
Surface Properties of Saponin—Chitosan Mixtures

 , , , , , ,
, , , , , ,  and
and 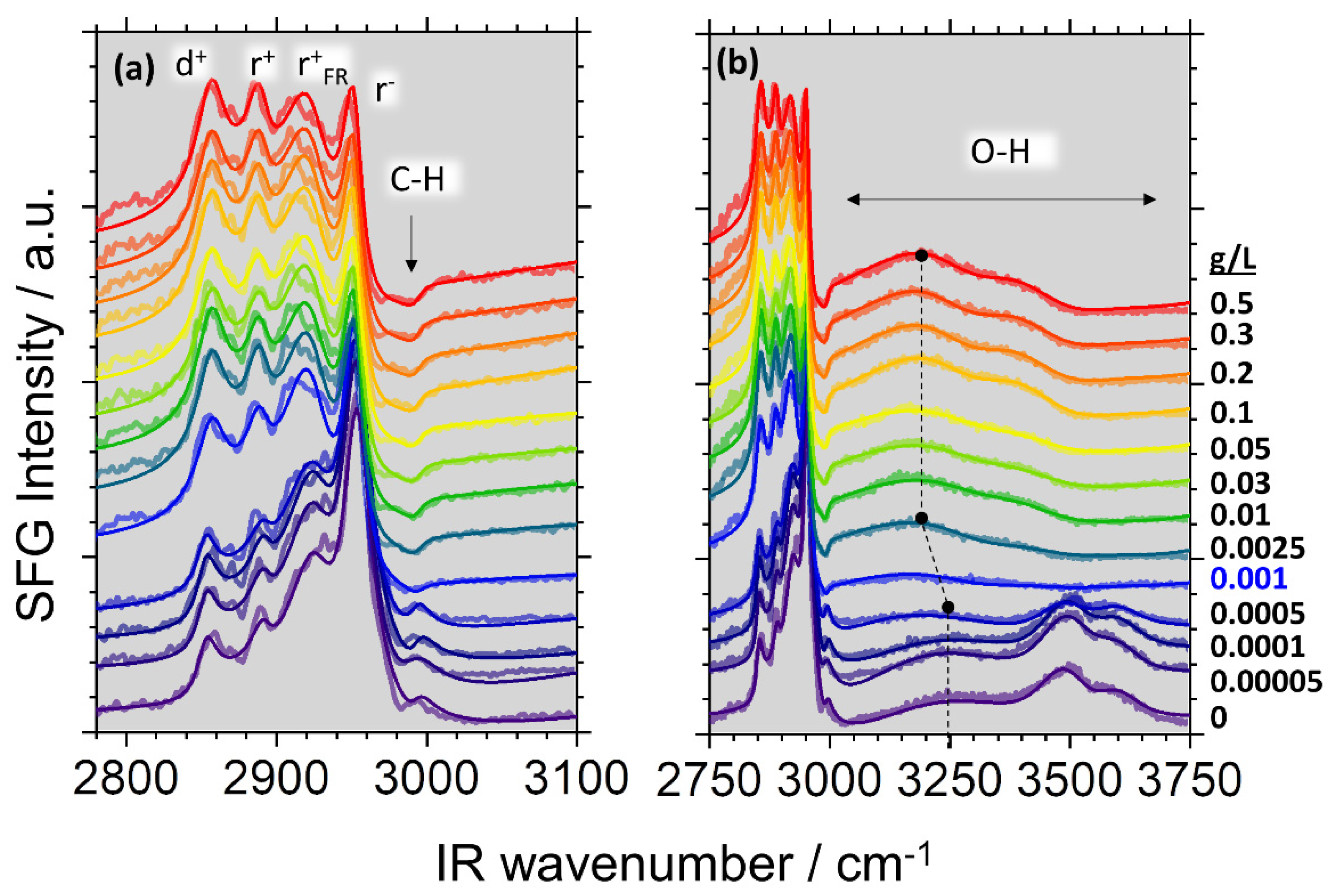{kind=link}
{kind=link}
{kind=link}
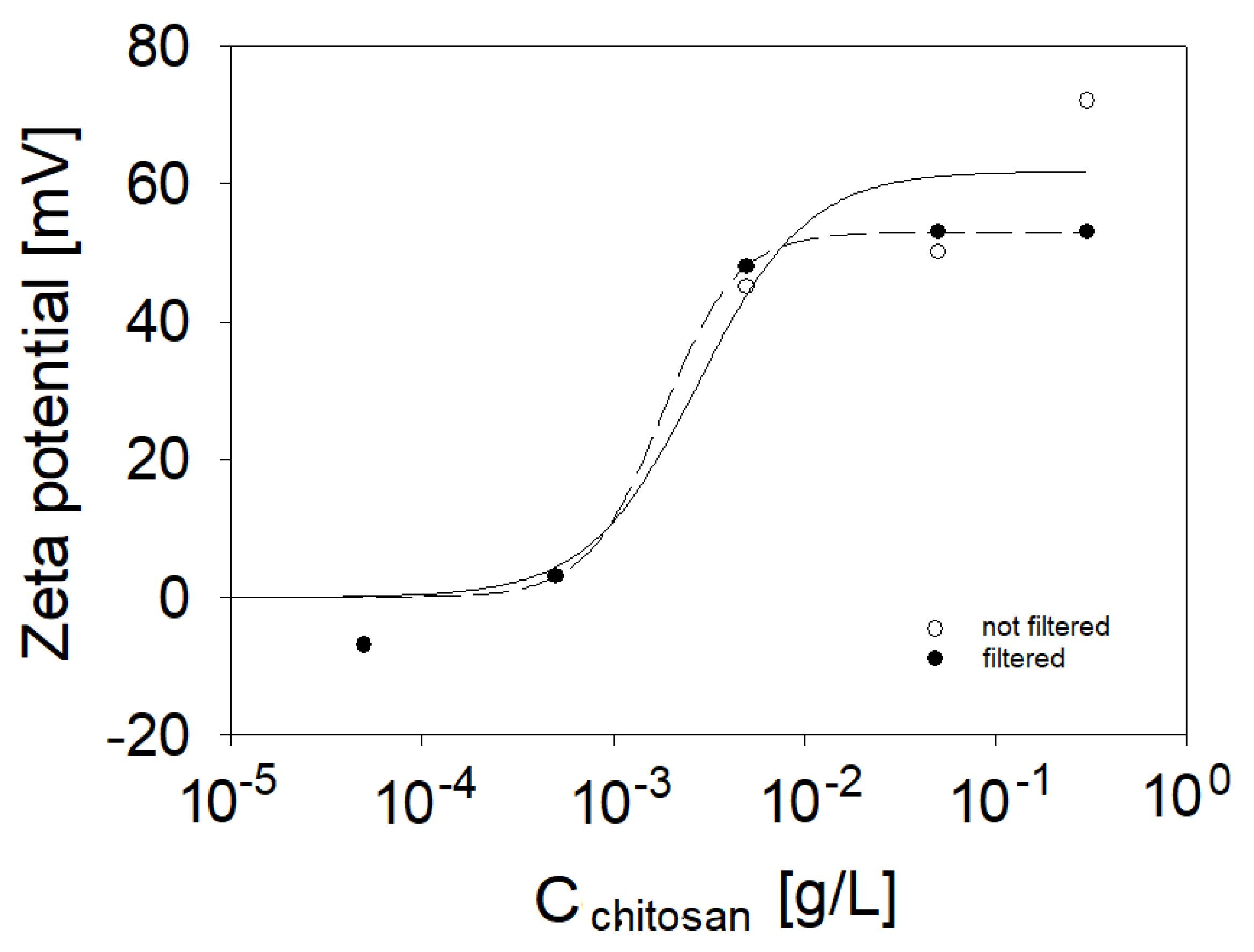
{kind=link}
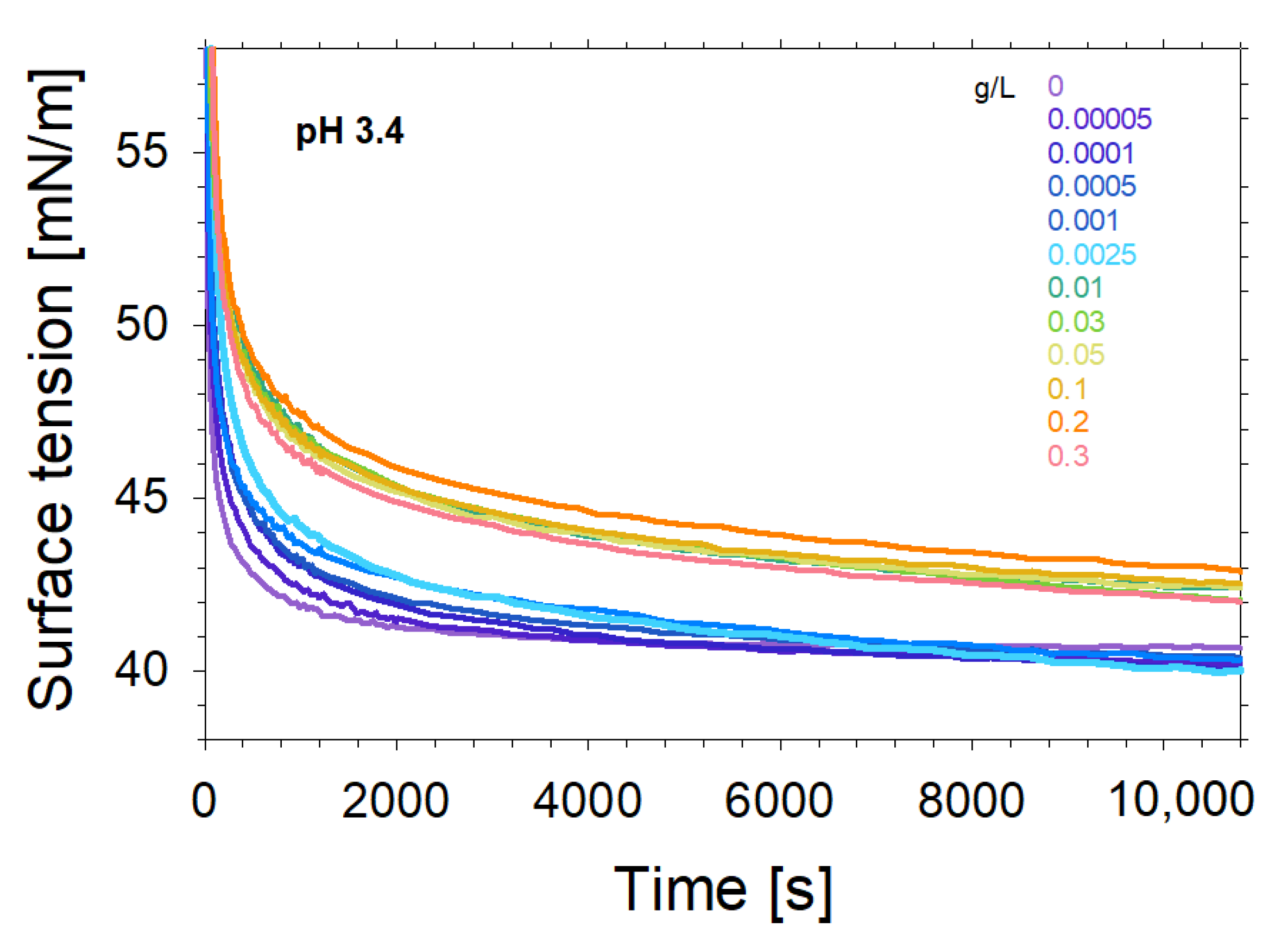
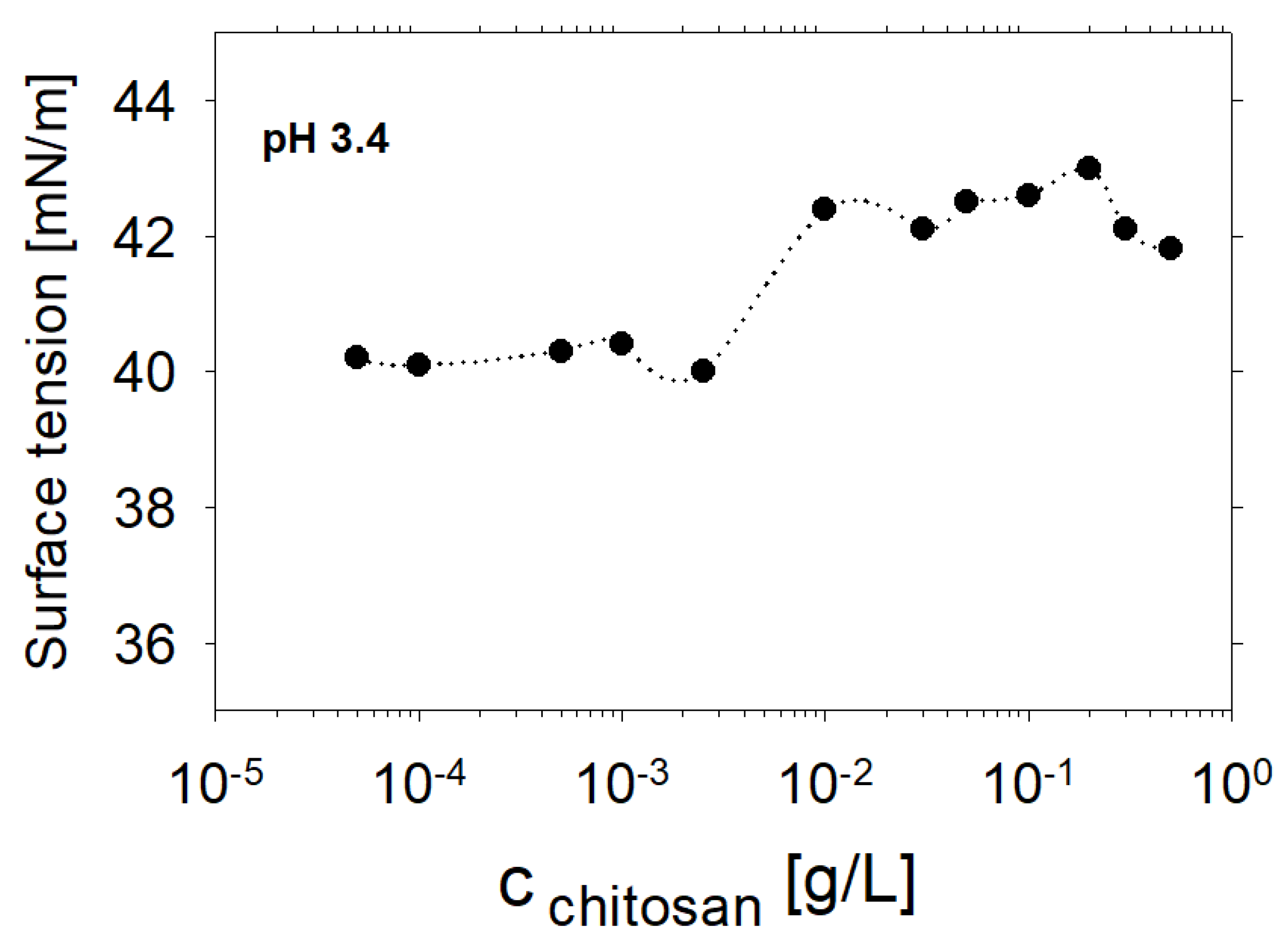{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
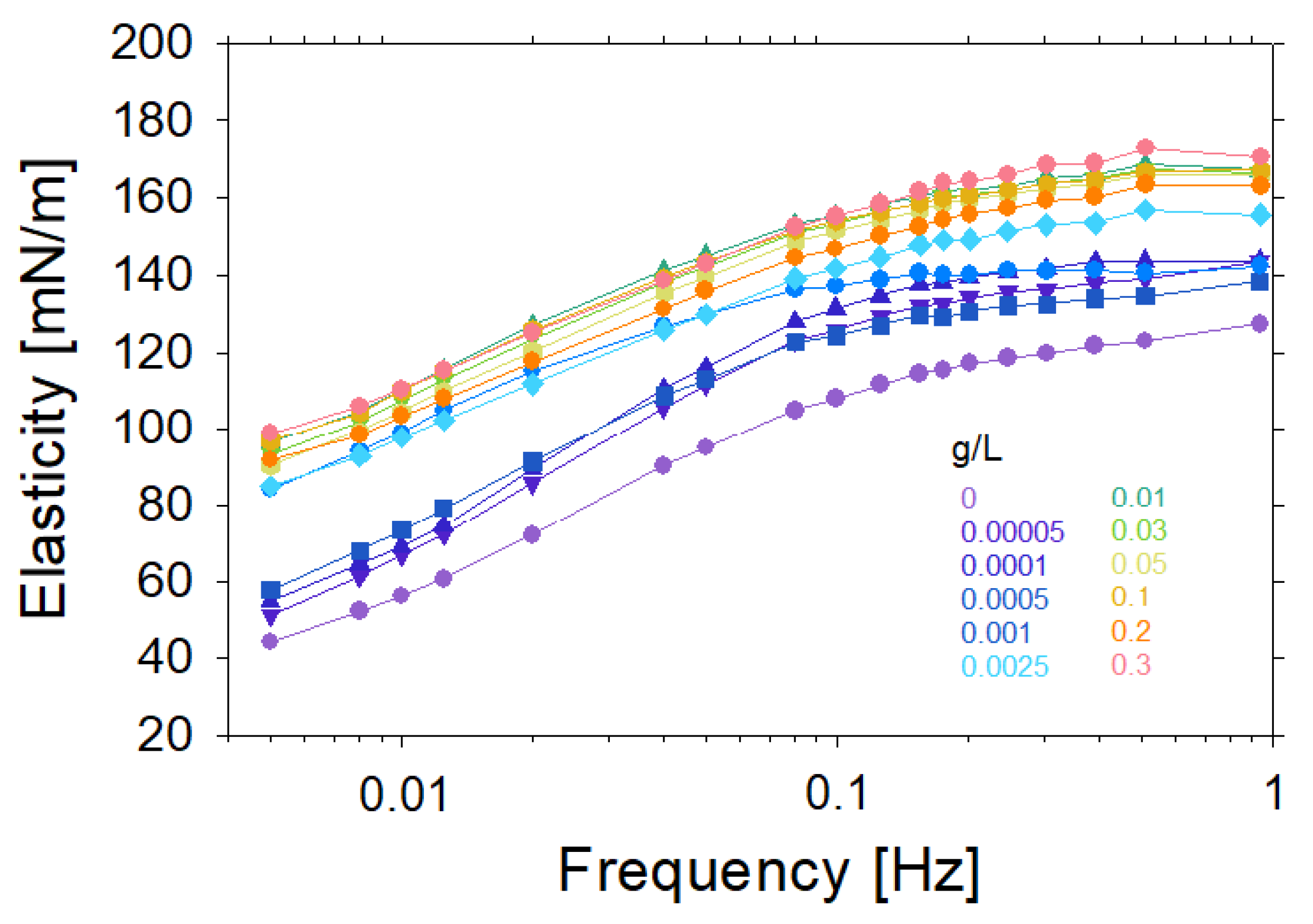
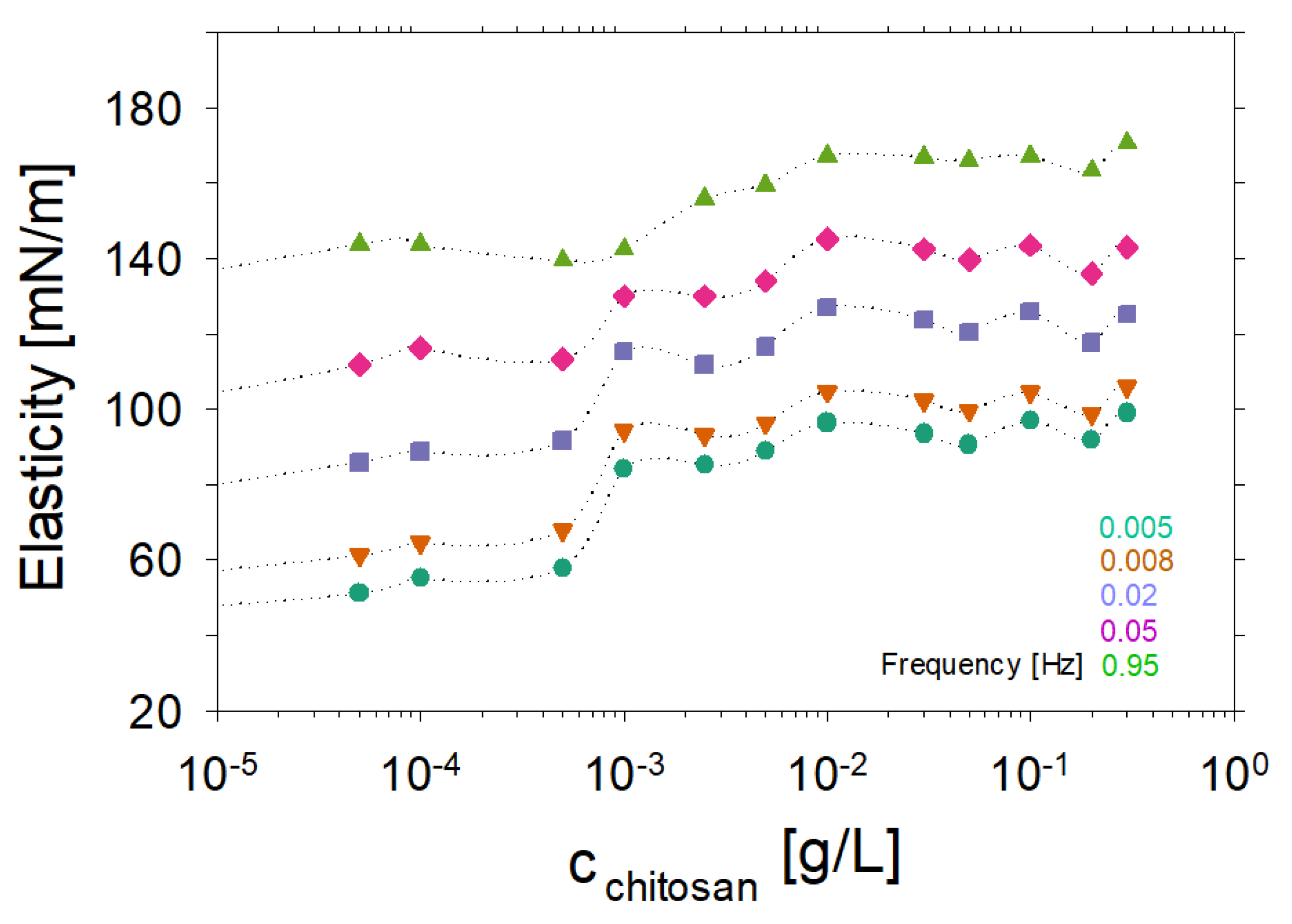
2. Experimental Results
3. Discussion and Summary
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.3. Methods
4.3.1. Sum-Frequency Generation Spectroscopy
4.3.2. Surface Activity and Dilational Surface Elasticity
4.3.3. Zeta-Potential
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lapitsky, Y.; Zahir, T.; Shoichet, M.S. Modular biodegradable biomaterials from surfactant and polyelectrolyte mixtures. Biomacromolecules 2008, 9, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.; Lee, H.; Liu, R.C.W.; Allen, C. Intermolecular interactions and morphology of aqueous polymer/surfactant mixtures containing cationic Chitosan and nonionic sorbitan esters. Biomacromolecules 2008, 9, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhu, Y.; Ran, X.; Wang, M.; Su, Y.; Cheng, T. Therapeutic Potential of Chitosan and Its Derivatives in Regenerative Medicine. J. Surg. Res. 2006, 133, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Jarek, E.; Ravera, F.; Liggieri, L.; Warszynski, P.; Krzan, M. Surface properties and foamability of saponin and saponin-chitosan systems. Colloids Surf. B Biointerfaces 2019, 181, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Oleszek, W.; Hamed, A. Saponin-Based Surfactants. In Surfactants from Renewable Resources; Kjellin, M., Johansson, I., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; pp. 239–249. ISBN 9780470760413. [Google Scholar] [CrossRef]
- Raafat, D.; Sahl, H.G. Chitosan and its antimicrobial potential—A critical literature survey. Microbiol. Biotechnol. 2009, 2, 186–201. [Google Scholar] [CrossRef]
- Wina, E.; Muetzel, S.; Becker, K. The impact of saponins or saponin-containing plant materials on ruminant production—A review. J. Agric. Food Chem. 2005, 53, 8093–8105. [Google Scholar] [CrossRef]
- Suzuki, R.; Ohno, H.; Murakami, T.; Shirataki, Y. Improving quality control of yucca extracts used as food additives by screening antimicrobial activity using NMR metabolomics. J. Nat. Med. 2020, 74, 306–310. [Google Scholar] [CrossRef]
- Guclu-Ustundag, Ö.; Mazza, G. Saponins, Properties, applications and processing. Crit. Rev. Food Sci. Nutr. 2007, 47, 231–258. [Google Scholar] [CrossRef]
- Nakabayashi, T.; Takakusagi, Y.; Iwabata, K.; Sakaguchi, K. Foam fractionation of protein, Correlation of protein adsorption onto bubbles with a pH-induced conformational transition. Anal. Biochem. 2011, 419, 173–179. [Google Scholar] [CrossRef]
- Jenkins, K.J.; Atwal, A.S. Effects of dietary saponins on fecal bile acids and neutral sterols, and availability of vitamins A and E in the chick. J. Nutr. Biochem. 1994, 5, 134–137. [Google Scholar] [CrossRef]
- Southon, S.; Wright, A.J.A.; Johnson, I.T.; Gee, J.M.; Price, K.; Fairweather-Tait, S.J. The Effect of Saponins on Mineral Availability; AFRC Institute of Food Research: Norwitch, UK, 1988; pp. 413–415. [Google Scholar]
- Tamura, Y.; Miyakoshi, M.; Yamamoto, M. Application of Saponin-Containing Plants in Foods and Cosmetics. chapter 5. In Alternative Medicine; Sakagami, H., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0903-7. [Google Scholar]
- Paul, T.J.; Taylor, T.A.; Rajendra Santosh, A.B. The potential of saponin from Jamaica’s Blighia sapida (ackee) as a substitute for sodium lauryl sulphate in toothpaste. Med. Hypotheses 2020, 137, 109555. [Google Scholar] [CrossRef] [PubMed]
- Cheeke, P.R. Actual and potential applications of and saponins in human and animal nutrition. J. Anim. Sci. 2000, 77, 1. [Google Scholar] [CrossRef]
- Devlieghere, F.; Vermeulen, A.; Debevere, J. Chitosan, Antimicrobial activity, interactions with food components and applicability as a coating on fruit and vegetables. Food Microbiol. 2004, 21, 703–714. [Google Scholar] [CrossRef]
- Kim, I.Y.; Seo, S.J.; Moon, H.S.; Yo, M.-K.; Park, I.-Y.; Kim, B.-C.; Cho, C.-S. Chitosan and its derivatives for tissue engineering applications. Biotechnol. Adv. 2008, 26, 1–21. [Google Scholar] [CrossRef]
- Dziza, K.; Santini, E.; Liggieri, L.; Jarek, E.; Krzan, M.; Fischer, T.L.; Ravera, F. Interfacial Properties and Emulsification of Biocompatible Liquid-Liquid Systems. Coatings 2020, 10, 397. [Google Scholar] [CrossRef]
- Monteux, C.; Fuller, G.G.; Bergeron, V. Shear and dilational surface rheology of oppositely charged polyelectrolyte/surfactant microgels adsorbed at the air-water interface. influence on foam stability. J. Phys. Chem. B 2004, 108, 16473–16482. [Google Scholar] [CrossRef]
- Golemanov, K.; Tcholakova, S.; Denkov, N.; Pelan, E.; Stoyanov, S.D. Surface shear rheology of saponin adsorption layers. Langmuir 2012, 28, 12071–12084. [Google Scholar] [CrossRef]
- Glikman, D.; García Rey, N.; Richert, M.; Meister, K.; Braunschweig, B. pH effects on the molecular structure and charging state of β-Escin biosurfactants at the air-water interface. J. Colloid Interface Sci. 2022, 607, 1754–1761. [Google Scholar] [CrossRef]
- Kagiyama, Y.; Miyamae, T. Interface Structure of Escin at Air-Water Interface probed by Sum Frequency Generation Spectroscopy. J. Raman. Spectrosc. 2022, 53, 1820. [Google Scholar] [CrossRef]
- Engelhardt, K.; Peukert, W.; Braunschweig, B. Vibrational sum-frequency generation at protein modified air-water interfaces, Effects of molecular structure and surface charging. Curr. Opin. Colloid Interface Sci. 2014, 19, 207–215. [Google Scholar] [CrossRef]
- Schulze-Zachau, F.; Braunschweig, B. Structure of Polystyrenesulfonate/Surfactant Mixtures at Air-Water Interfaces and Their Role as Building Blocks for Macroscopic Foam. Langmuir 2017, 33, 3499–3508. [Google Scholar] [CrossRef] [PubMed]
- Richert, M.E.; García Rey, N.; Braunschweig, B. Charge-Controlled Surface Properties of Native and Fluorophore-Labeled Bovine Serum Albumin at the Air-Water Interface. J. Phys. Chem. B 2018, 122, 10377–10383. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, K.; Weichsel, U.; Kraft, E.; Segets, D.; Peukert, W.; Braunschweig, B. Mixed layers of β-lactoglobulin and SDS at air-water interfaces with tunable intermolecular interactions. J. Phys. Chem. B 2014, 118, 4098–4105. [Google Scholar] [CrossRef] [PubMed]
- Tyrode, E.; Hedberg, J. A comparative study of the CD and CH stretching spectral regions of typical surfactants systems using VSFS, Orientation analysis of the terminal CH 3 and CD 3 groups. J. Phys. Chem. C 2012, 116, 1080–1091. [Google Scholar] [CrossRef]
- Lu, R.; Gan, W.; Wu, B.H.; Chen, H.; Wang, H.F. Vibrational polarization spectroscopy of CH stretching modes of the methylene group at the vapor/liquid interfaces with sum frequency generation. J. Phys. Chem. B 2004, 108, 7297–7306. [Google Scholar] [CrossRef]
- Lu, R.; Gan, W.; Wu, B.H.; Zhang, Z.; Guo, Y.; Wang, H.F. C-H stretching vibrations of methyl, methylene and methine groups at the vapor/Alcohol (n = 1–8) interfaces. J. Phys. Chem. B 2005, 109, 14118–14129. [Google Scholar] [CrossRef]
- Moghimipour, E.; Handali, S. Saponin, Properties, Methods of Evaluation and Applications. Annu. Res. Rev. Biol. 2015, 5, 207–220. [Google Scholar] [CrossRef]
- Scatena, L.F.; Brown, M.G.; Richmond, G.L. Water at Hydrophobic Surfaces: Weak Hydrogen Bonding and Strong Orientation Effects. Science 2001, 292, 908–912. [Google Scholar] [CrossRef]
- Gonella, G.; Lütgebaucks, C.; de Beer, A.G.F.; Roke, S. Second Harmonic and Sum-Frequency Generation from Aqueous Interfaces Is Modulated by Interference. J. Phys. Chem. C 2016, 120, 9165–9173. [Google Scholar] [CrossRef]
- Engelhardt, K.; Lexis, M.; Gochev, G.; Konnerth, C.; Miller, R.; Willenbacher, N.; Peukert, W.; Braunschweig, B. pH Effects on the Molecular Structure of β-Lactoglobulin Modified Air–Water Interfaces and Its Impact on Foam Rheology. Langmuir 2013, 29, 11646–11655. [Google Scholar] [CrossRef]
- Guckeisen, T.; Hosseinpour, S.; Peukert, W. Isoelectric Points of Proteins at the Air/Liquid Interface and in Solution. Langmuir 2019, 35, 5004–5012. [Google Scholar] [CrossRef] [PubMed]
- Richmond, G.L. Molecular Bonding and Interactions at Aqueous Surfaces as Probed by Vibrational Sum Frequency Spectroscopy. Chem. Rev. 2002, 102, 2693–2724. [Google Scholar] [CrossRef] [PubMed]
- Duffey, K.C.; Shih, O.; Wong, N.L.; Drisdell, W.S.; Saykally, R.J.; Cohen, R.C. Evaporation kinetics of aqueous acetic acid droplets: Effects of soluble organic aerosol components on the mechanism of water evaporation. Phys. Chem. Chem. Phys. 2013, 15, 11634–11639. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.; Backus, E.H.G.; Nagata, Y.; Bonn, M. Both Inter- and Intramolecular Coupling of O-H Groups Determine the Vibrational Response of the Water/Air Interface. J. Phys. Chem. Lett. 2016, 7, 4591–4595. [Google Scholar] [CrossRef]
- Das, S.; Imoto, S.; Sun, S.; Nagata, Y.; Backus, E.H.G.; Bonn, M. Nature of Excess Hydrated Proton at the Water-Air Interface. J. Am. Chem. Soc. 2020, 142, 945–952. [Google Scholar] [CrossRef]
- Gragson, D.E.; Richmond, G.L. Investigations of the Structure and Hydrogen Bonding of Water Molecules at Liquid Surfaces by Vibrational Sum Frequency Spectroscopy. J. Phys. Chem. B 1998, 102, 3847–3861. [Google Scholar] [CrossRef]
- Shen, Y.R.; Ostroverkhov, V. Sum-frequency vibrational spectroscopy on water interfaces, Polar orientation of water molecules at interfaces. Chem. Rev. 2006, 106, 1140–1154. [Google Scholar] [CrossRef]
- Sovago, M.; Campen, R.K.; Wurpel, G.W.H.; Müller, M.; Bakker, H.J.; Bonn, M. Vibrational response of hydrogen-bonded interfacial water is dominated by intramolecular coupling. Phys. Rev. Lett. 2008, 100, 173901. [Google Scholar] [CrossRef]
- Ohto, T.; Backus, E.H.G.; Hsieh, C.S.; Sulpizi, M.; Bonn, M.; Nagata, Y. Lipid Carbonyl Groups Terminate the Hydrogen Bond Network of Membrane-Bound Water. J. Phys. Chem. Lett. 2015, 6, 4499–4503. [Google Scholar] [CrossRef]
- Nagata, Y.; Mukamel, S. Vibrational sum-frequency generation spectroscopy at the water/lipid interface, Molecular dynamics simulation study. J. Am. Chem. Soc. 2010, 132, 6434–6442. [Google Scholar] [CrossRef]
- Nojima, Y.; Suzuki, Y.; Yamaguchi, S. Weakly Hydrogen-Bonded Water Inside Charged Lipid Monolayer Observed with Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy. J. Phys. Chem. C 2017, 121, 2173–2180. [Google Scholar] [CrossRef]
- Mondal, J.A.; Nihonyanagi, S.; Yamaguchi, S.; Tahara, T. Three distinct water structures at a zwitterionic lipid/water interface revealed by heterodyne-detected vibrational sum frequency generation. J. Am. Chem. Soc. 2012, 134, 7842–7850. [Google Scholar] [CrossRef] [PubMed]
- Tielrooij, K.J.; Paparo, D.; Piatkowski, L.; Bakker, H.J.; Bonn, M. Dielectric relaxation dynamics of water in model membranes probed by terahertz spectroscopy. Biophys. J. 2009, 97, 2484–2492. [Google Scholar] [CrossRef]
- Zhao, W.; Moilanen, D.E.; Fenn, E.E.; Fayer, M.D. Water at the surfaces of aligned phospholipid multibilayer model membranes probed with ultrafast vibrational spectroscopy. J. Am. Chem. Soc. 2008, 130, 13927–13937. [Google Scholar] [CrossRef]
- Schnurbus, M.; Hardt, M.; Steinforth, P.; Carrascosa-Tejedor, J.; Winnall, S.; Gutfreund, P.; Schönhoff, M.; Campbell, R.A.; Braunschweig, B. Responsive Material and Interfacial Properties through Remote Control of Polyelectrolyte–Surfactant Mixtures. ACS Appl. Mater. Interfaces 2022, 14, 4656–4667. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Zachau, F.; Braunschweig, B. CnTAB/polystyrene sulfonate mixtures at air-water interfaces, Effects of alkyl chain length on surface activity and charging state. Phys. Chem. Chem. Phys. 2019, 21, 7847–7856. [Google Scholar] [CrossRef] [PubMed]
- Varga, I.; Campbell, R.A. General physical description of the behavior of oppositely charged polyelectrolyte/surfactant mixtures at the air/water interface. Langmuir 2017, 33, 5915–5924. [Google Scholar] [CrossRef]
- Guzmán, E.; Llamas, S.; Maestro, A.; Fernandez-Pena, L.; Akanno, A.; Miller, R.; Ortega, F.; Rubio, R.G. Polymer-surfactant systems in bulk and at fluid interfaces. Adv. Colloid Interface Sci. 2016, 233, 38–64. [Google Scholar] [CrossRef]
- Richert, M.E.; Gochev, G.G.; Braunschweig, B. Specific Ion Effects of Trivalent Cations on the Structure and Charging State of β-Lactoglobulin Adsorption Layers. Langmuir 2019, 35, 11299–11307. [Google Scholar] [CrossRef]
- Sthoer, A.; Tyrode, E. Interactions of Na+ Cations with a Highly Charged Fatty Acid Langmuir Monolayer, Molecular Description of the Phase Transition. J. Phys. Chem. C 2019, 123, 23037–23048. [Google Scholar] [CrossRef]
- Hosseinpour, S.; Roeters, S.J.; Bonn, M.; Peukert, W.; Woutersen, S.; Weidner, T. Structure and Dynamics of Interfacial Peptides and Proteins from Vibrational Sum-Frequency Generation Spectroscopy. Chem. Rev. 2020, 120, 3420–3465. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, K. Surface activity of saponin from Quillaja bark at the air/water and oil/water interfaces. Colloids Surf. B Biointerfaces 2013, 108, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Stanimirova, R.; Marinova, K.; Tcholakova, S.; Denkov, N.D.; Stoyanov, S.; Pelan, E. Surface rheology of saponin adsorption layers. Langmuir 2011, 27, 12486–12498. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Dungan, S.R. Micellar Properties of Quillaja Saponin. 1. Effects of Temperature, Salt, and pH on Solution Properties. J. Agric. Food Chem. 1997, 45, 1587–1595. [Google Scholar] [CrossRef]
- Chen, Z.; Gracias, D.H.; Somorjai, G.A. Sum frequency generation (SFG)—Surface vibrational spectroscopy studies of buried interfaces, Catalytic reaction intermediates on transition metal crystal surfaces at high reactant pressures, Polymer surface structures at the solid-gas and solid-liquid. Appl. Phys. B Lasers Opt. 1999, 68, 549–557. [Google Scholar] [CrossRef][Green Version]
- Chen, Z.; Shen, Y.R.; Somorjai, G.A. Studies of polymer surfaces by sum frequency generation vibrational spectroscopy. Annu. Rev. Phys. Chem. 2002, 53, 437–465. [Google Scholar] [CrossRef]
- Vidal, F.; Tadjeddine, A. Sum-frequency generation spectroscopy of interfaces. Rep. Prog. Phys. 2005, 68, 1095–1127. [Google Scholar] [CrossRef]
- Rey, N.G.; Dlott, D.D. Studies of electrochemical interfaces by broadband sum frequency generation. J. Electroanal Chem. 2017, 800, 114–125. [Google Scholar] [CrossRef]
- Chowdhury, A.U.; Muralidharan, N.; Daniel, C.; Amin, R.; Belharouak, I. Probing the electrolyte/electrode interface with vibrational sum frequency generation spectroscopy, A review. J. Power Sources 2021, 506, 230173. [Google Scholar] [CrossRef]
- Han, H.L.; Horowitz, Y.; Somorjai, G.A. A Review on in Situ Sum Frequency Generation Vibrational Spectroscopy Studies of Liquid-Solid Interfaces in Electrochemical Systems. In Encyclopedia of Interfacial Chemistry, Surface Science and Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–12. [Google Scholar] [CrossRef]
- Ding, B.; Chen, Z. Sum Frequency Generation Vibrational Spectroscopy. Encycl. Biophys. 2013, 19, 2512–2516. [Google Scholar] [CrossRef]
- Jubb, A.M.; Hua, W.; Allen, H.C. Environmental chemistry at vapor/water interfaces, Insights from vibrational sum frequency generation spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, 109–130. [Google Scholar] [CrossRef] [PubMed]
- Nihonyanagi, S.; Mondal, J.A.; Yamaguchi, S.; Tahara, T. Structure and dynamics of interfacial water studied by heterodyne-detected vibrational sum-frequency generation. Annu. Rev. Phys. Chem. 2013, 64, 579–603. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.R. Basic theory of surface sum-frequency generation. J. Phys. Chem. C 2012, 116, 15505–15509. [Google Scholar] [CrossRef]
- Wang, H.F.; Gan, W.; Lu, R.; Rao, Y.; Wu, B.H. Quantitative spectral and orientational analysis in surface sum frequency generation vibrational spectroscopy (SFG-VS). Int. Rev. Phys. Chem. 2005, 24, 191–256. [Google Scholar] [CrossRef]
- Bell, G.R.; Bain, C.D.; Ward, R.N. Sum-frequency vibrational spectroscopy of soluble surfactants at the air/water interface. J. Chem. Soc.-Faraday Trans. 1996, 92, 515–523. [Google Scholar] [CrossRef]
- Garciá Rey, N.; Weißenborn, E.; Schulze-Zachau, F.; Gochev, G.; Braunschweig, B. Quantifying Double-Layer Potentials at Liquid-Gas Interfaces from Vibrational Sum-Frequency Generation. J. Phys. Chem. C 2019, 123, 1279–1286. [Google Scholar] [CrossRef]
- Tyrode, E.; Johnson, C.M.; Baldelli, S.; Leygraf, C.; Rutland, M.W. A vibrational sum frequency spectroscopy study of the liquid-gas interface of acetic acid-water mixtures, 2. Orientation analysis. J. Phys. Chem. B 2005, 109, 329–341. [Google Scholar] [CrossRef]
- Tyrode, E.; Corkery, R. Charging of Carboxylic Acid Monolayers with Monovalent Ions at Low Ionic Strengths, Molecular Insight Revealed by Vibrational Sum Frequency Spectroscopy. J. Phys. Chem. C 2018, 122, 28775–28786. [Google Scholar] [CrossRef]
- Hore, D.K.; Tyrode, E. Probing Charged Aqueous Interfaces Near Critical Angles, Effect of Varying Coherence Length. J. Phys. Chem. C 2019, 123, 16911–16920. [Google Scholar] [CrossRef]
- Pool, R.E.; Versluis, J.; Backus, E.H.G.; Bonn, M. Comparative study of direct and phase-specific vibrational sum-frequency generation spectroscopy, Advantages and limitations. J. Phys. Chem. B 2011, 115, 15362–15369. [Google Scholar] [CrossRef]
- Tian, C.S.; Shen, Y.R. Structure and charging of hydrophobic material/water interfaces studied by phase-sensitive sum-frequency vibrational spectroscopy. Proc. Natl. Acad. Sci. USA. 2009, 106, 15148–15153. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Shen, Y.R.; Waychunas, G.A. The interfacial structure of water/protonated α-Al2O3 () as a function of pH. J. Phys. Condens. Matter. 2012, 24, 124101. [Google Scholar] [CrossRef] [PubMed]
- Ravera, F.; Loglio, G.; Kovalchuk, V.I. Interfacial dilational rheology by oscillating bubble/drop methods. Curr. Opin. Colloid Interface Sci. 2010, 15, 217–228. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzan, M.; Rey, N.G.; Jarek, E.; Czakaj, A.; Santini, E.; Ravera, F.; Liggieri, L.; Warszynski, P.; Braunschweig, B. Surface Properties of Saponin—Chitosan Mixtures. Molecules 2022, 27, 7505. https://doi.org/10.3390/molecules27217505
Krzan M, Rey NG, Jarek E, Czakaj A, Santini E, Ravera F, Liggieri L, Warszynski P, Braunschweig B. Surface Properties of Saponin—Chitosan Mixtures. Molecules. 2022; 27(21):7505. https://doi.org/10.3390/molecules27217505
Chicago/Turabian StyleKrzan, Marcel, Natalia García Rey, Ewelina Jarek, Agnieszka Czakaj, Eva Santini, Francesca Ravera, Libero Liggieri, Piotr Warszynski, and Björn Braunschweig. 2022. "Surface Properties of Saponin—Chitosan Mixtures" Molecules 27, no. 21: 7505. https://doi.org/10.3390/molecules27217505
APA StyleKrzan, M., Rey, N. G., Jarek, E., Czakaj, A., Santini, E., Ravera, F., Liggieri, L., Warszynski, P., & Braunschweig, B. (2022). Surface Properties of Saponin—Chitosan Mixtures. Molecules, 27(21), 7505. https://doi.org/10.3390/molecules27217505