
New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molecular Docking, and In Silico ADME Profile Studies




Abstract
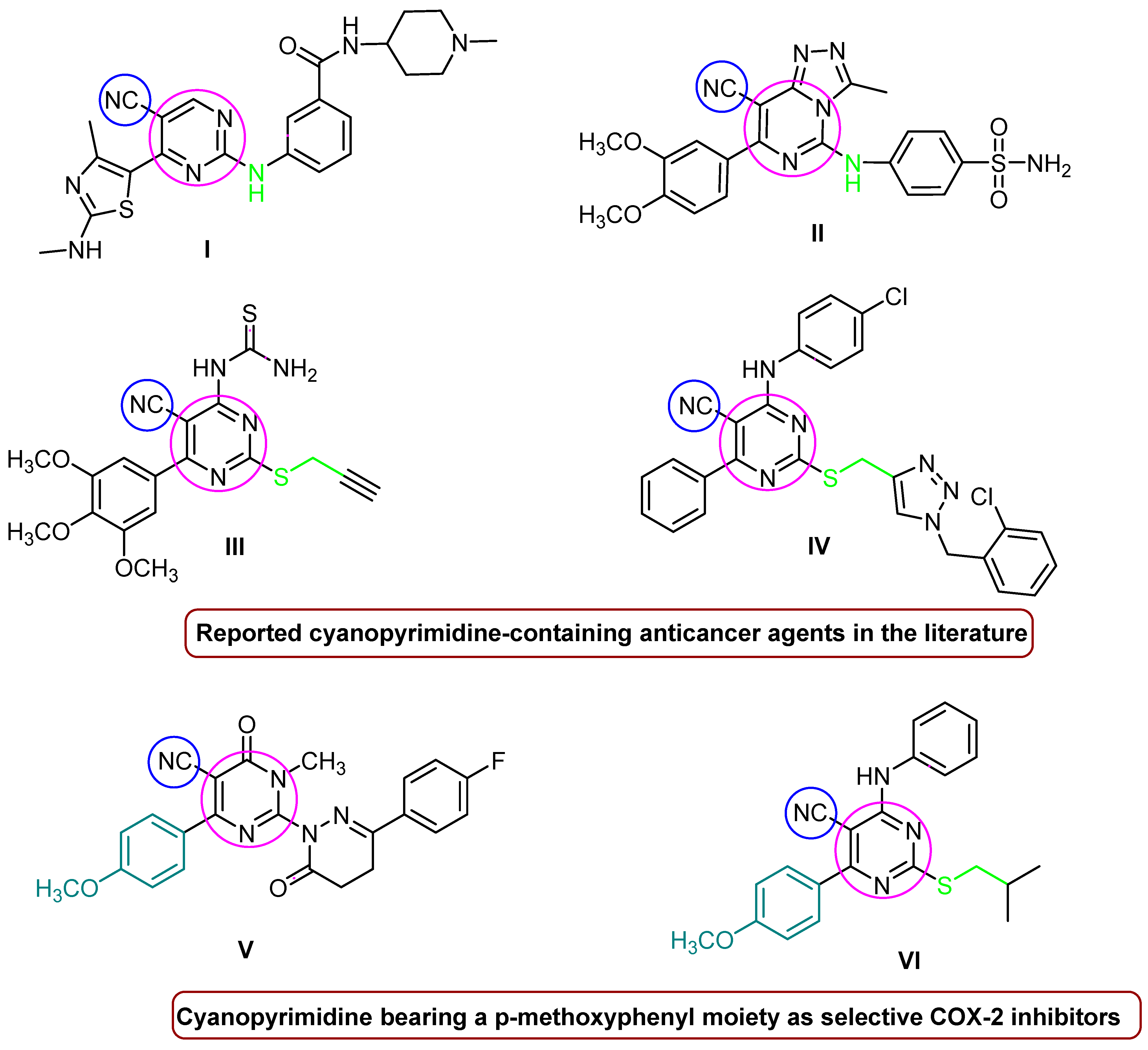
1. Introduction
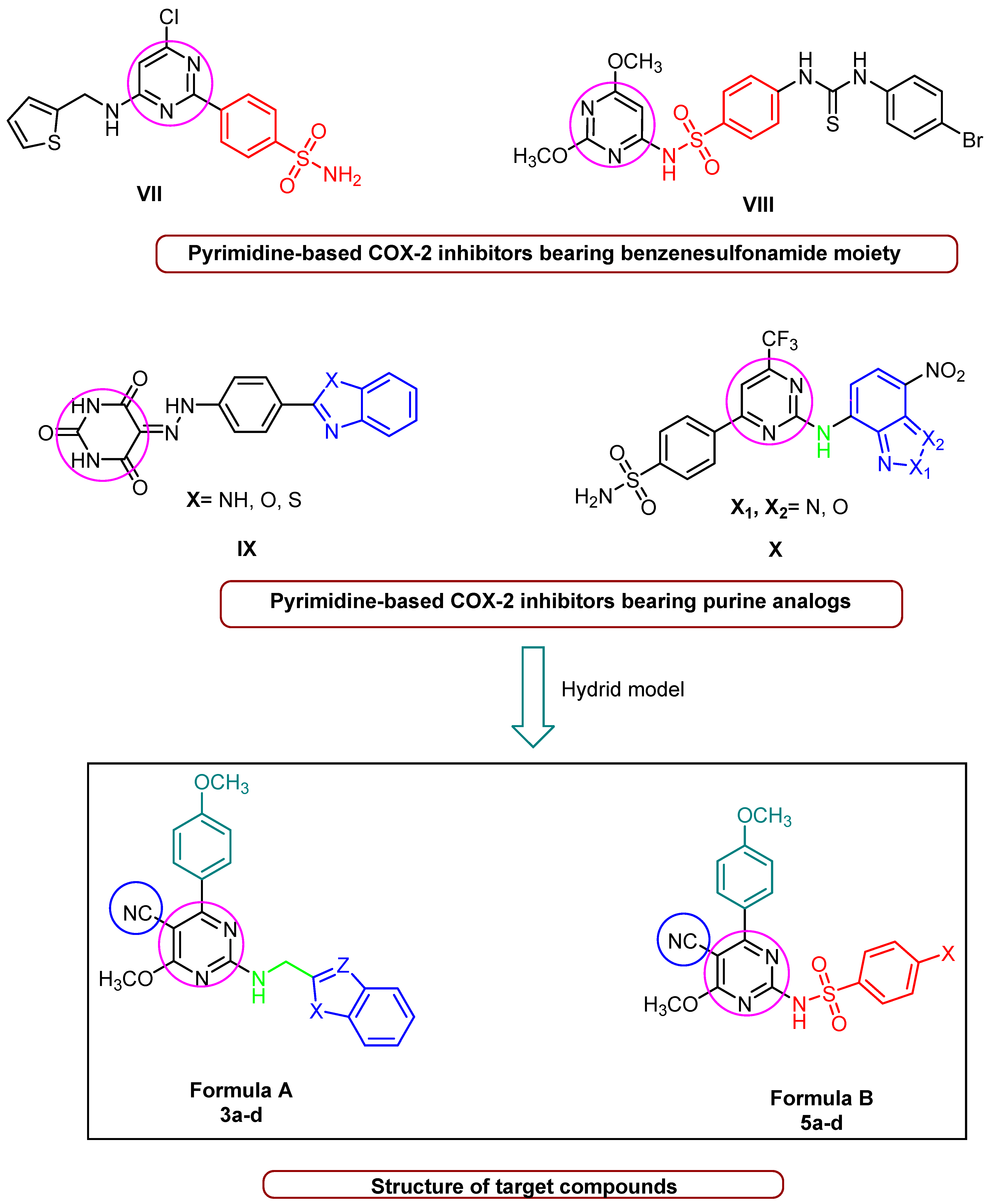
The Rationale of Molecular Design
2. Results and Discussions
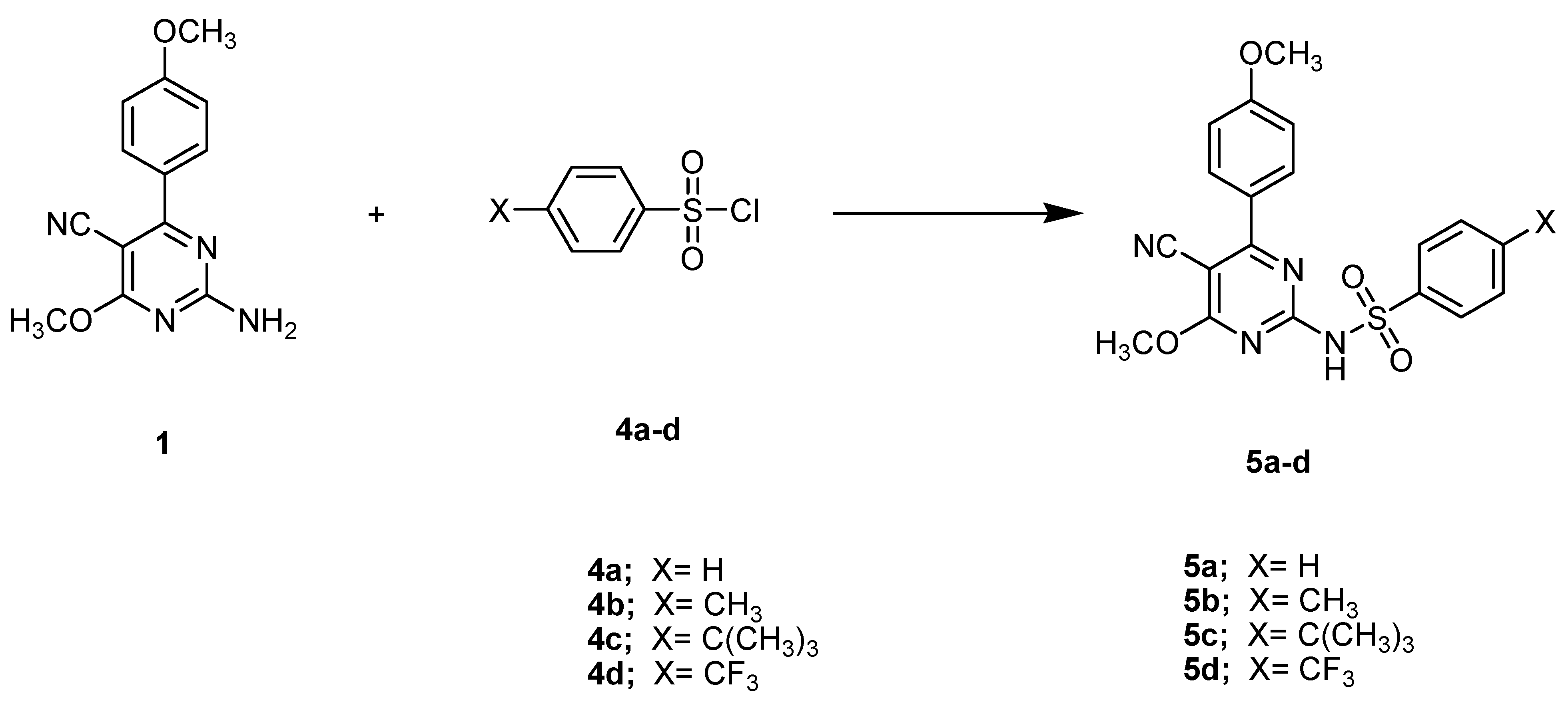
2.1. Structural Elucidation
2.2. Biological Evaluation
2.2.1. In-Vitro COX-2 Inhibition
2.2.2. Anticancer Screening (MTT Assay)
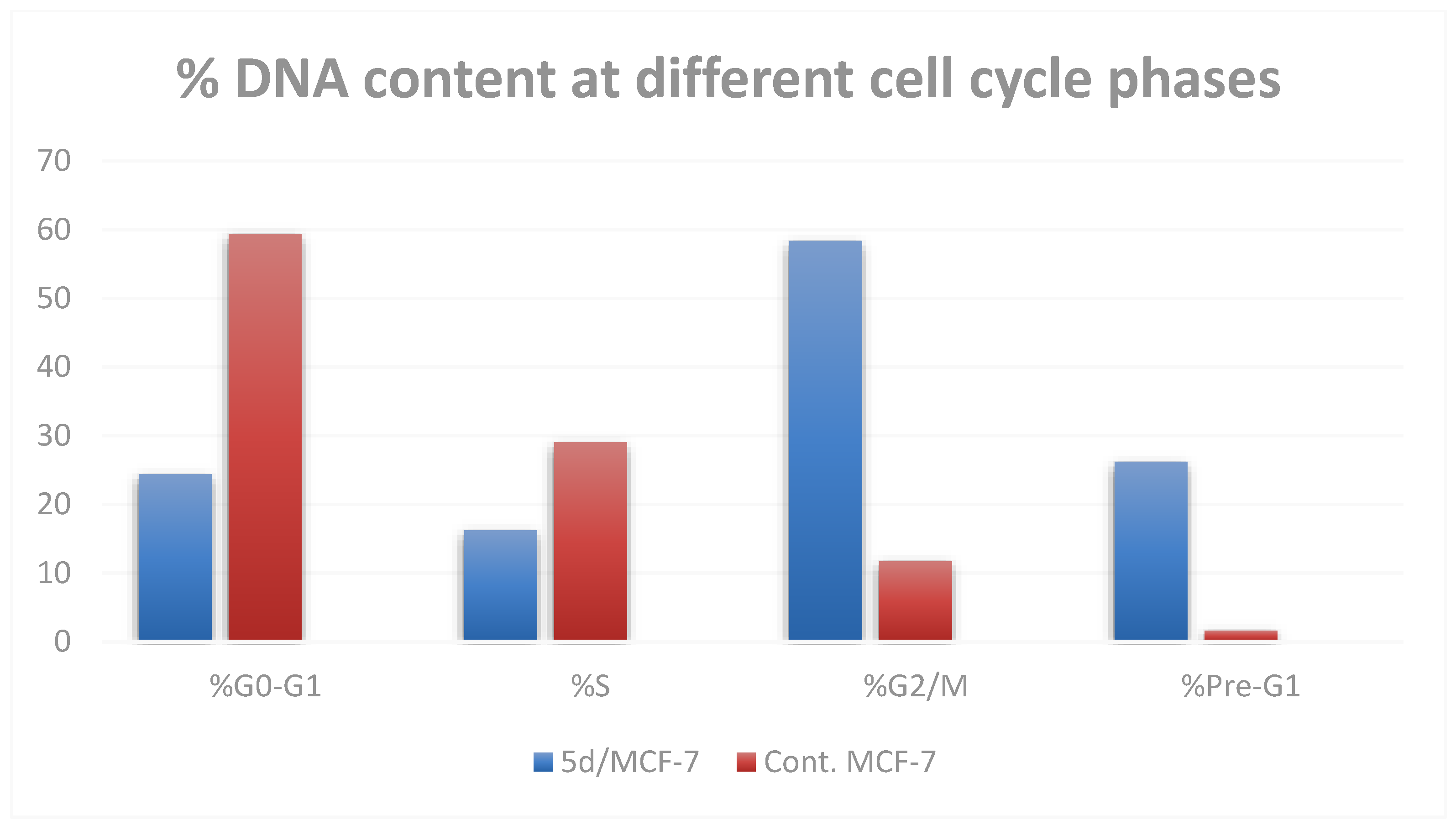
2.2.3. Cell Cycle Analysis
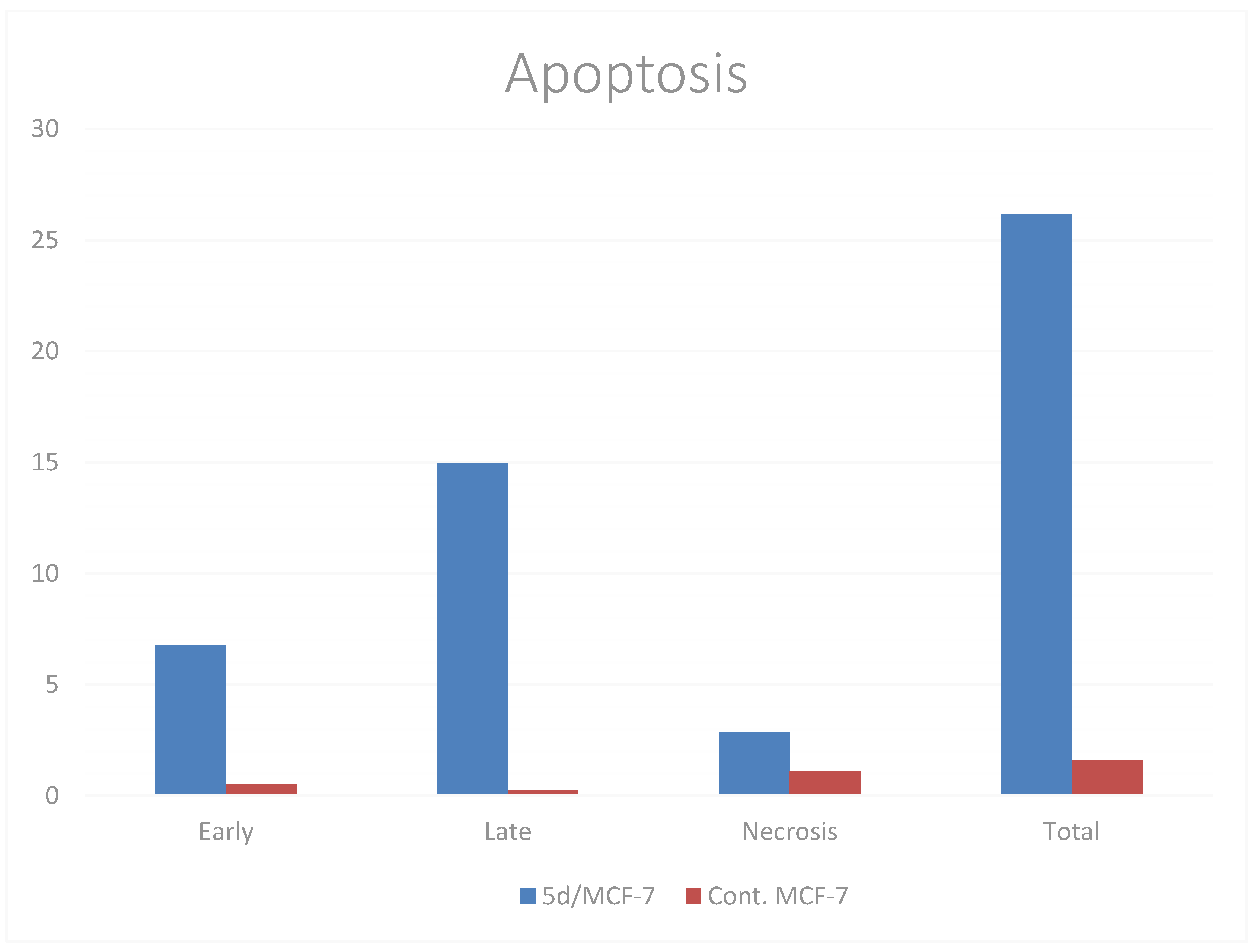
2.2.4. Apoptosis Determination by Annexin-V Assay
2.3. In Silico Studies
2.3.1. Docking Studies of Compounds 3b, 5b and 5d
2.3.2. In Silico Physicochemical Features, Pharmacokinetics Profiles, and Drug-Likeness Data of 3b, 5b, and 5d Compared to Celecoxib
3. Experimental
3.1. Materials
3.2. Instrumentation
3.3. Procedure for Synthesis of Substrate Compound 1 [34]
3.4. General Procedure for Synthesis of Compounds 3a–3d
3.4.1. 2-((1H-Benzo[d]imidazol-2-yl)methylamino)-4-methoxy-6-(4-methoxyphenyl)pyrimidine-5-carbonitrile (3a)
3.4.2. 2-(Benzo[d]oxazol-2-Ylmethylamino)-4-methoxy-6-(4-methoxyphenyl)pyrimidine-5-carbonitrile (3b)
3.4.3. 2-(Benzo[d]thiazol-2-ylmethylamino)-4-methoxy-6-(4-methoxyphenyl)pyrimidine-5-carbonitrile (3c)
3.4.4. 2-(Benzo[b]thiophen-2-ylmethylamino)-4-methoxy-6-(4-methoxyphenyl)pyrimidine-5-carbonitrile (3d)
3.5. General Procedure for Synthesis of Compounds 5a–5d
3.5.1. N-(5-Cyano-4-methoxy-6-(4-methoxyphenyl)pyrimidin-2-yl)benzenesulfonamide (5a)
3.5.2. N-(5-Cyano-4-methoxy-6-(4-methoxyphenyl)pyrimidin-2-yl)-4-methylbenzene-sulfonamide (5b)
3.5.3. 4-Tert-Butyl-N-(5-cyano-4-methoxy-6-(4-methoxyphenyl)pyrimidin-2-yl)benzene sulfonamide (5c)
3.5.4. N-(5-Cyano-4-methoxy-6-(4-methoxyphenyl)pyrimidin-2-yl)-4-(trifluoromethyl)benzene sulfonamide (5d)
3.6. Cell Lines
3.7. Cytotoxicity Protocol
3.8. Cell Cycle Analysis
3.9. Apoptosis Assay
3.10. COX-2 Inhibition Assay
3.11. Statistical Analysis
3.12. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fitzpatrick, F. Cyclooxygenase enzymes: Regulation and function. Curr. Pharm. Des. 2004, 10, 577–588. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Kam, P.C.A.; So, A. COX-3: Uncertainties and controversies. Anaesth. Intensive Care. 2009, 20, 50–53. [Google Scholar] [CrossRef]
- Regulski, M.; Regulska, K.; Prukała, W.; Piotrowska, H.; Stanisz, B.; Murias, M. COX-2 inhibitors: A novel strategy in the management of breast cancer. Drug Discov. Today 2016, 21, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Phillips, D.C.; Szekely-Szucs, K.; Elghazi, L.; Desmots, F.; Houghton, J.A. Cyclooxygenase-2 inhibition sensitizes human colon carcinoma cells to TRAIL-induced apoptosis through clustering of DR5 and concentrating death-inducing signaling complex components into ceramide-enriched caveolae. Cancer Res. 2005, 65, 11447–11458. [Google Scholar] [CrossRef][Green Version]
- Sivula, A.; Talvensaari-Mattila, A.; Lundin, J.; Joensuu, H.; Haglund, C.; Ristimäki, A.; Turpeenniemi-Hujanen, T. Association of cyclooxygenase-2 and matrix metalloproteinase-2 expression in human breast cancer. Breast Cancer Res. Treat. 2005, 89, 215–220. [Google Scholar] [CrossRef]
- Xu, H.-B.; Shen, F.-M.; Lv, Q.-Z. Celecoxib enhanced the cytotoxic effect of cisplatin in chemo-resistant gastric cancer xenograft mouse models through a cyclooxygenase-2-dependent manner. Eur. J. Pharmacol. 2016, 776, 1–8. [Google Scholar] [CrossRef]
- Rioux, N.; Castonguay, A. Prevention of NNK-induced lung tumorigenesis in A/J mice by acetylsalicylic acid and NS-398. Cancer Res. 1998, 58, 5354–5360. [Google Scholar]
- Castonguay, A.; Rioux, N.; Duperron, C.; Jalbert, G. Inhibition of lung tumorigenesis by NSAIDS: A working hypothesis. Exp. Lung Res. 1998, 24, 605–615. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef]
- Bertolini, A.; Ottani, A.; Sandrini, M. Dual acting anti-inflammatory drugs: A reappraisal. Pharmacol. Res. 2001, 44, 437–450. [Google Scholar] [CrossRef]
- Galal, S.A.; Khairat, S.H.; Ragab, F.A.; Abdelsamie, A.S.; Ali, M.M.; Soliman, S.M.; Mortier, J.; Wolber, G.; El Diwani, H.I. Design, synthesis and molecular docking study of novel quinoxalin-2(1H)-ones as anti-tumor active agents with inhibition of tyrosine kinase receptor and studying their cyclooxygenase-2 activity. Eur. J. Med. Chem. 2014, 86, 122–132. [Google Scholar] [CrossRef]
- Li, S.; Jiang, M.; Wang, L.; Yu, S. Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: Recent advancement. Biomed. Pharmacother. 2020, 129, 110389. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, R.-H.; Zhang, H.; Wang, Y.-C.; Yang, D.; Zhao, Y.-L.; Yan, G.-Y.; Xu, G.-B.; Guan, H.-Y.; Zhou, Y.-H.; et al. Design, synthesis, and biological evaluation of 2, 4-diamino pyrimidine derivatives as potent FAK inhibitors with anti-cancer and anti-angiogenesis activities. Eur. J. Med. Chem. 2021, 222, 113573. [Google Scholar] [CrossRef]
- Yousefi, R.; Alavian-Mehr, M.-M.; Mokhtari, F.; Panahi, F.; Mehraban, M.H.; Khalafi-Nezhad, A. Pyrimidine-fused heterocycle derivatives as a novel class of inhibitors for α-glucosidase. J. Enzyme Inhib. Med. Chem. 2013, 28, 1228–1235. [Google Scholar] [CrossRef]
- Ahmed, M.; Qadir, M.A.; Hameed, A.; Imran, M.; Muddassar, M. Screening of curcumin-derived isoxazole, pyrazoles, and pyrimidines for their anti-inflammatory, antinociceptive, and cyclooxygenase-2 inhibition. Chem. Biol. Drug Des. 2018, 91, 338–343. [Google Scholar] [CrossRef]
- Devi, M.; Jaiswal, S.; Jain, S.; Kaur, N.; Dwivedi, J. Synthetic and Biological Attributes of Pyrimidine Derivatives: A Recent Update. Curr. Org. Synth. 2021, 18, 790–825. [Google Scholar] [CrossRef]
- Omar, A.M.; Abd El Razik, H.A.; Hazzaa, A.A.; El-Attar, M.A.; El Demellawy, M.A.; Abdel Wahab, A.E.; El Hawash, S.A.M. New pyrimidines and triazolopyrimidines as antiproliferative and antioxidants with cyclooxygenase-1/2 inhibitory potential. Future Med. Chem. 2019, 11, 1583–1603. [Google Scholar] [CrossRef]
- Akhtar, W.; Nainwal, L.M.; Khan, M.F.; Verma, G.; Chashoo, G.; Bakht, A.; Iqbal, M.; Akhtar, M.; Shaquiquzzaman, M.; Alam, M.M. Synthesis, COX-2 inhibition and metabolic stability studies of 6-(4-fluorophenyl)-pyrimidine-5-carbonitrile derivatives as anticancer and anti-inflammatory agents. J. Fluor. Chem. 2020, 236, 109579. [Google Scholar] [CrossRef]
- Zhelev, N.; Trifonov, D.; Wang, S.; Hassan, M.; El Serafi, I.; Mitev, V. From Roscovitine to CYC202 to Seliciclib–from bench to bedside: Discovery and development. BioDiscovery 2013, 10, e8956. [Google Scholar]
- Martins, P.; Jesus, J.; Santos, S.; Raposo, L.R.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules 2015, 20, 16852–16891. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Shi, S.; Foley, D.W.; Lam, F.; Abbas, A.Y.; Liu, X.; Huang, S.; Jiang, X.; Baharin, N.; Fischer, P.M.; et al. Synthesis, structure–activity relationship and biological evaluation of 2, 4, 5-trisubstituted pyrimidine CDK inhibitors as potential anti-tumour agents. Eur. J. Med. Chem. 2013, 70, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Fathalla, O.A.E.-F.M.; Ismail, M.A.; Anwar, M.M.; Abouzid, K.A.; Ramadan, A.A.K. Novel 2-thiopyrimidine derivatives as CDK2 inhibitors: Molecular modeling, synthesis, and anti-tumor activity evaluation. Med. Chem. Res. 2013, 22, 659–673. [Google Scholar] [CrossRef]
- Ma, L.-Y.; Zheng, Y.-C.; Wang, S.-Q.; Wang, B.; Wang, Z.-R.; Pang, L.-P.; Zhang, M.; Wang, J.-W.; Ding, L.; Li, J.; et al. Design, synthesis, and structure–activity relationship of novel LSD1 inhibitors based on pyrimidine–thiourea hybrids as potent, orally active antitumor agents. J. Med. Chem. 2015, 58, 1705–1716. [Google Scholar] [CrossRef]
- Ma, L.-Y.; Pang, L.-P.; Wang, B.; Zhang, M.; Hu, B.; Xue, D.-Q.; Shao, K.-P.; Zhang, B.-L.; Liu, Y.; Zhang, E.; et al. Design and synthesis of novel 1, 2, 3-triazole-pyrimidine hybrids as potential anticancer agents. Eur. J. Med. Chem. 2014, 86, 368–380. [Google Scholar] [CrossRef]
- Akhtar, W.; Verma, G.; Khan, M.F.; Shaquiquzzaman, M.; Rana, A.; Anwer, T.; Akhter, M.; Alam, M.M. Synthesis of hybrids of dihydropyrimidine and pyridazinone as potential anti-breast cancer agents. Mini Rev. Med. Chem. 2018, 18, 369–379. [Google Scholar] [CrossRef]
- Akhtar, W.; Nainwal, L.M.; Kaushik, S.K.; Akhtar, M.; Shaquiquzzaman, M.; Almalki, F.; Saifullah, K.; Marella, A.; Alam, M.M. Methylene-bearing sulfur-containing cyanopyrimidine derivatives for treatment of cancer: Part-II. Arch. Pharm. 2020, 353, e1900333. [Google Scholar] [CrossRef]
- Supuran, C.; Casini, A.; Mastrolorenzo, A.; Scozzafava, A. COX-2 Selective Inhibitors, Carbonic Anhydrase Inhibition and Anticancer Properties of Sulfonamides Belonging to This Class of Pharmacological Agents. Mini Rev. Med. Chem. 2004, 4, 625–632. [Google Scholar] [CrossRef]
- Orjales, A.; Mosquera, R.; Lopez, B.; Olivera, R.; Labeaga, L.; Núñez, M.T. Novel 2-(4-methylsulfonylphenyl) pyrimidine derivatives as highly potent and specific COX-2 inhibitors. Bioorg. Med. Chem. 2008, 16, 2183–2199. [Google Scholar] [CrossRef]
- Ghorab, M.M.; El-Gaby, M.S.A.; Alsaid, M.S.; Elshaier, Y.A.M.M.; Soliman, A.M.; El-Senduny, F.F.; Badria, F.A.; Sherif, A.Y.A. Novel thiourea derivatives bearing sulfonamide moiety as anticancer agents through COX-2 inhibition. Anti-Cancer Agents Med. Chem. 2017, 17, 1411–1425. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Ahmad, W.; Al-Sanea, M.M.; Elshemy, H.A. New pyrimidine-benzoxazole/benzimidazole hybrids: Synthesis, antioxidant, cytotoxic activity, in vitro cyclooxygenase and phospholipase A2-V inhibition. Bioorg. Chem. 2019, 92, 103218. [Google Scholar] [CrossRef]
- Yatam, S.; Gundla, R.; Jadav, S.S.; reddy Pedavenkatagari, N.; Chimakurthy, J.; Kedam, T. Focused library design and synthesis of 2-mercapto benzothiazole linked 1, 2, 4-oxadiazoles as COX-2/5-LOX inhibitors. J. Mol. Struct. 2018, 1159, 193–204. [Google Scholar] [CrossRef]
- Perez, M.A.; Soto, J.L. Synthesis of 6-alkoxy-2-amino-5-cyanopyrimidines through sodium alkoxide-induced regiospecific cyclization of 1, 3-dicarbonitriles. Synthesis 1981, 12, 955–958. [Google Scholar] [CrossRef]
- Tietz, O.; Sharma, S.K.; Kaur, J.; Way, J.; Marshall, A.; Wuest, M.; Wuest, F. Synthesis of three 18 F-labelled cyclooxygenase-2 (COX-2) inhibitors based on a pyrimidine scaffold. Org. Biomol. Chem. 2013, 11, 8052–8064. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Lipinski, C.; Lombardo, F.; Dominy, B.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Meerloo, J.V.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Cancer Cell Culture. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [PubMed]
- Sağlık, B.N.; Osmaniye, D.; Levent, S.; Çevik, U.A.; Çavuşoğlu, B.K.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis and biological assessment of new selective COX-2 inhibitors including methyl sulfonyl moiety. Eur. J. Med. Chem. 2021, 209, 112918. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | COX-2 Inhibition (%) a | COX-2 IC50 (µM) b | ||||
|---|---|---|---|---|---|---|
| 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | ||
| 3a | 71.20 ± 1.13 | 62.51 ± 0.95 | 41.14 ± 0.55 | 32.03 ± 0.61 | 22.29 ± 0.45 | 0.23 ± 0.01 |
| 3b | 40.31 ± 0.62 | 33.29 ± 0.77 | 22.39 ± 0.59 | 77.01 ± 0.03 | 61.28 ± 0.96 | 0.20 ± 0.01 |
| 3c | 74.31 ± 1.03 | 57.14 ± 0.92 | 37.58 ± 0.60 | 32.52 ± 0.53 | 20.98 ± 0.40 | 0.29 ± 0.01 |
| 3d | 38.27 ± 0.63 | 31.61 ± 0.54 | 21.51 ± 0.49 | 70.28 ± 1.11 | 60.35 ± 0.98 | 0.34 ± 0.01 |
| 5a | 40.12 ± 0.57 | 30.25 ± 0.49 | 23.55 ± 0.51 | 72.17 ± 1.31 | 61.28 ± 0.84 | 0.26 ± 0.01 |
| 5b | 40.21 ± 0.58 | 36.22 ± 0.44 | 24.74 ± 0.31 | 75.25 ± 1.11 | 59.46 ± 1.04 | 0.18 ± 0.01 |
| 5c | 42.15 ± 0.93 | 28.67 ± 0.51 | 20.31 ± 0.52 | 73.23 ± 1.21 | 64.30 ± 1.05 | 0.26 ± 0.01 |
| 5d | 41.80 ± 0.91 | 35.87 ± 0.72 | 23.15 ± 0.25 | 76.14 ± 1.05 | 60.67 ± 1.09 | 0.16 ± 0.01 |
| Celecoxib | 75.27 ± 1.20 | 61.21 ± 0.98 | 58.21 ± 0.82 | 38.25 ± 0.70 | 19.31 ± 0.51 | 0.17 ± 0.01 |
| Nimesulide | 69.52 ± 1.02 | 36.39 ± 0.84 | 29.43 ± 0.55 | 24.03 ± 0.42 | 17.08 ± 0.38 | 1.68 ± 0.22 |
| Cytotoxicity IC50 (nM) a | WI-38 IC50 (µM) a | ||||
|---|---|---|---|---|---|
| Compound | MCF7 | A549 | A498 | HepG2 | |
| 3b | 3 ± 0.1 | 19 ± 0.52 | 4 ± 0.15 | 22 ± 0.62 | 94.71 ± 2.5 |
| 5b | 2 ± 0.05 | 3 ± 0.07 | 1 ± 0.05 | 12 ± 0.38 | 89.62 ± 2.75 |
| 5d | 1 ± 0.03 | 1 ± 0.05 | 2 ± 0.08 | 9 ± 0.34 | 91.29 ± 0.47 |
| Doxorubicin | 9 ± 0.26 | 13 ± 0.42 | 7 ± 0.23 | 25 ± 0.55 | 8.83 ± 0.09 |
| Compounds | %G0-G1 | %S | %G2/M | %Pre-G1 |
|---|---|---|---|---|
| 5d/MCF-7 | 24.39 ± 3.01 ** | 16.23 ± 1.23 * | 58.37 ± 0.52 *** | 26.16 ± 1.53 |
| cont. MCF-7 | 59.38 ± 2.26 | 29.02 ± 1.81 | 11.71 ± 0.62 | 1.62 ± 0.06 |
| Compounds | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 5d/MCF-7 | 26.16 ± 1.13 *** | 6.78 ± 0.25 | 14.96 ± 1.10 | 2.83 ± 0.47 |
| cont. MCF-7 | 1.62 ± 0.06 | 0.53 ± 0.01 | 0.25 ± 0.005 | 1.07 ± 0.04 |
| Cpd No. | Binding Scores (kcal Mol−1) | Ligand Atom | Residue | Binding Interaction | Bond Length (Å) |
|---|---|---|---|---|---|
| 3b | −5.64 | C21 | Leu352 | H-bond | 3.05 |
| 4-CH3O-phenyl | Arg120 | Arene cation | 4.29 | ||
| 5b | −4.93 | NH | Leu352 | H-bond | 2.94 |
| O of SO2 | Arg513 | H-bond | 3.21 | ||
| 5d | −3.76 | C35 | Leu352 | H-bond | 3.10 |
| O32 of SO2 | Arg513 | H-bond | 2.68 | ||
| O34 of SO2 | His90 | H-bond | 3.24 | ||
| 4-CH3O-phenyl | Arg120 | Arene cation | 4.19 | ||
| Celecoxib | −6.13 | NH2 | Leu352 | H-bond | 3.55 |
| NH2 | Gln192 | H-bond | 3.39 | ||
| O of SO2 | Arg513 | H-bond | 3.15 | ||
| Phenyl ring | Ser352 | Arene-H | 3.80 |
| Molecule | GI Absorption | BBB Permeant | Pgp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|---|---|
| (3b) | High | No | No | Yes | No | Yes | Yes | No |
| (5b) | High | No | No | Yes | No | Yes | No | Yes |
| (5d) | Low | No | No | Yes | Yes | Yes | No | No |
| Celecoxib | High | No | No | Yes | No | Yes | No | No |
| Molecule | a MW < 500 | b Log Po/w < 5 | c HBA < 10 | d HBD < 5 | e TPSA Ǻ2 < 160 | f Log S | g NRB < 5 | #Heavy Atoms |
|---|---|---|---|---|---|---|---|---|
| (3b) | 387.39 | 3.44 | 7 | 1 | 106.09 | 5.46 * | 6 | 29 |
| (5b) | 410.45 | 4.03 | 7 | 1 | 122.58 | 5.32 * | 6 | 29 |
| (5d) | 464.42 | 5.89 | 10 | 1 | 122.58 | 5.86 * | 7 | 32 |
| Celecoxib | 381.37 | 5.75 | 7 | 1 | 86.36 | 4.89 * | 4 | 26 |
| Molecule | Lipinski #Violations | Ghose #Violations | Veber #Violations | Egan #Violations | Muegge #Violations | PAINS #Alerts | Brenk #Alerts |
|---|---|---|---|---|---|---|---|
| (3b) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (5b) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (5d) | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| Celecoxib | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AL-Ghulikah, H.A.; El-Sebaey, S.A.; Bass, A.K.A.; El-Zoghbi, M.S. New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molecular Docking, and In Silico ADME Profile Studies. Molecules 2022, 27, 7485. https://doi.org/10.3390/molecules27217485
AL-Ghulikah HA, El-Sebaey SA, Bass AKA, El-Zoghbi MS. New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molecular Docking, and In Silico ADME Profile Studies. Molecules. 2022; 27(21):7485. https://doi.org/10.3390/molecules27217485
Chicago/Turabian StyleAL-Ghulikah, Hanan A., Samiha A. El-Sebaey, Amr K. A. Bass, and Mona S. El-Zoghbi. 2022. "New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molecular Docking, and In Silico ADME Profile Studies" Molecules 27, no. 21: 7485. https://doi.org/10.3390/molecules27217485
APA StyleAL-Ghulikah, H. A., El-Sebaey, S. A., Bass, A. K. A., & El-Zoghbi, M. S. (2022). New Pyrimidine-5-Carbonitriles as COX-2 Inhibitors: Design, Synthesis, Anticancer Screening, Molecular Docking, and In Silico ADME Profile Studies. Molecules, 27(21), 7485. https://doi.org/10.3390/molecules27217485