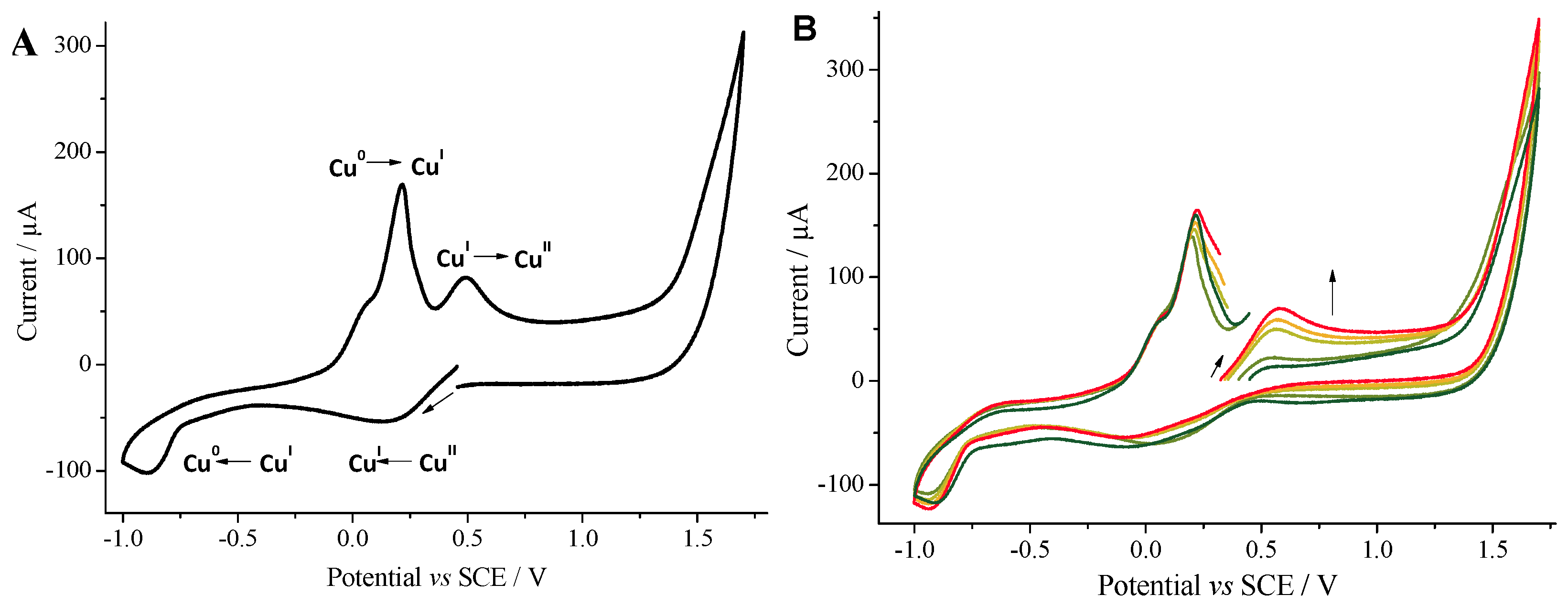
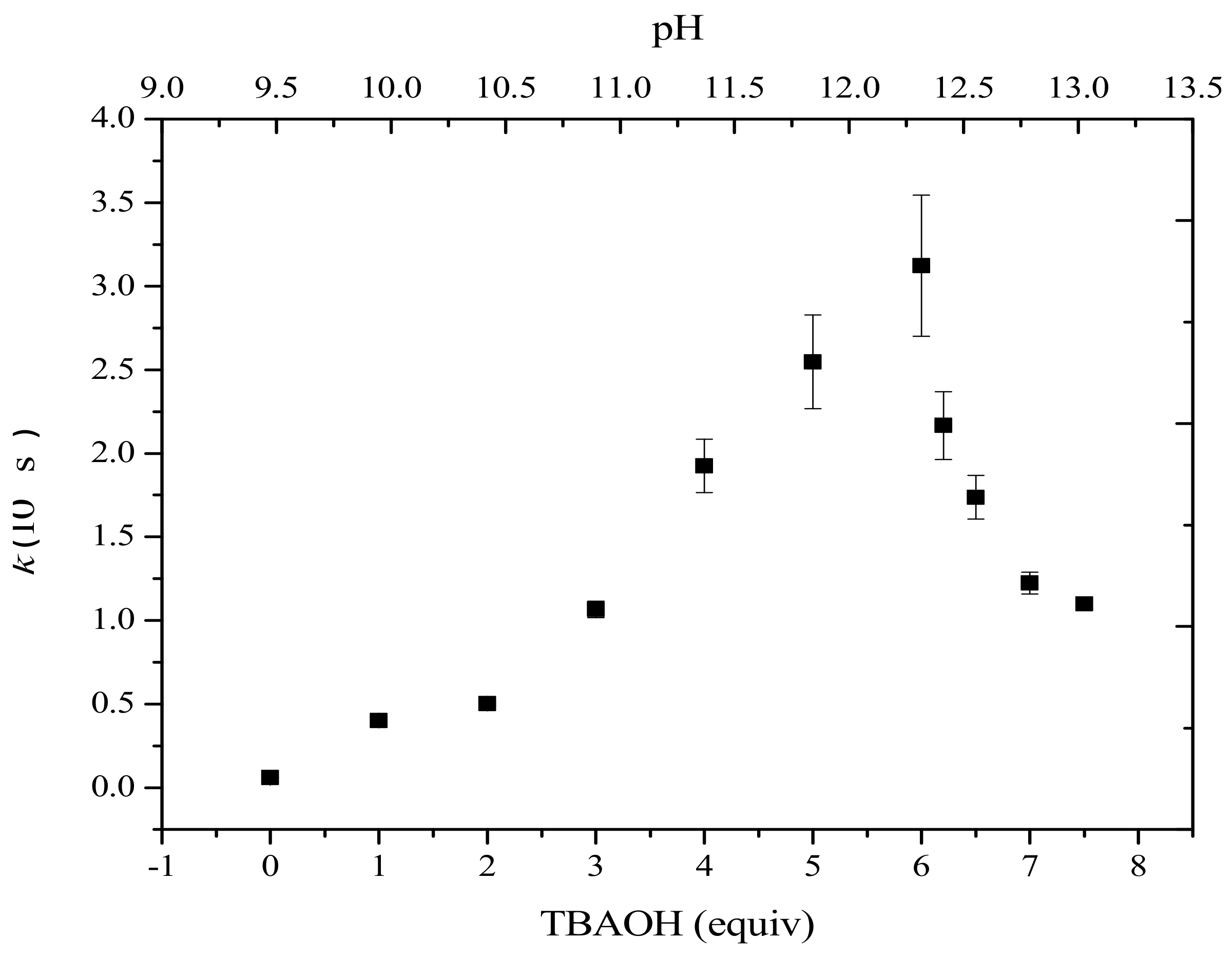
2.2. Cyclic Voltammetry Monitoring of the Homocoupling Reaction
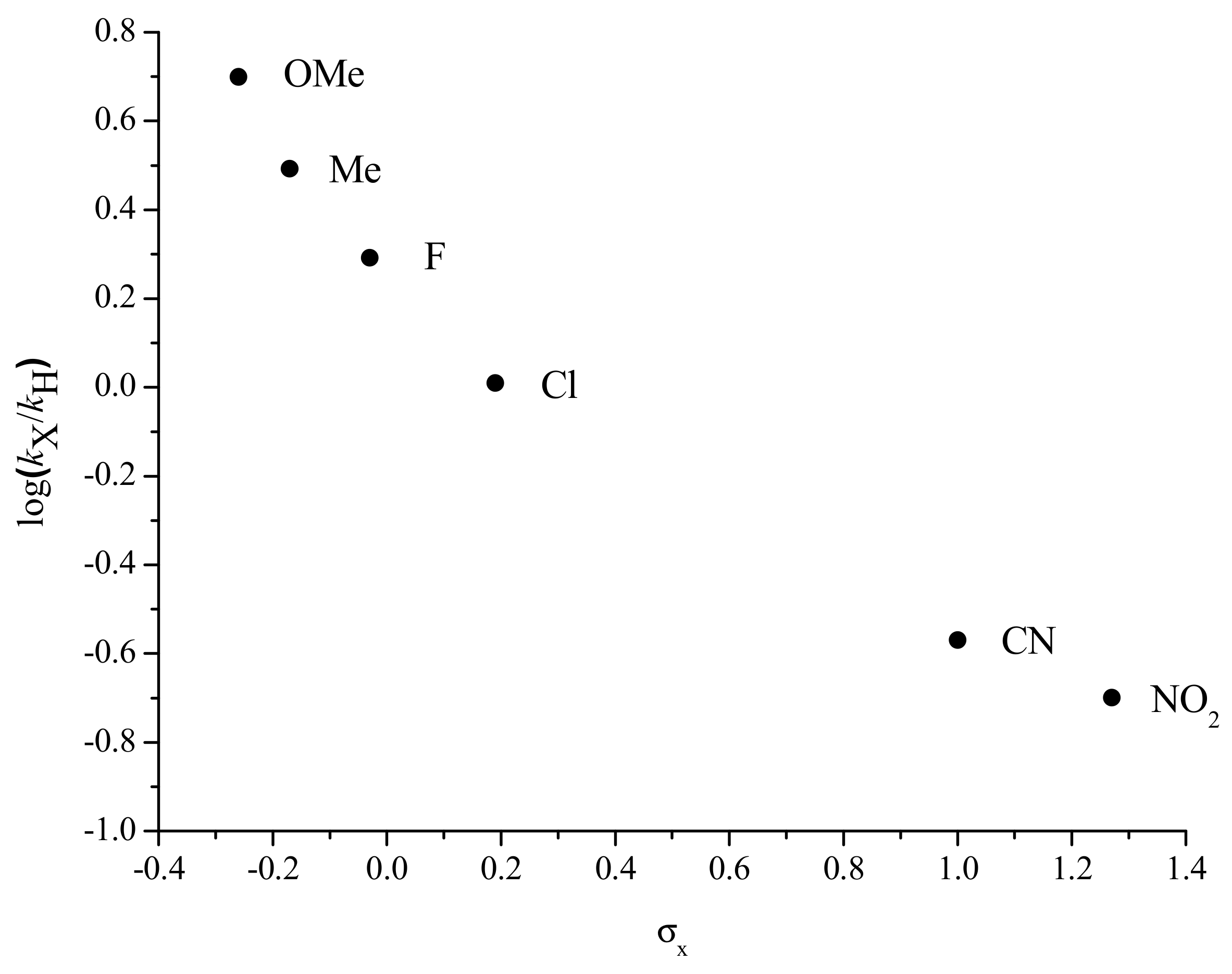
2.3. Electronic Effects on the Kinetics of the Reaction: Hammett Plot
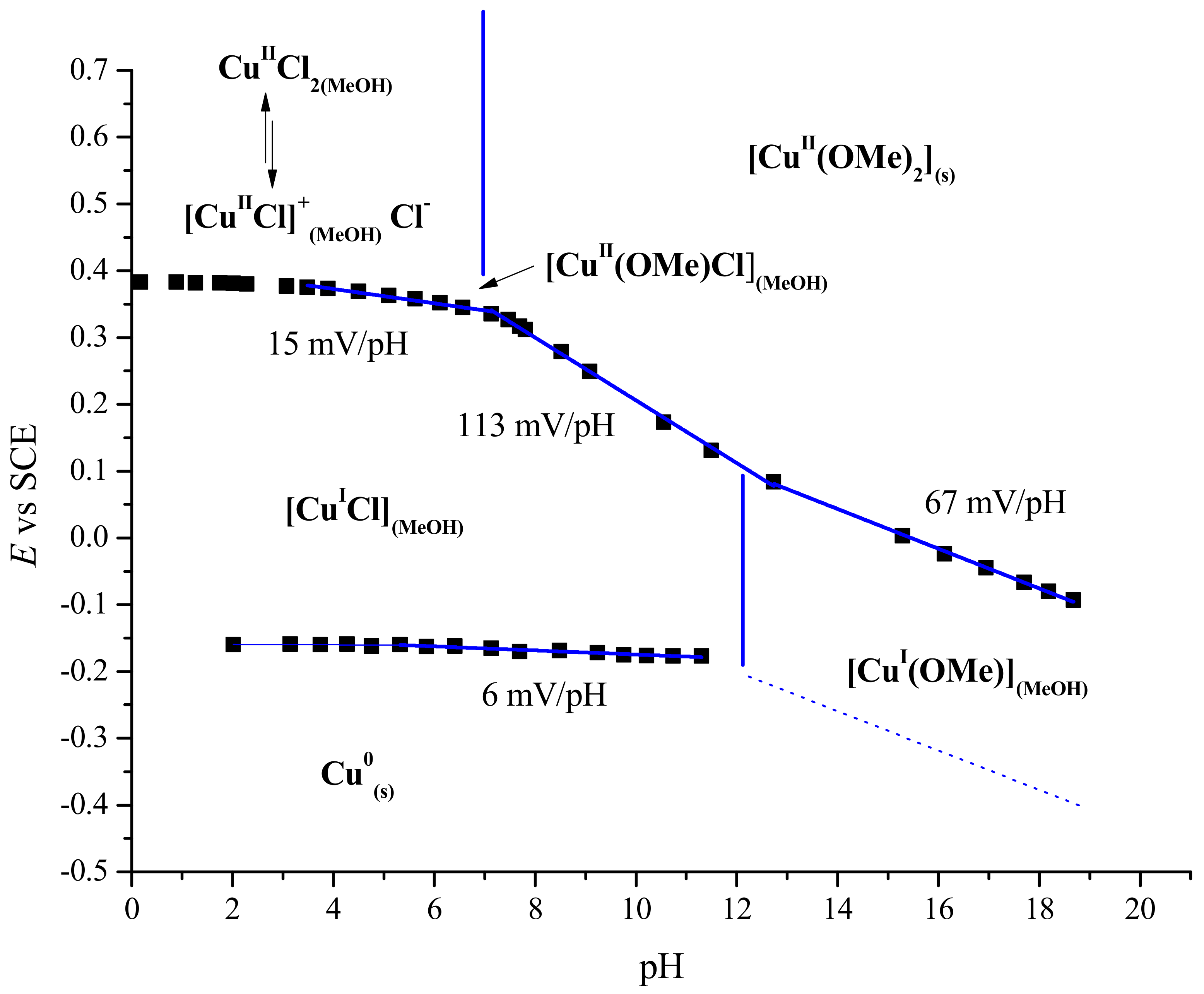
2.4. Structure of Cu(II) in Methanol
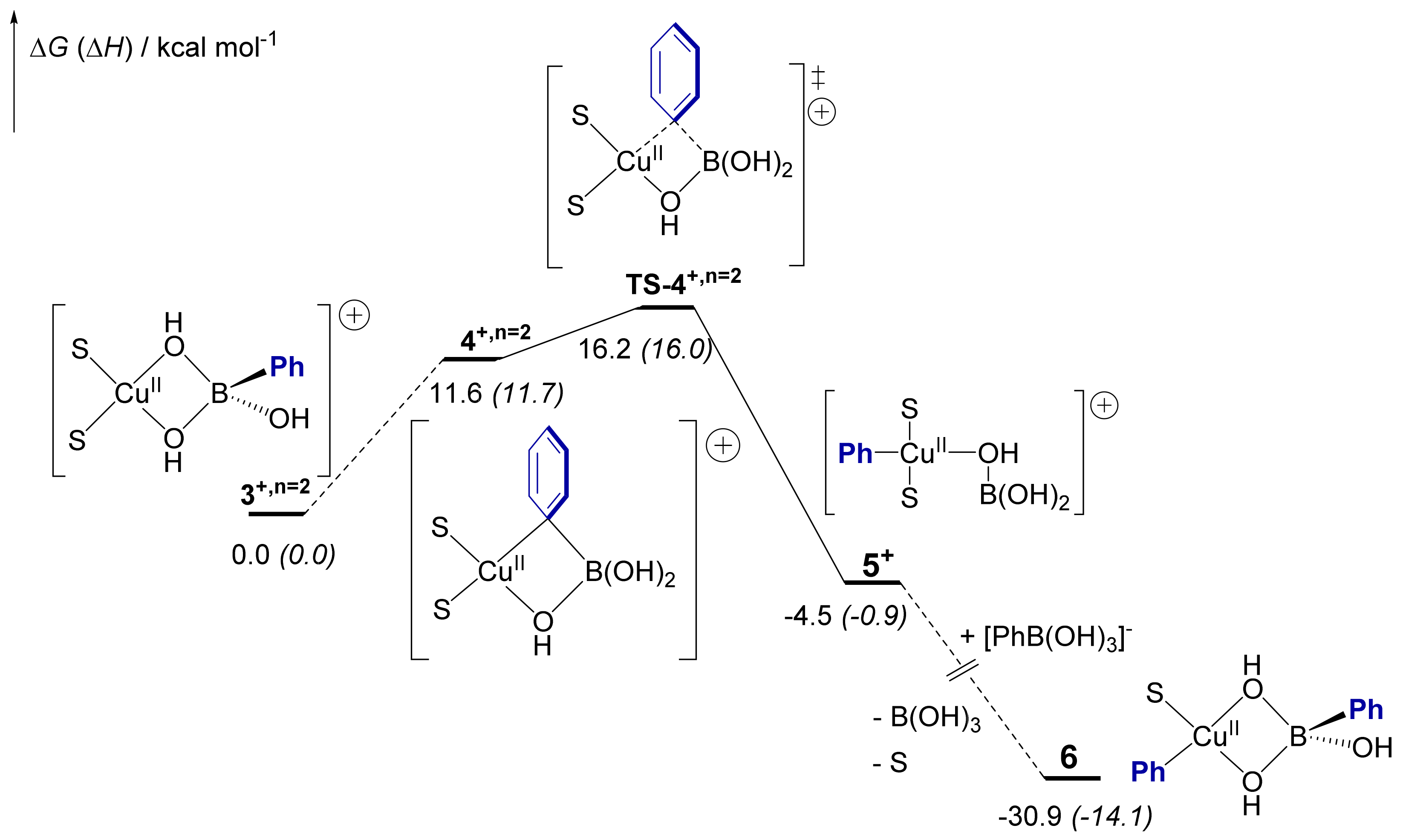
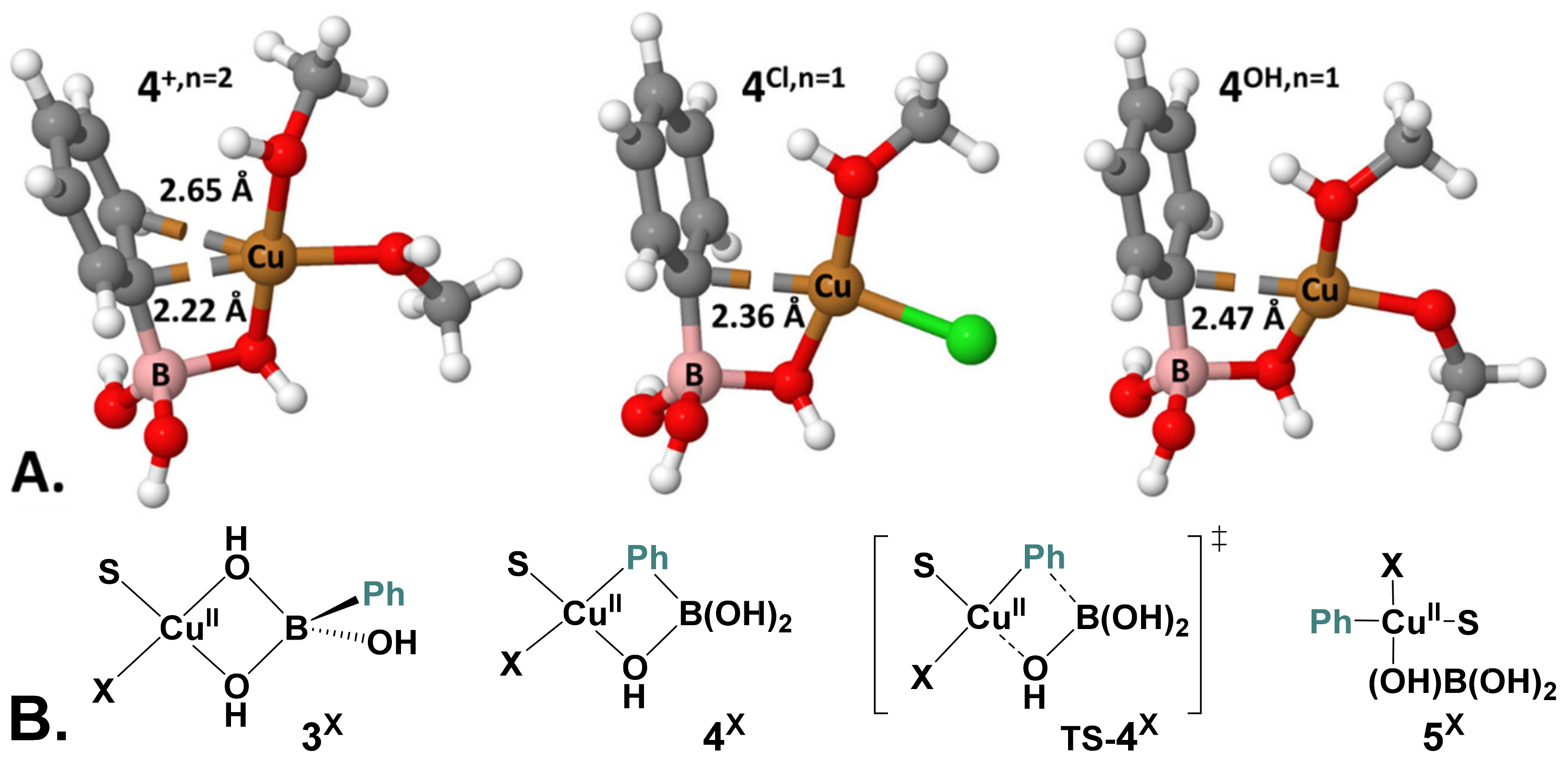
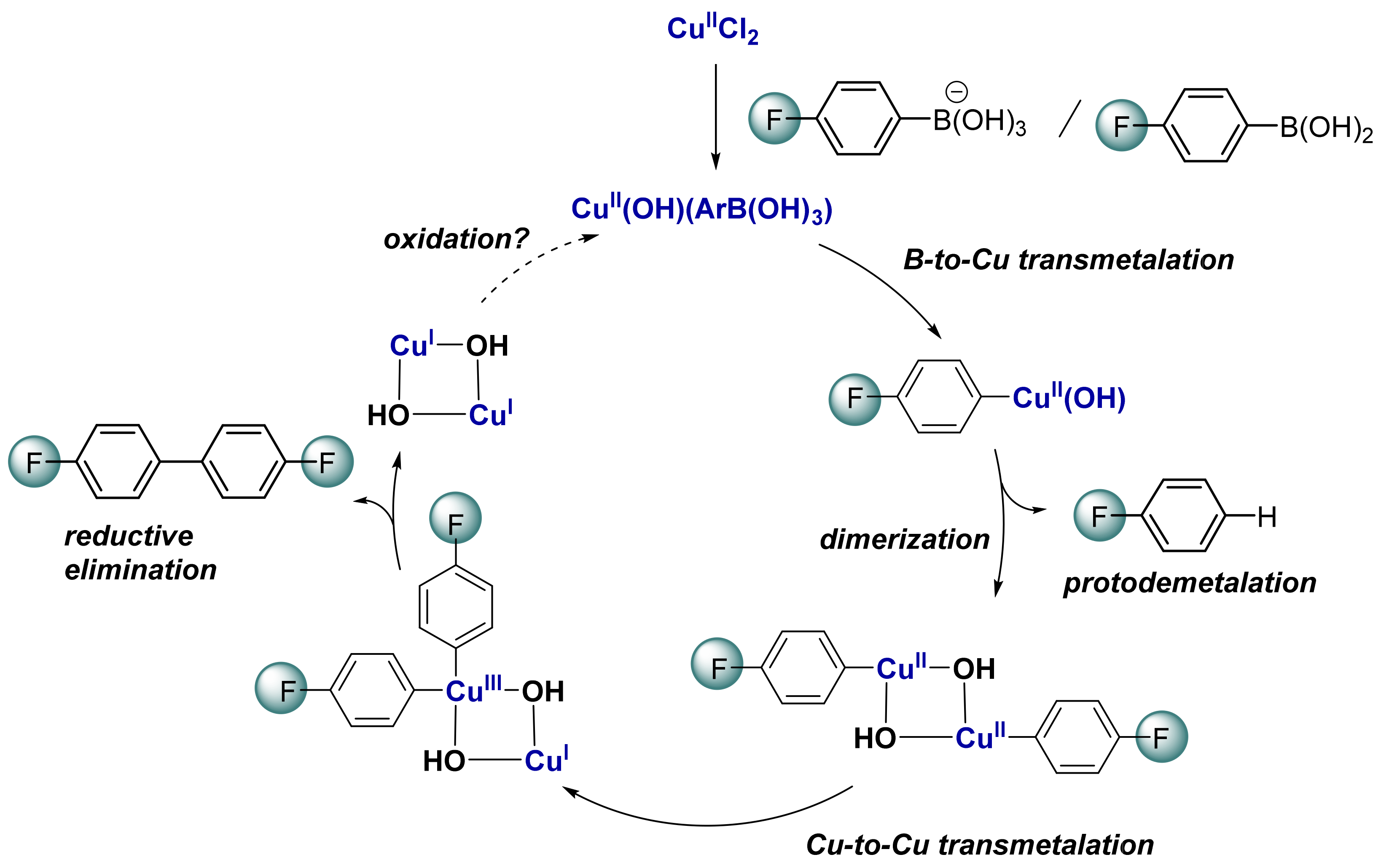
2.5. Computational Mechanistic Investigation: B-to-Cu(II) Transmetalation
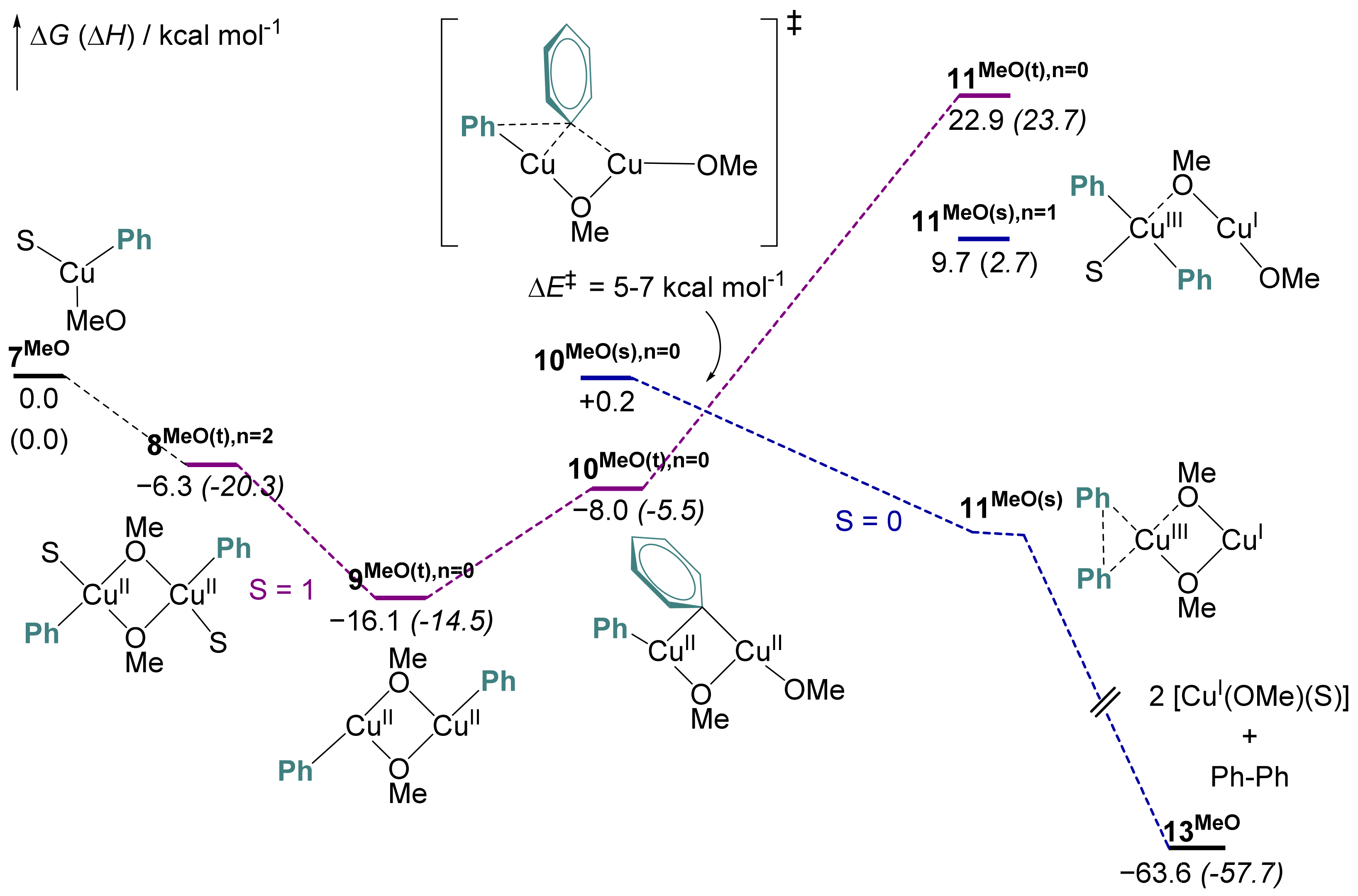
2.6. Computational Mechanistic Investigation: Cu(II)-Cu(II) Transmetalation and Reductive Elimination
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ullmann, F.; Bielecki, J. Ueber Synthesen in der Biphenylreihe. Ber. Dtsch. Chem. Ges. 1901, 34, 2174–2185. [Google Scholar] [CrossRef]
- Ullmann, F. Ueber eine neue Bildungsweise von Diphenylaminderivaten. Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar] [CrossRef]
- Ullmann, F.; Sponagel, P. Ueber die Phenylirung von Phenolen. Ber. Dtsch. Chem. Ges. 1905, 38, 2211–2212. [Google Scholar] [CrossRef]
- Goldberg, I. Ueber Phenylirungen bei Gegenwart von Kupfer als Katalysator. Ber. Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. The Complementary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 2012, 31, 7753–7808. [Google Scholar] [CrossRef]
- Kirai, N.; Yamamoto, Y. Homocoupling of Arylboronic Acids Catalyzed by 1,10-Phenanthroline-Ligated Copper Complexes in Air. Eur. J. Org. Chem. 2009, 2009, 1864–1867. [Google Scholar] [CrossRef]
- Cao, Y.-N.; Tian, X.-C.; Chen, X.-X.; Yao, Y.-X.; Gao, F.; Zhou, X.-L. Rapid Ligand-Free Base-Accelerated Copper-Catalyzed Homocoupling Reaction of Arylboronic Acids. Synlett 2017, 28, 601–606. [Google Scholar]
- Kaboudin, B.; Haruki, T.; Yokomatsu, T. CuSO4-Mediated Homocoupling of Arylboronic Acids under Ligand- and Base-Free Conditions in Air. Synthesis 2011, 2011, 91–96. [Google Scholar] [CrossRef]
- Cheng, G.; Luo, M. Homocoupling of Arylboronic Acids Catalyzed by CuCl in Air at Room Temperature. Eur. J. Org. Chem. 2011, 2011, 2519–2523. [Google Scholar] [CrossRef]
- Yang, C.-T.; Zhang, Z.-Q.; Liu, Y.-C.; Liu, L. Copper-Catalyzed Cross-Coupling Reaction of Organoboron Compounds with Primary Alkyl Halides and Pseudohalides. Angew. Chem. Int. 2011, 50, 3904–3907. [Google Scholar] [CrossRef]
- Villalobos, J.M.; Srogl, J.; Liebeskind, L.S. A New Paradigm for Carbon−Carbon Bond Formation: Aerobic, Copper-Templated Cross-Coupling. J. Am. Chem. Soc. 2007, 129, 15734–15735. [Google Scholar] [CrossRef]
- Kang, S.-K.; Kim, J.-S.; Choi, S.-C. Copper- and Manganese-Catalyzed Cross-Coupling of Organostannanes with Organic Iodides in the Presence of Sodium Chloride. J. Org. Chem. 1997, 62, 4208–4209. [Google Scholar] [CrossRef]
- Miyake, Y.; Wu, M.; Rahman, M.J.; Kuwatani, Y.; Iyoda, M. Efficient Construction of Biaryls and Macrocyclic Cyclophanes via Electron-Transfer Oxidation of Lipshutz Cuprates. J. Org. Chem. 2006, 71, 6110–6117. [Google Scholar] [CrossRef]
- Strieter, E.R.; Bhayana, B.; Buchwald, S.L. Mechanistic Studies on the Copper-Catalyzed N-Arylation of Amides. J. Am. Chem. Soc. 2009, 131, 78–88. [Google Scholar] [CrossRef]
- Lefèvre, G.; Franc, G.; Tlili, A.; Adamo, C.; Taillefer, M.; Ciofini, I.; Jutand, A. Contribution to the Mechanism of Copper-Catalyzed C–N and C–O Bond Formation. Organometallics 2012, 31, 7694–7707. [Google Scholar] [CrossRef]
- Mansour, M.; Giacovazzi, R.; Ouali, A.; Taillefer, M.; Jutand, A. Activation of aryl halides by Cu0/1,10-phenanthroline: Cu0 as precursor of CuI catalyst in cross-coupling reactions. Chem. Commun. 2008, 6051–6053. [Google Scholar] [CrossRef]
- Stollenz, M.; Meyer, F. Mesitylcopper—A Powerful Tool in Synthetic Chemistry. Organometallics 2012, 31, 7708–7727. [Google Scholar] [CrossRef]
- Piers, E.; Yee, J.G.K.; Gladstone, P.L. CuCl-Mediated Intramolecular Oxidative Coupling of Aryl- and Alkenyltrimethylstannane Functions. Org. Lett. 2000, 2, 481–484. [Google Scholar] [CrossRef]
- Demir, A.S.; Reis, Ö.; Emrullahoglu, M. Role of Copper Species in the Oxidative Dimerization of Arylboronic Acids: Synthesis of Symmetrical Biaryls. J. Org. Chem. 2003, 68, 10130–10134. [Google Scholar] [CrossRef]
- Zhou, Y.; You, W.; Smith, K.B.; Brown, M.K. Copper-Catalyzed Cross-Coupling of Boronic Esters with Aryl Iodides and Application to the Carboboration of Alkynes and Allenes. Angew. Chem. Int. 2014, 53, 3475–3479. [Google Scholar] [CrossRef]
- Goj, L.A.; Blue, E.D.; Delp, S.A.; Gunnoe, T.B.; Cundari, T.R.; Petersen, J.L. Single-Electron Oxidation of Monomeric Copper(I) Alkyl Complexes: Evidence for Reductive Elimination through Bimolecular Formation of Alkanes. Organometallics 2006, 25, 4097–4104. [Google Scholar] [CrossRef]
- Vantourout, J.C.; Miras, H.N.; Isidro-Llobet, A.; Sproules, S.; Watson, A.J.B. Spectroscopic Studies of the Chan–Lam Amination: A Mechanism-Inspired Solution to Boronic Ester Reactivity. J. Am. Chem. Soc. 2017, 139, 4769–4779. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Fyfe, J.W.B.; Vantourout, J.C.; Watson, A.J.B. Mechanistic Development and Recent Applications of the Chan–Lam Amination. Chem. Rev. 2019, 119, 12491–12523. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, J.C.; Law, R.P.; Isidro-Llobet, A.; Atkinson, S.J.; Watson, A.J.B. Chan–Evans–Lam Amination of Boronic Acid Pinacol (BPin) Esters: Overcoming the Aryl Amine Problem. J. Org. Chem. 2016, 81, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, J.C.; Li, L.; Bendito-Moll, E.; Chabbra, S.; Arrington, K.; Bode, B.E.; Isidro-Llobet, A.; Kowalski, J.A.; Nilson, M.G.; Wheelhouse, K.M.P.; et al. Mechanistic Insight Enables Practical, Scalable, Room Temperature Chan–Lam N-Arylation of N-Aryl Sulfonamides. ACS Catal. 2018, 8, 9560–9566. [Google Scholar] [CrossRef]
- Bell, N.L.; Xu, C.; Fyfe, J.W.B.; Vantourout, J.C.; Brals, J.; Chabbra, S.; Bode, B.E.; Cordes, D.B.; Slawin, A.M.Z.; McGuire, T.M.; et al. Cu(OTf)2-Mediated Cross-Coupling of Nitriles and N-Heterocycles with Arylboronic Acids to Generate Nitrilium and Pyridinium Products**. Angew. Chem. Int. 2021, 60, 7935–7940. [Google Scholar] [CrossRef]
- Adamo, C.; Amatore, C.; Ciofini, I.; Jutand, A.; Lakmini, H. Mechanism of the Palladium-Catalyzed Homocoupling of Arylboronic Acids: Key Involvement of a Palladium Peroxo Complex. J. Am. Chem. Soc. 2006, 128, 6829–6836. [Google Scholar] [CrossRef]
- Lakmini, H.; Ciofini, I.; Jutand, A.; Amatore, C.; Adamo, C. Pd-Catalyzed Homocoupling Reaction of Arylboronic Acid: Insights from Density Functional Theory. J. Phys. Chem. A 2008, 112, 12896–12903. [Google Scholar] [CrossRef]
- Jutand, A. Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef]
- Opekar, F.; Beran, P. Rotating disk electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1976, 69, 1–105. [Google Scholar] [CrossRef]
- Amatore, C.; Le Duc, G.; Jutand, A. Mechanism of Palladium-Catalyzed Suzuki–Miyaura Reactions: Multiple and Antagonistic Roles of Anionic “Bases” and Their Countercations. Chem. Eur. J. 2013, 19, 10082–10093. [Google Scholar] [CrossRef]
- Schreck, J.O. Nonlinear Hammett relationships. J. Chem. Educ. 1971, 48, 103. [Google Scholar] [CrossRef]
- Salmon, P.S.; Neilson, G.W.; Enderby, J.E. The structure of Cu2+ aqueous solutions. J. Phys. C Solid State Phys. 1988, 21, 1335–1349. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Norjmaa, G.; Ujaque, G.; Lledós, A. Beyond Continuum Solvent Models in Computational Homogeneous Catalysis. Top. Catal. 2022, 65, 118–140. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Payard, P.-A.; Bohn, A.; Tocqueville, D.; Jaouadi, K.; Escoude, E.; Ajig, S.; Dethoor, A.; Gontard, G.; Perego, L.A.; Vitale, M.; et al. Role of dppf Monoxide in the Transmetalation Step of the Suzuki–Miyaura Coupling Reaction. Organometallics 2021, 40, 1120–1128. [Google Scholar] [CrossRef]
- Thomas Andy, A.; Denmark Scott, E. Pre-transmetalation intermediates in the Suzuki-Miyaura reaction revealed: The missing link. Science 2016, 352, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Zahrt, A.F.; Delaney, C.P.; Denmark, S.E. Elucidating the Role of the Boronic Esters in the Suzuki–Miyaura Reaction: Structural, Kinetic, and Computational Investigations. J. Am. Chem. Soc. 2018, 140, 4401–4416. [Google Scholar] [CrossRef] [PubMed]
- Payard, P.-A.; Perego, L.A.; Ciofini, I.; Grimaud, L. Taming Nickel-Catalyzed Suzuki-Miyaura Coupling: A Mechanistic Focus on Boron-to-Nickel Transmetalation. ACS Catal. 2018, 8, 4812–4823. [Google Scholar] [CrossRef]
- Bose, S.; Dutta, S.; Koley, D. Entering Chemical Space with Theoretical Underpinning of the Mechanistic Pathways in the Chan–Lam Amination. ACS Catal. 2022, 12, 1461–1474. [Google Scholar] [CrossRef]
- Del Pozo, J.; Pérez-Iglesias, M.; Álvarez, R.; Lledós, A.; Casares, J.A.; Espinet, P. Speciation of ZnMe2, ZnMeCl, and ZnCl2 in Tetrahydrofuran (THF), and Its Influence on Mechanism Calculations of Catalytic Processes. ACS Catal. 2017, 7, 3575–3583. [Google Scholar] [CrossRef]
- Peltzer, R.M.; Eisenstein, O.; Nova, A.; Cascella, M. How Solvent Dynamics Controls the Schlenk Equilibrium of Grignard Reagents: A Computational Study of CH3MgCl in Tetrahydrofuran. J. Phys. Chem. B 2017, 121, 4226–4237. [Google Scholar] [CrossRef]
- Rio, J.; Perrin, L.; Payard, P.-A. Structure—Reactivity Relationship of Organo-zinc and -zincate Reagents: Key Elements towards Molecular Understanding. Eur. J. Org. Chem. 2022, e202200906. [Google Scholar] [CrossRef]
- Lozano-Lavilla, O.; Gómez-Orellana, P.; Lledós, A.; Casares, J.A. Transmetalation Reactions Triggered by Electron Transfer between Organocopper Complexes. Inorg. Chem. 2021, 60, 11633–11639. [Google Scholar] [CrossRef]
- King, A.E.; Ryland, B.L.; Brunold, T.C.; Stahl, S.S. Kinetic and Spectroscopic Studies of Aerobic Copper(II)-Catalyzed Methoxylation of Arylboronic Esters and Insights into Aryl Transmetalation to Copper(II). Organometallics 2012, 31, 7948–7957. [Google Scholar] [CrossRef]
- Gray, R.D. Kinetics of oxidation of copper(I) by molecular oxygen in perchloric acid-acetonitrile solutions. J. Am. Chem. Soc. 1969, 91, 56–62. [Google Scholar] [CrossRef]
- Nord, H.; Sæland, E.; Morpurgo, A.; Caglieris, A. Kinetics of the Autoxidation of Cuprous Chloride in Hydrochloric Acid Solution. Acta Chem. Scand. 1955, 9, 430–437. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0.; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013; Available online: http://nbo6.chem.wisc.edu/ (accessed on 7 October 2022).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X, n | 3X | 4X | TS-4X | 5X |
|---|---|---|---|---|
| Cl, n = 1 | 0.0 (0.0) | 9.9 (11.4) | 17.2 (18.5) | 0.0 (3.7) |
| MeO, n = 1 | 0.0 (0.0) | 11.9 (11.8) | 23.7 (22.7) | 6.5 (8.5) |
| MeO, n = 0 | 0.0 (0.0) | 9.5 (19.1) | 18.0 (16.9) | 0.6 (−3.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salamé, A.; Rio, J.; Ciofini, I.; Perrin, L.; Grimaud, L.; Payard, P.-A. Copper-Catalyzed Homocoupling of Boronic Acids: A Focus on B-to-Cu and Cu-to-Cu Transmetalations. Molecules 2022, 27, 7517. https://doi.org/10.3390/molecules27217517
Salamé A, Rio J, Ciofini I, Perrin L, Grimaud L, Payard P-A. Copper-Catalyzed Homocoupling of Boronic Acids: A Focus on B-to-Cu and Cu-to-Cu Transmetalations. Molecules. 2022; 27(21):7517. https://doi.org/10.3390/molecules27217517
Chicago/Turabian StyleSalamé, Aude, Jordan Rio, Ilaria Ciofini, Lionel Perrin, Laurence Grimaud, and Pierre-Adrien Payard. 2022. "Copper-Catalyzed Homocoupling of Boronic Acids: A Focus on B-to-Cu and Cu-to-Cu Transmetalations" Molecules 27, no. 21: 7517. https://doi.org/10.3390/molecules27217517
APA StyleSalamé, A., Rio, J., Ciofini, I., Perrin, L., Grimaud, L., & Payard, P.-A. (2022). Copper-Catalyzed Homocoupling of Boronic Acids: A Focus on B-to-Cu and Cu-to-Cu Transmetalations. Molecules, 27(21), 7517. https://doi.org/10.3390/molecules27217517