


MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines
 ,
,
Abstract
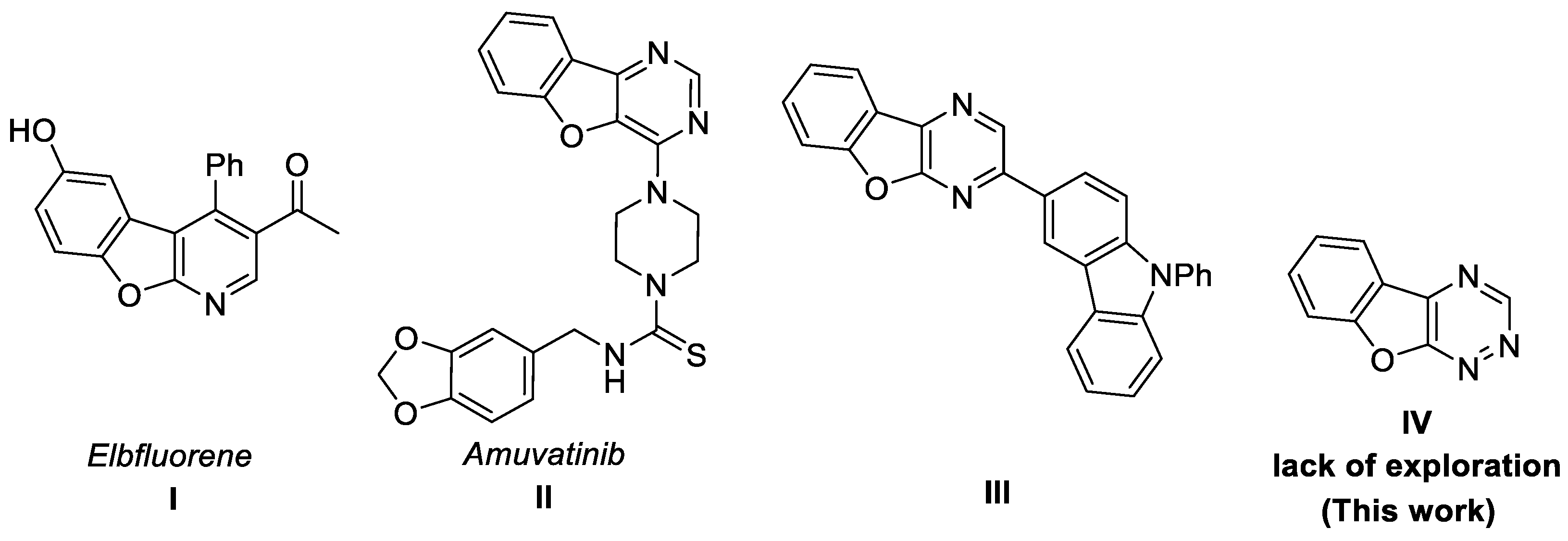
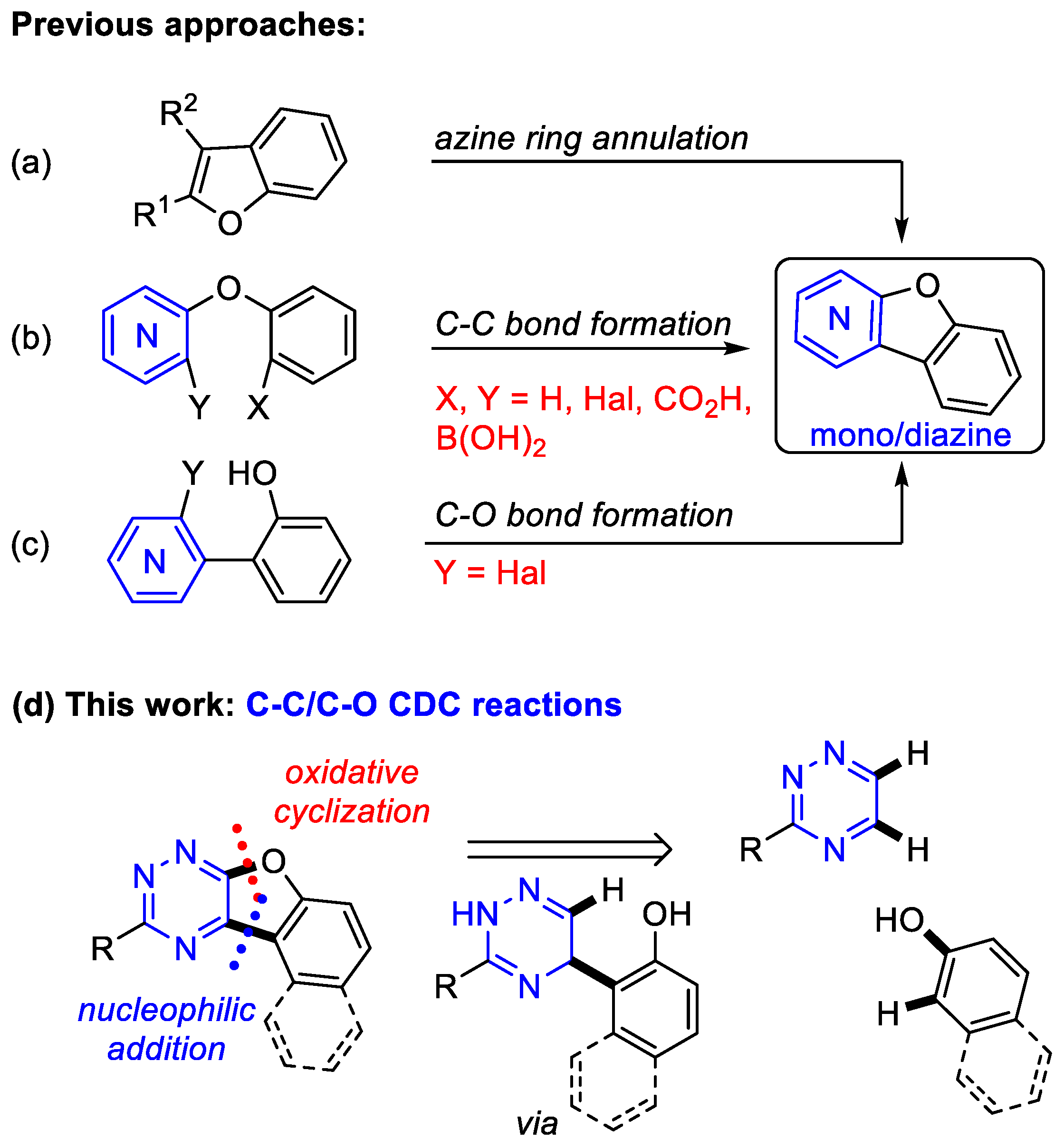
1. Introduction
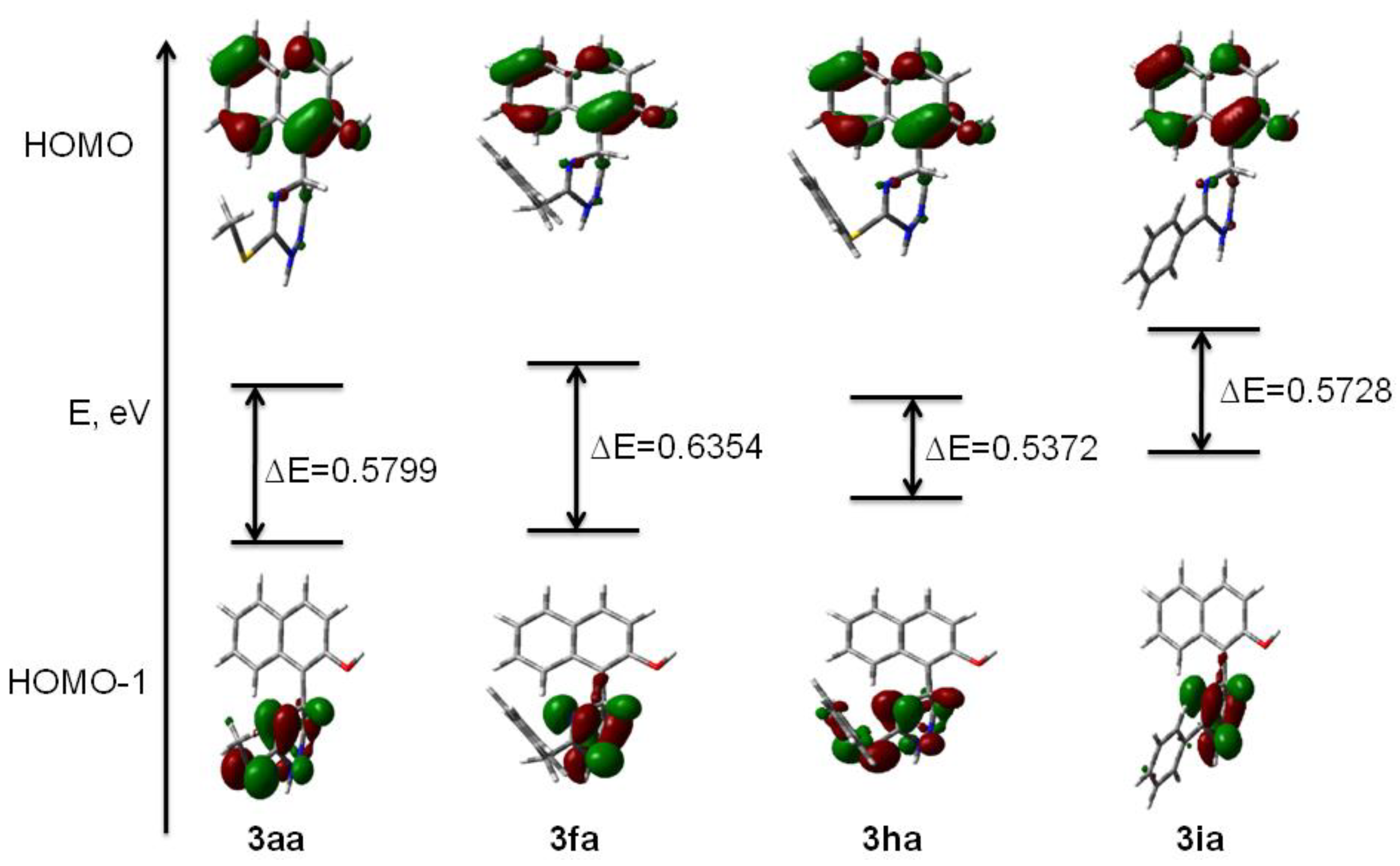
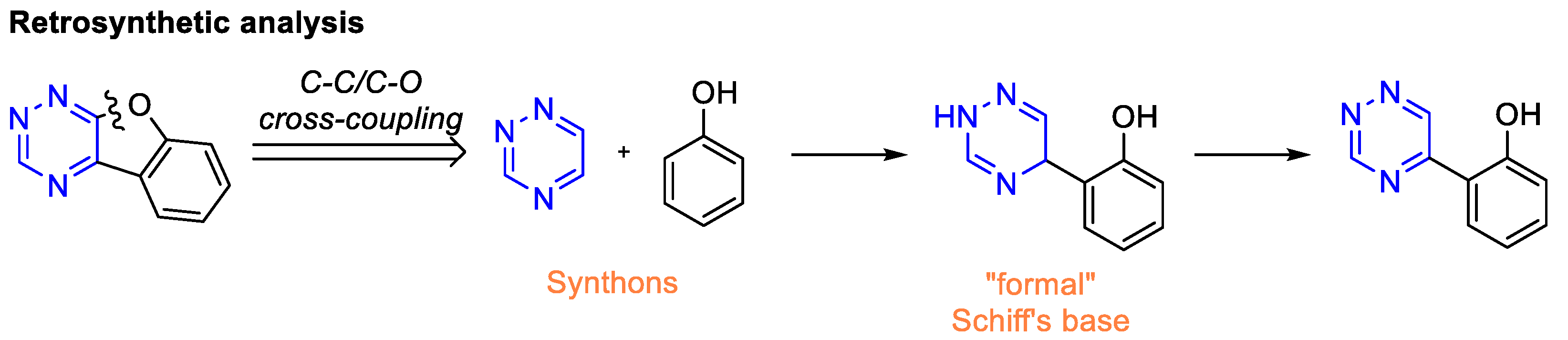
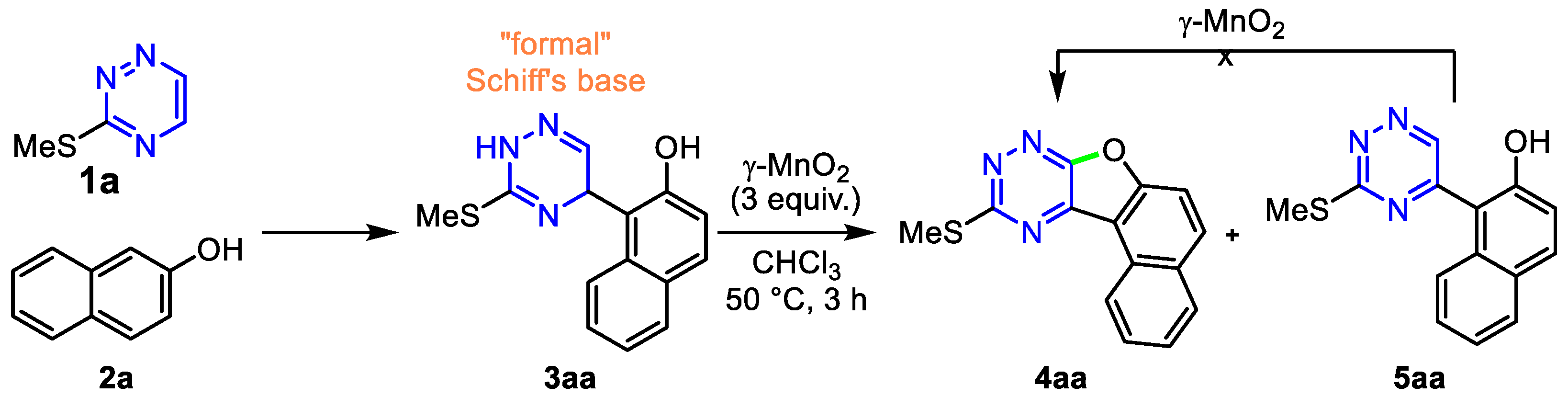
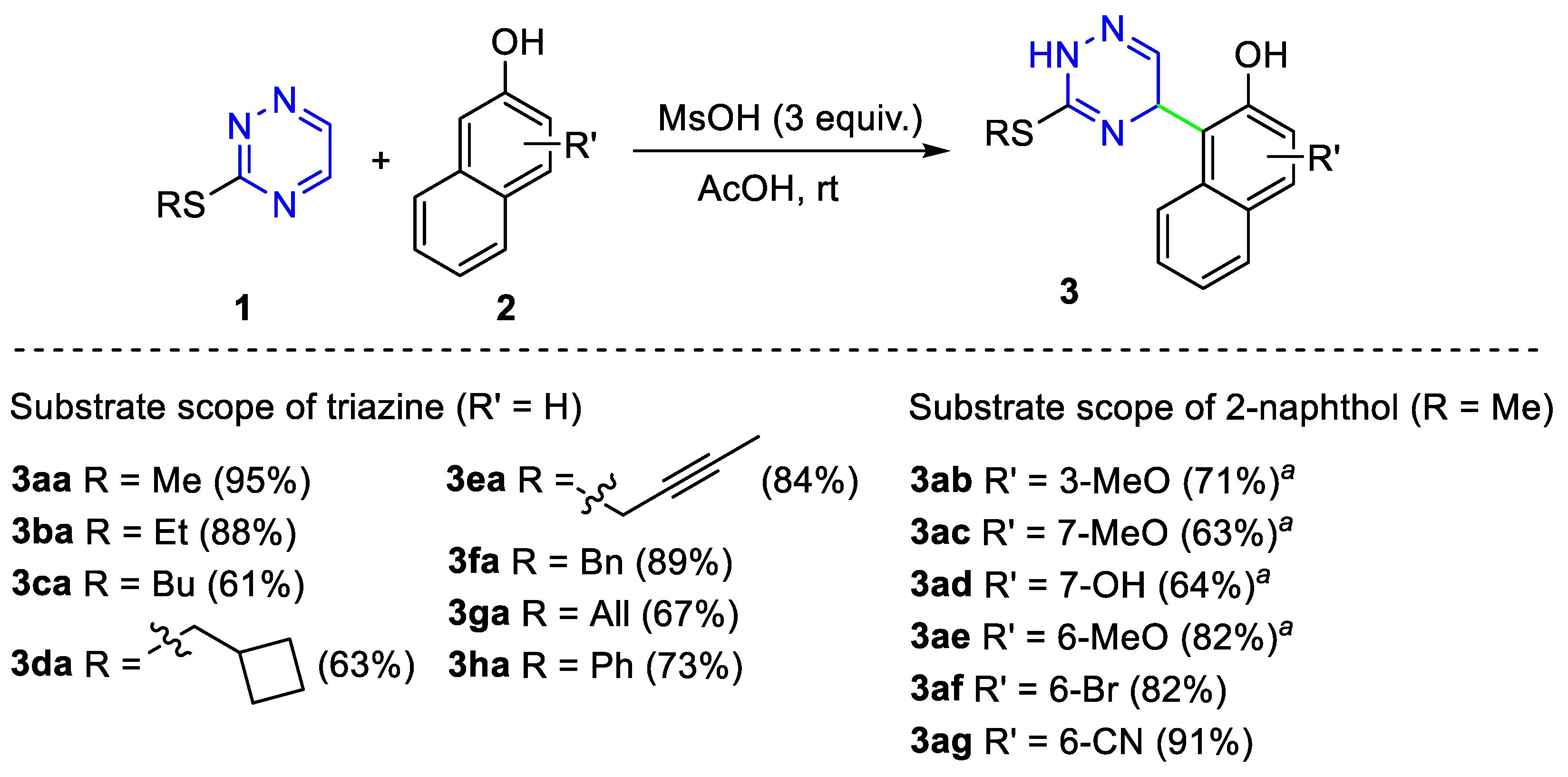
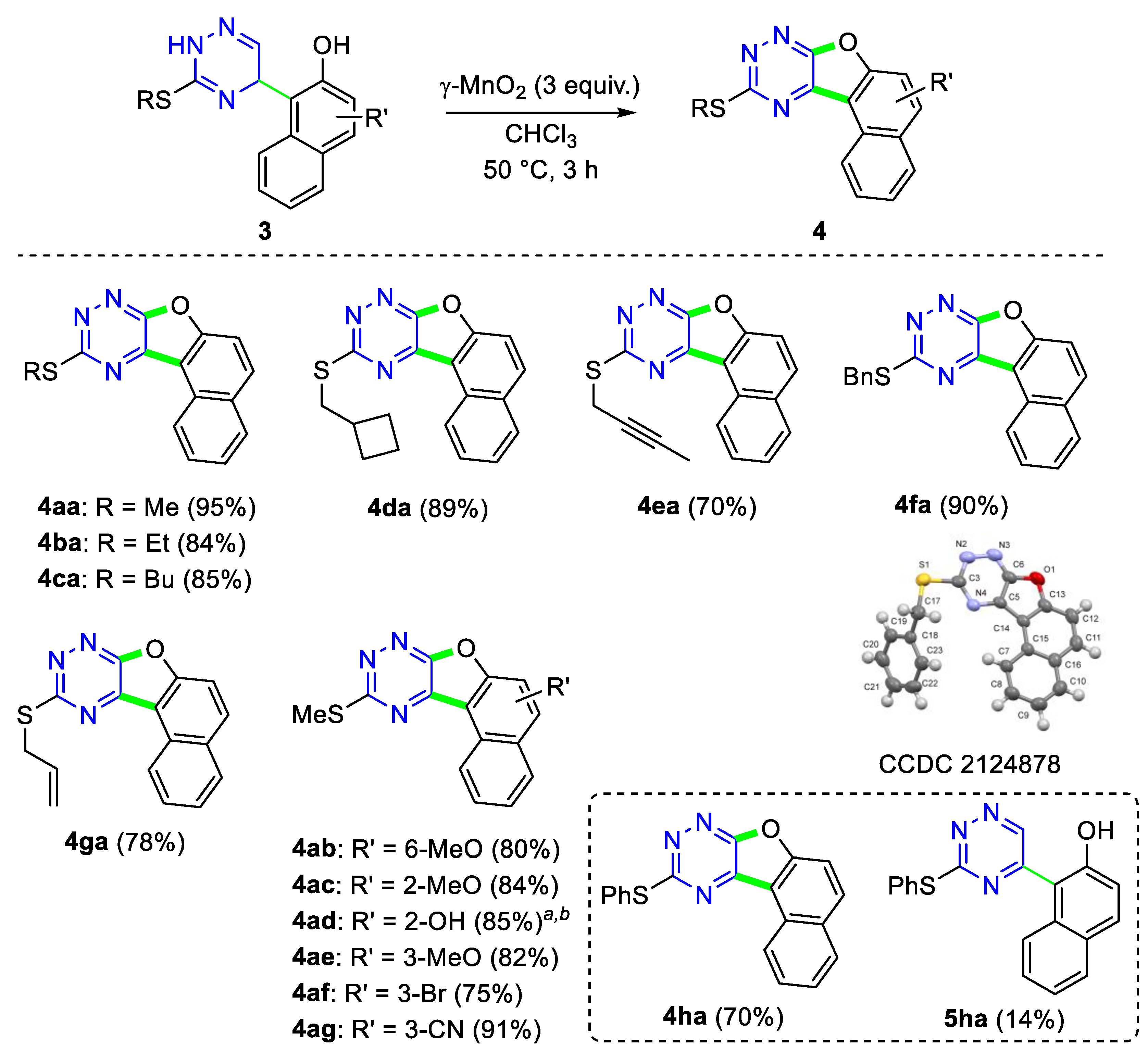
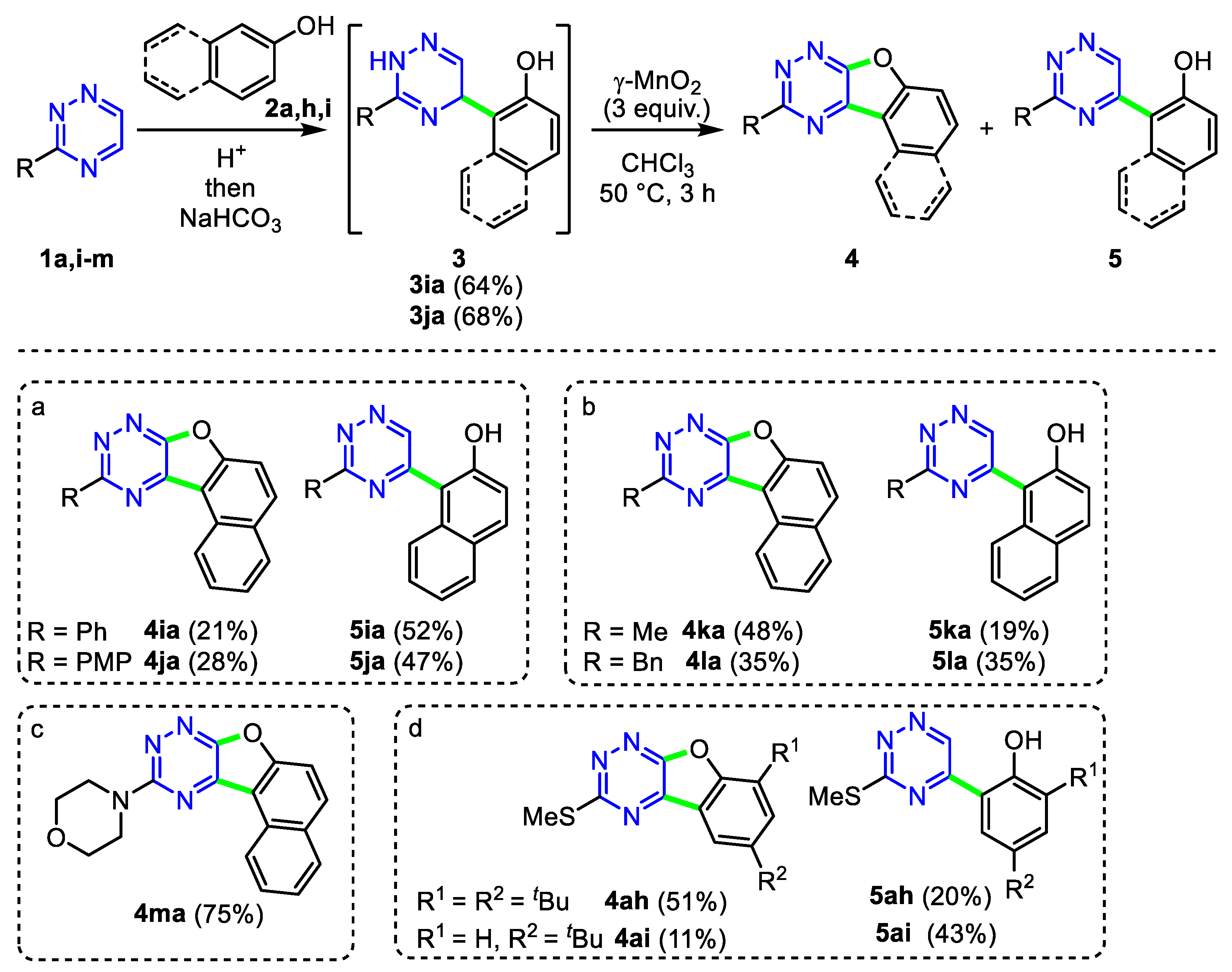
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of S-substituted 3-thio-1,2,4-triazines 1a-f
3.1.1. Synthesis of 3-(phenylthio)-1,2,4-triazine 1h
3.1.2. Synthesis of 3-phenyl- and 3-(4-methoxyphenyl)-1,2,4-triazines 1i and 1j
3.1.3. Synthesis of 3-methyl- and 3-benzyl-1,2,4-triazine 1k and 1l
3.1.4. Synthesis of 4-(1,2,4-triazin-3-yl)morpholine 1m
3.2. General Procedure for the Synthesis of Dihydrotriazines 3
3.2.1. Method A
3.2.2. Method B
3.3. General Procedure for the Synthesis of Naphthofuro-Fused Triazines 4
3.3.1. 10-(Methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4aa
3.3.2. 10-(Ethylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ba
3.3.3. 10-(Butylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ca
3.3.4. 10-((Cyclobutylmethyl)thio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4da
3.3.5. 10-(But-2-yn-1-ylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ea
3.3.6. 10-(Benzylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4fa
3.3.7. 10-(Allylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ga
3.3.8. 10-(Phenylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ha
3.3.9. 1-(3-(Phenylthio)-1,2,4-triazin-5-yl)naphthalen-2-ol 5ha
3.3.10. 10-Phenylnaphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ia
3.3.11. 1-(3-Phenyl-1,2,4-triazin-5-yl)naphthalen-2-ol 5ia
3.3.12. 10-(4-Methoxyphenyl)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ja
3.3.13. 1-(3-(4-Methoxyphenyl)-1,2,4-triazin-5-yl)naphthalen-2-ol 5ja
3.3.14. 6-Methoxy-10-(methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ab
3.3.15. 2-Methoxy-10-(methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ac
3.3.16. 10-(Methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazin-2-ol 4ad
3.3.17. 3-Methoxy-10-(methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ae
3.3.18. 3-Bromo-10-(methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4af
3.3.19. 10-(Methylthio)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine-3-carbonitrile 4ag
3.3.20. Synthesis of 10-alkyl naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazines 4ka and 4la
3.3.21. 10-Methylnaphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ka
3.3.22. 1-(3-Methyl-1,2,4-triazin-5-yl)naphthalen-2-ol 5ka
3.3.23. 10-Benzylnaphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4la
3.3.24. 1-(3-Methyl-1,2,4-triazin-5-yl)naphthalen-2-ol 5la
3.3.25. 10-Morpholinonaphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 4ma
3.3.26. Synthesis of Benzofuro-Fused Triazines 4ah and 4ai
3.3.27. 6,8-Di-tert-butyl-3-(methylthio)benzofuro[3,2-e][1,2,4]triazine 4ah
3.3.28. 2,4-Di-tert-butyl-6-(3-(methylthio)-1,2,4-triazin-5-yl)phenol 5ah
3.3.29. 6-(tert-Butyl)-3-(methylthio)benzofuro[3,2-e][1,2,4]triazine 4ai
3.3.30. 4-(tert-Butyl)-2-(3-(methylthio)-1,2,4-triazin-5-yl)phenol 5ai
3.3.31. 40 mmol Scaled Synthesis of 3ad
3.4. Further Modifications of Compound 4aa
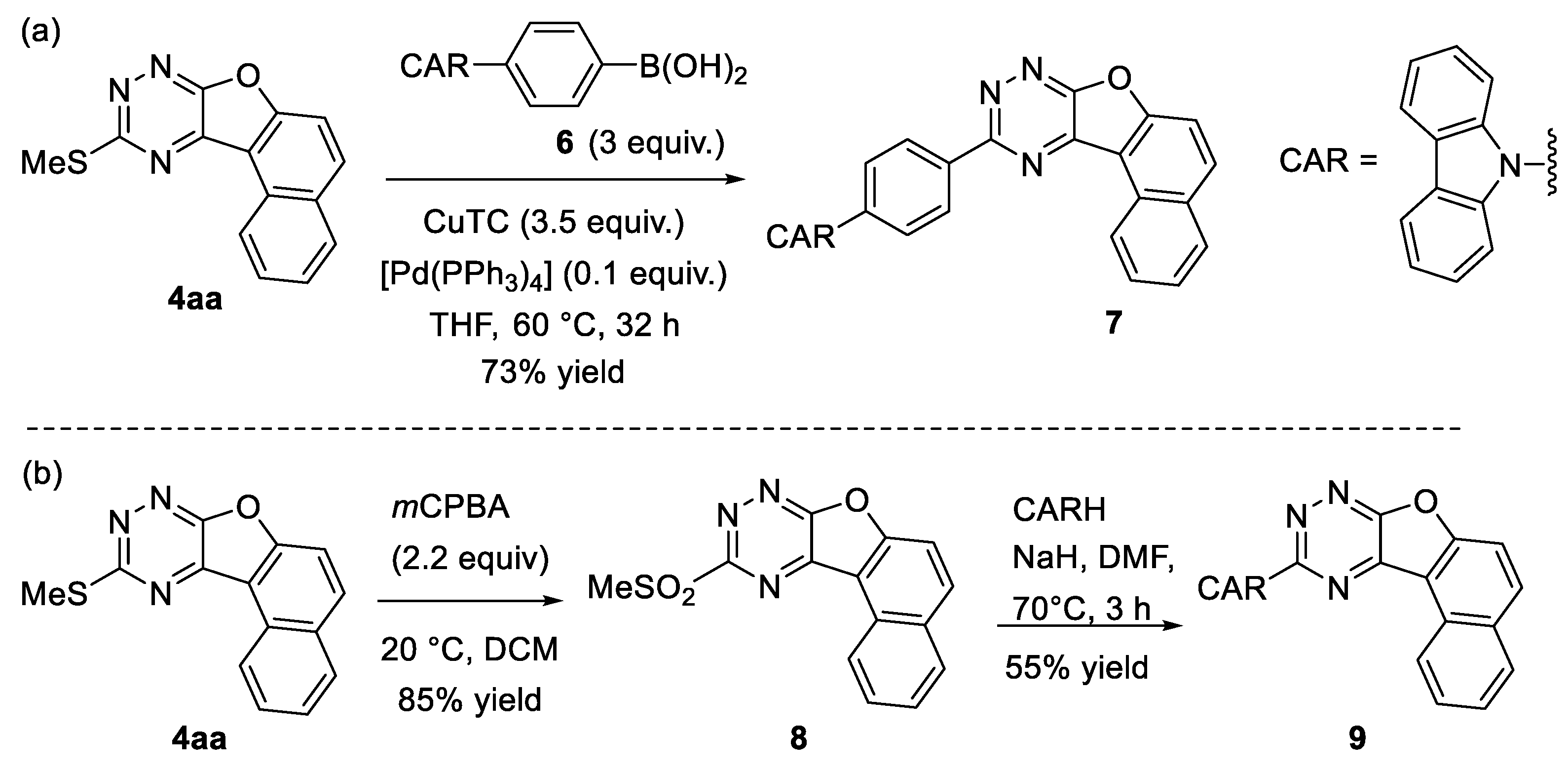
3.4.1. 10-(4-(Carbazol-9-yl)phenyl)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 7
3.4.2. 10-(Methylsulfonyl)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 8
3.4.3. 10-(Carbazol-9-yl)naphtho[1′,2′:4,5]furo[3,2-e][1,2,4]triazine 9
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, N.; Hopkinson, M.N.; Wencel-Delord, J.; Glorius, F. Beyond Directing Groups: Transition-Metal-Catalyzed C-H Activation of Simple Arenes. Angew. Chem. Int. Ed. 2012, 51, 10236–10254. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, S.R.; Sanford, M.S. Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Direct C−H transformation via iron catalysis. Chem. Rev. 2011, 111, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef]
- Rit, R.K.; Yadav, M.R.; Ghosh, K.; Sahoo, A.K. Reusable directing groups [8-aminoquinoline, picolinamide, sulfoximine] in C(sp3)–H bond activation: Present and future. Tetrahedron 2015, 71, 4450–4459. [Google Scholar] [CrossRef]
- Bag, S.; Patra, T.; Modak, A.; Deb, A.; Maity, S.; Dutta, U.; Dey, A.; Kancherla, R.; Maji, A.; Hazra, A.; et al. Remote para-C–H Functionalization of Arenes by a D-Shaped Biphenyl Template-Based Assembly. J. Am. Chem. Soc. 2015, 137, 11888–11891. [Google Scholar] [CrossRef]
- Yoshikai, N. Cobalt-Catalyzed C-C Bond-Forming Reactions via Chelation-Assisted CH Activation. Yuki Gosei Kagaku Kyokaishi 2014, 72, 1198–1206. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. New Trends in Enantioselective Cross-Dehydrogenative Coupling. Catalysts 2020, 10, 529. [Google Scholar] [CrossRef]
- Bosque, I.; Chinchilla, R.; Gonzalez-Gomez, J.C.; Guijarro, D.; Alonso, F. Cross-dehydrogenative coupling involving benzylic and allylic C–H bonds. Org. Chem. Front. 2020, 7, 1717–1742. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, L.; Chen, T.; Han, L. Cross-Dehydrogenative Alkynylation: A Powerful Tool for the Synthesis of Internal Alkynes. ChemSusChem 2020, 13, 4776–4794. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K. Recent advances in oxidative C–C coupling reaction of amides with carbon nucleophiles. Tetrahedron Lett. 2017, 58, 4655–4662. [Google Scholar] [CrossRef]
- Li, C.-J. Cross-dehydrogenative coupling (CDC): Exploring C−C bond formations beyond functional group transformations. Acc. Chem. Res. 2009, 42, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, Z.; Li, C.-J. Cross-dehydrogenative coupling: A sustainable reaction for C–C bond formations. Green Chem. 2021, 23, 6789–6862. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Kang, H.; Li, J.; Li, C.-J. En route to intermolecular cross-dehydrogenative coupling reactions. J. Org. Chem. 2019, 84, 12705–12721. [Google Scholar] [CrossRef]
- Rohit, K.R.; Radhika, S.; Saranya, S.; Anilkumar, G. Manganese-Catalysed Dehydrogenative Coupling—An Overview. Adv. Synth. Catal. 2020, 362, 1602–1650. [Google Scholar] [CrossRef]
- Voigt, B.; Meijer, L.; Lozach, O.; Schächtele, C.; Totzke, F.; Hilgeroth, A. Novel CDK inhibition profiles of structurally varied 1-aza-9-oxafluorenes. Bioorg. Med. Chem. Lett. 2005, 15, 823–825. [Google Scholar] [CrossRef]
- Tell, V.; Mahmoud, K.A.; Wichapong, K.; Schächtele, C.; Totzke, F.; Sippl, W.; Hilgeroth, A. Novel aspects in structure–activity relationships of profiled 1-aza-9-oxafluorenes as inhibitors of Alzheimer’s disease-relevant kinases cdk1, cdk5 and gsk3β. MedChemComm 2012, 3, 1413–1418. [Google Scholar] [CrossRef]
- Tell, V.; Holzer, M.; Herrmann, L.; Mahmoud, K.A.; Schächtele, C.; Totzke, F.; Hilgeroth, A. Multitargeted drug development: Discovery and profiling of dihydroxy substituted 1-aza-9-oxafluorenes as lead compounds targeting Alzheimer disease relevant kinases. Bioorg. Med. Chem. Lett. 2012, 22, 6914–6918. [Google Scholar] [CrossRef]
- Schade, N.; Koch, P.; Ansideri, F.; Krystof, V.; Holzer, M.; Hilgeroth, A. Evaluation of Novel Substituted Furopyridines as Inhibitors of Protein Kinases Related to Tau Pathology in Alzheimer’s Disease. Med. Chem. 2021, 17, 844–855. [Google Scholar] [CrossRef]
- Marciano, R.; David, H.B.; Akabayov, B.; Rotblat, B. The Amuvatinib Derivative, N-(2H-1,3-Benzodioxol-5-yl)-4-{thieno [3,2-d]pyrimidin-4-yl}piperazine-1-carboxamide, Inhibits Mitochondria and Kills Tumor Cells under Glucose Starvation. Int. J. Mol. Sci. 2020, 21, 1041. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.S.H.; Abdel Aziz, Y.M.; Elgawish, M.S.; Said, M.M.; Abouzid, K.A.M. Design, synthesis, biological evaluation and molecular modeling study of new thieno [2,3-d]pyrimidines with anti-proliferative activity on pancreatic cancer cell lines. Bioorg. Chem. 2020, 94, 103472. [Google Scholar] [CrossRef] [PubMed]
- Phillip, C.J.; Zaman, S.; Shentu, S.; Balakrishnan, K.; Zhang, J.; Baladandayuthapani, V.; Taverna, P.; Redkar, S.; Wang, M.; Stellrecht, C.M.; et al. Targeting MET kinase with the small-molecule inhibitor amuvatinib induces cytotoxicity in primary myeloma cells and cell lines. J. Hematol. Oncol. 2013, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J. Targeting receptor tyrosine kinase MET in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef]
- Huang, C.-C.; Xue, M.-M.; Wu, F.-P.; Yuan, Y.; Liao, L.-S.; Fung, M.-K. Deep-blue and hybrid-white organic light emitting diodes based on a twisting carbazole-benzofuro [2,3-b] pyrazine fluorescent emitter. Molecules 2019, 24, 353. [Google Scholar] [CrossRef] [PubMed]
- Salman, G.A.; Nisa, R.U.; Iaroshenko, V.O.; Iqbal, J.; Ayub, K.; Langer, P. Pyrrole versus quinoline formation in the palladium catalyzed reaction of 2-alkynyl-3-bromothiophenes and 2-alkynyl-3-bromofurans with anilines. A combined experimental and computational study. Org. Biomol. Chem. 2012, 10, 9464–9473. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-aryl-7-methoxybenzo[b]furo [3,2-d]pyrimidin-4-amines and their N-arylbenzo[b]thieno [3,2-d]pyrimidin-4-amine analogues as dual inhibitors of CLK1 and DYRK1A kinases. Eur. J. Med. Chem. 2013, 59, 283–295. [Google Scholar] [CrossRef]
- Rao, Y.; Li, Z.; Yin, G. Clean and efficient assembly of functionalized benzofuro [2,3-c] pyridines via metal-free one-pot domino reactions. Green Chem. 2014, 16, 2213–2218. [Google Scholar] [CrossRef]
- Brikci-Nigassa, N.M.; Bentabed-Ababsa, G.; Erb, W.; Chevallier, F.; Picot, L.; Vitek, L.; Fleury, A.; Thiéry, V.; Souab, M.; Robert, T.; et al. 2-Aminophenones, a common precursor to N-aryl isatins and acridines endowed with bioactivities. Tetrahedron 2018, 74, 1785–1801. [Google Scholar] [CrossRef]
- Yonekura, K.; Shinoda, M.; Yonekura, Y.; Tsuchimoto, T. Indium-Catalyzed Annulation of o-Acylanilines with Alkoxyheteroarenes: Synthesis of Heteroaryl[b]quinolines and Subsequent Transformation to Cryptolepine Derivatives. Molecules 2018, 23, 838. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, Y.; Tang, J.; Zhang, L.; Liu, Q.; Li, Q.; Gao, G.; You, J. Ir-Catalyzed Cascade C–H Fusion of Aldoxime Ethers and Heteroarenes: Scope and Mechanisms. ACS Catal. 2020, 10, 203–209. [Google Scholar] [CrossRef]
- Bouarfa, S.; Bentabed-Ababsa, G.; Erb, W.; Picot, L.; Thiéry, V.; Roisnel, T.; Dorcet, V.; Mongin, F. Iodothiophenes and Related Compounds as Coupling Partners in Copper-Mediated N-Arylation of Anilines. Synthesis 2021, 53, 1271–1284. [Google Scholar] [CrossRef]
- Rong, B.; Xu, G.; Yan, H.; Zhang, S.; Wu, Q.; Zhu, N.; Fang, Z.; Duan, J.; Guo, K. Synthesis of benzofuro-and benzothieno [2,3-c]pyridines via copper-catalyzed [4+2] annulation of ketoxime acetates with acetoacetanilide. Org. Chem. Front. 2021, 8, 2939–2943. [Google Scholar] [CrossRef]
- Sadeghzadeh, P.; Pordel, M.; Davoodnia, A. Synthesis of 3H-[1]Benzofuro [2,3-b]imidazo [4,5-f]quinolines as New Fluorescent Heterocyclic Systems for Dye-Sensitized Solar Cells. Russ. J. Org. Chem. 2021, 57, 440–447. [Google Scholar] [CrossRef]
- Yue, W.S.; Li, J.J. A concise synthesis of all four possible benzo [4,5]furopyridines via palladium-mediated reactions. Org. Lett. 2002, 4, 2201–2203. [Google Scholar] [CrossRef]
- Sun, W.; Wang, M.; Zhang, Y.; Wang, L. Synthesis of Benzofuro [3,2-b]pyridines via Palladium-Catalyzed Dual C–H Activation of 3-Phenoxypyridine 1-Oxides. Org. Lett. 2015, 17, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, R.M.; Hickey, A.; Reen, F.J.; O’Gara, F.; McGlacken, G.P. Synthesis of Benzofuroquinolines via Phosphine-Free Direct Arylation of 4-Phenoxyquinolines in Air. Eur. J. Org. Chem. 2018, 2018, 6140–6149. [Google Scholar] [CrossRef]
- Shanahan, R.M.; Hickey, A.; Bateman, L.M.; Light, M.E.; McGlacken, G.P. One-Pot Cross-Coupling/C–H Functionalization Reactions: Quinoline as a Substrate and Ligand through N–Pd Interaction. J. Org. Chem. 2020, 85, 2585–2596. [Google Scholar] [CrossRef]
- Rathod, P.K.; Jonnalagadda, S.; Panaganti, L. A simple and efficient synthesis of benzofuroquinolines via the decarboxylative cross-coupling. Tetrahedron Lett. 2021, 66, 152808. [Google Scholar] [CrossRef]
- Nakamura, S.; Tohnai, N.; Nishii, Y.; Hinoue, T.; Miura, M. Effect of Substitution Pattern of tert-Butyl Groups in a Bisbenzofuropyrazine Core π-System on Optical Properties: Unique Mechanochromic Fluorescence Behavior. ChemPhotoChem 2019, 3, 46–53. [Google Scholar] [CrossRef]
- Singh, R.; Bhatia, H.; Prakash, P.; Debroye, E.; Dey, S.; Dehaen, W. Tandem Nenitzescu reaction/nucleophilic aromatic substitution to form novel pyrido fused indole frameworks. Eur. J. Org. Chem. 2021, 2021, 4865–4875. [Google Scholar] [CrossRef]
- Qi, X.; Xiang, H.; He, Q.; Yang, C. Synthesis of multisubstituted 2-aminopyrroles/pyridines via chemoselective Michael addition/intramolecular cyclization reaction. Org. Lett. 2014, 16, 4186–4189. [Google Scholar] [CrossRef] [PubMed]
- Miliutina, M.; Janke, J.; Hassan, S.; Zaib, S.; Iqbal, J.; Lecka, J.; Sévigny, J.; Villinger, A.; Friedrich, A.; Lochbrunner, S.; et al. A domino reaction of 3-chlorochromones with aminoheterocycles. Synthesis of pyrazolopyridines and benzofuropyridines and their optical and ecto-5′-nucleotidase inhibitory effects. Org. Biomol. Chem. 2018, 16, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jing, C.; Hang, D.; Fan, H.; Duan, L.; Fang, S.; Yan, L. Synthesis, characterization, and photoelectric properties of iridium(III) complexes containing an N hetero-dibenzofuran C^N ligand. RSC Adv. 2021, 11, 11004–11010. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fitzgerald, A.E.; Mani, N.S. Facile assembly of fused benzo [4,5]furo heterocycles. J. Org. Chem. 2008, 73, 2951–2954. [Google Scholar] [CrossRef] [PubMed]
- Cramp, S.; Dyke, H.J.; Higgs, C.; Clark, D.E.; Gill, M.; Savy, P.; Jennings, N.; Price, S.; Lockey, P.M.; Norman, D.; et al. Identification and hit-to-lead exploration of a novel series of histamine H4 receptor inverse agonists. Bioorg. Med. Chem. Lett. 2010, 20, 2516–2519. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-L.; Steglich, W. Synthesis of some benzofuronaphthyridines and benzofuronaphthyridine derivatives. J. Heterocycl. Chem. 1993, 30, 909–912. [Google Scholar] [CrossRef]
- Xiao, X.; Lai, M.; Song, Z.; Geng, M.; Ding, J.; Xie, H.; Zhang, A. Design, synthesis and pharmacological evaluation of bicyclic and tetracyclic pyridopyrimidinone analogues as new KRASG12C inhibitors. Eur. J. Med. Chem. 2021, 213, 113082. [Google Scholar] [CrossRef]
- Ondachi, P.W.; Comins, D.L. Synthesis of Fused-Ring Nicotine Derivatives from (S)-Nicotine. J. Org. Chem. 2010, 75, 1706–1716. [Google Scholar] [CrossRef]
- Kumar, K.S.; Adepu, R.; Kapavarapu, R.; Rambabu, D.; Krishna, G.R.; Reddy, C.M.; Priya, K.K.; Parsa, K.V.L.; Pal, M. AlCl3 induced C-arylation/cyclization in a single pot: A new route to benzofuran fused N-heterocycles of pharmacological interest. Tetrahedron Lett. 2012, 53, 1134–1138. [Google Scholar] [CrossRef]
- Eid, M.M.; Kadry, A.M.; Hassan, R.A. Synthesis and reactions of some 6-(2-hydroxy-1-naphthyl)-1,2,4-triazines. J. Heterocycl. Chem. 1988, 25, 1117–1118. [Google Scholar] [CrossRef]
- Seitz, G.; Richter, J. Donorsubstituierte Benzonitrile als Seitenkettendienophile bei der intramolekularen [4+2]-Cycloaddition mit inversem Elektronenbedarf. Chem. Ber. 1989, 122, 2177–2181. [Google Scholar] [CrossRef]
- Chupakhin, O.N.; Rusinov, G.L.; Beresnev, D.G.; Charushin, V.N.; Neunhoeffer, H. A simple one pot synthesis of condensed 1,2,4-triazines by using the tandem aN-SNipso and SNH-SNipsa reactions. J. Heterocycl. Chem. 2001, 38, 901–907. [Google Scholar] [CrossRef]
- Fatykhov, R.F.; Savchuk, M.I.; Starnovskaya, E.S.; Bobkina, M.V.; Kopchuk, D.S.; Nosova, E.V.; Zyryanov, G.V.; Khalymbadzha, I.A.; Chupakhin, O.N.; Charushin, V.N.; et al. Nucleophilic substitution of hydrogen–the Boger reaction sequence as an approach towards 8-(pyridin-2-yl) coumarins. Mendeleev Commun. 2019, 29, 299–300. [Google Scholar] [CrossRef]
- Savchuk, M.I.; Kopchuk, D.S.; Taniya, O.S.; Nikonov, I.L.; Egorov, I.N.; Santra, S.; Zyryanov, G.V.; Chupakhin, O.N.; Charushin, V.N. 5-Aryl-6-arylthio-2, 2′-bipyridine and 6-Arylthio-2, 5-diarylpyridine Fluorophores: Pot, Atom, Step Economic (PASE) Synthesis and Photophysical Studies. J. Fluoresc. 2021, 31, 1099–1111. [Google Scholar] [CrossRef]
- Raw, S.A.; Taylor, R.J.K. Highly substituted pyridines via tethered imine–enamine (TIE) methodology. Chem. Commun. 2004, 508–509. [Google Scholar] [CrossRef]
- Papadopoulou, M.V.; Taylor, E.C. Intramolecular Diels-Alder reactions of 1,2,4-triazines. Synthesis of 3-alkylpyridines via Raney nickel desulfurization of thieno [2,3-b] pyridines. Tetrahedron 2021, 89, 132158. [Google Scholar] [CrossRef]
- Moseev, T.D.; Nikiforov, E.A.; Varaksin, M.V.; Starnovskaya, E.S.; Savchuk, M.I.; Nikonov, I.L.; Kopchuk, D.S.; Zyryanov, G.V.; Chupakhin, O.N.; Charushin, V.N. Novel Pentafluorophenyl-and Alkoxyphenyl-Appended 2, 2′-Bipyridine Push–Pull Fluorophores: A Convenient Synthesis and Photophysical Studies. Synthesis 2021, 53, 3597–3607. [Google Scholar] [CrossRef]
- Lipińska, T.; Branowska, D.; Rykowski, A. 1,2,4-triazines in organic synthesis. 8. Intramolecular diels-alder reaction of 5-acyl-1, 2, 4-triazineoxime ethers. New route of synthesis of alkylhetarylketones. Chem. Heterocycl. Compd. 1999, 35, 334–342. [Google Scholar] [CrossRef]
- Krinochkin, A.P.; Kopchuk, D.S.; Kim, G.A.; Shevyrin, V.A.; Santra, S.; Rahman, M.; Taniya, O.S.; Zyryanov, G.V.; Rusinov, V.L.; Chupakhin, O.N. Water-soluble luminescent lanthanide complexes based on C6-DTTA-appended 5-aryl-2, 2′-bipyridines. Polyhedron 2020, 181, 114473. [Google Scholar] [CrossRef]
- Fernández, S.Y.; Raw, S.A.; Taylor, R.J.K. Improved methodologies for the preparation of highly substituted pyridines. J. Org. Chem. 2005, 70, 10086–10095. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ren, N.; Ma, X.; Nie, J.; Zhang, F.-G.; Ma, J.-A. Silver-Catalyzed [3 + 3] Dipolar Cycloaddition of Trifluorodiazoethane and Glycine Imines: Access to Highly Functionalized Trifluoromethyl-Substituted Triazines and Pyridines. ACS Catal. 2019, 9, 4600–4608. [Google Scholar] [CrossRef]
- Yamanaka, H.; Sagi, M.; Wada, K.; Konno, S. Studies on as-triazine derivatives. XV, Intramolecular reverse-electron demand Diels-Alter reaction of 1,2,4-triazine derivatives. Heterocycles 1990, 30, 1009–1021. [Google Scholar] [CrossRef]
- Taylor, E.C.; Pont, J.L. Intramolecular Diels-Alder reactions of 1,2,4-triazines. Synthesis of condensed pyrimidines. J. Org. Chem. 1987, 52, 4287–4292. [Google Scholar] [CrossRef]
- Taylor, E.C.; Pont, J.L.; Warner, J.C. Heterodienophilic intramolecular Diels-Alder reactions of 1,2,4-triazines: Synthesis of novel polycyclic condensed pyrazines and lumazines. Tetrahedron 1987, 43, 5159–5168. [Google Scholar] [CrossRef]
- Taylor, E.C.; French, L.G. Intramolecular Diels-Alder reactions of 1,2,4-triazines. Routes to condensed pyrazines via cycloaddition of nitrile dienophiles. J. Org. Chem. 1989, 54, 1245–1249. [Google Scholar] [CrossRef]
- Khalymbadzha, I.A.; Chupakhin, O.N.; Fatykhov, R.F.; Charushin, V.N.; Schepochkin, A.V.; Kartsev, V.G. Transition-metal-free cross-dehydrogenative coupling of triazines with 5,7-dihydroxycoumarins. Synlett 2016, 27, 2606–2610. [Google Scholar] [CrossRef]
- Khalymbadzha, I.A.; Fatykhov, R.F.; Chupakhin, O.N.; Charushin, V.N.; Tseitler, T.A.; Sharapov, A.D.; Inytina, A.K.; Kartsev, V.G. Transition-Metal-Free C–C Coupling of 5, 7-Dihydroxybenzopyrones with Quinoxalones and Pteridinones. Synthesis 2018, 50, 2423–2431. [Google Scholar] [CrossRef]
- Utepova, I.A.; Nemytov, A.I.; Ishkhanian, V.A.; Chupakhin, O.N.; Charushin, V.N. Metal-free C–H/C–H coupling of 1,3-diazines and 1,2,4-triazines with 2-naphthols facilitated by Brønsted acids. Tetrahedron 2020, 76, 131391. [Google Scholar] [CrossRef]
- Fatykhov, R.; Khalymbadzha, I.; Chupakhin, O. Cross-Dehydrogenative Coupling Reactions between Phenols and Hetarenes: Modern Trends in Cross-Coupling Chemistry of Phenols. Adv. Synth. Catal. 2022, 364, 1052–1068. [Google Scholar] [CrossRef]
- Alphonse, F.-A.; Suzenet, F.; Keromnes, A.; Lebret, B.; Guillaumet, G. A general approach to selective functionalization of 1, 2, 4-triazines using organometallics in palladium-catalyzed cross-coupling and addition reactions. Synthesis 2004, 2004, 2893–2899. [Google Scholar] [CrossRef]
- Costa, S.P.G.; Oliveira, E.; Lodeiro, C.; Raposo, M.M.M. Heteroaromatic alanine derivatives bearing (oligo) thiophene units: Synthesis and photophysical properties. Tetrahedron Lett. 2008, 49, 5258–5261. [Google Scholar] [CrossRef][Green Version]
- Verma, V.; Singh, K.; Kumar, D.; Klapötke, T.M.; Stierstorfer, J.; Narasimhan, B.; Qazi, A.K.; Hamid, A.; Jaglan, S. Synthesis, antimicrobial and cytotoxicity study of 1,3-disubstituted-1H-naphtho [1,2-e][1,3]oxazines. Eur. J. Med. Chem. 2012, 56, 195–202. [Google Scholar] [CrossRef]
- Koleda, O.; Broese, T.; Noetzel, J.; Roemelt, M.; Suna, E.; Francke, R. Synthesis of benzoxazoles using electrochemically generated hypervalent iodine. J. Org. Chem. 2017, 82, 11669–11681. [Google Scholar] [CrossRef]
- Babbs, A.; Berg, A.; Chatzopoulou, M.; Davies, K.E.; Davies, S.G.; Edwards, B.; Elsey, D.J.; Emer, E.; Guiraud, S.; Harriman, S.; et al. 2-Arylbenzo[d]oxazole Phosphinate Esters as Second-Generation Modulators of Utrophin for the Treatment of Duchenne Muscular Dystrophy. J. Med. Chem. 2020, 63, 7880–7891. [Google Scholar] [CrossRef]
- Ravinaik, B.; Ramachandran, D.; Rao, M.V.B. Synthesis and anticancer evaluation of amide derivatives of 1, 3, 4-oxadiazole linked with benzoxazole. Russ. J. Gen. Chem. 2019, 89, 1003–1008. [Google Scholar] [CrossRef]
- Ferreira, R.C.M.; Raposo, M.M.M.; Costa, S.P.G. Heterocyclic amino acids as fluorescent reporters for transition metals: Synthesis and evaluation of novel furyl-benzoxazol-5-yl-l-alanines. N. J. Chem. 2018, 42, 3483–3492. [Google Scholar] [CrossRef]
- Varma, R.S.; Kumar, D. Manganese triacetate oxidation of phenolic schiffs bases: Synthesis of 2-arylbenzoxazoles. J. Heterocycl. Chem. 1998, 35, 1539–1540. [Google Scholar] [CrossRef]
- Ozokan, K.G.; Gumus, M.K.; Kaban, S. Synthesis of hetaryl-substituted benzoxazoles via oxidative cyclization of phenolic schiff’s bases. J. Heterocycl. Chem. 2008, 45, 1831–1834. [Google Scholar] [CrossRef]
- Bougrin, K.; Loupy, A.; Soufiaoui, M. Trois nouvelles voies de synthèse des dérivés 1,3-azoliques sous micro-ondes. Tetrahedron 1998, 54, 8055–8064. [Google Scholar] [CrossRef]
- Vereshchagin, L.I.; Gainulina, S.R.; Podskrebysheva, S.A.; Gaivoroskii, L.A.; Okhapkina, L.L.; Vorob’eva, V.G.; Latyshev, V.P. Reactivity of Manganese Oxides in the Oxidation of Alcohols. J. Org. Chem. USSR 1972, 8, 1143–1147. [Google Scholar]
- Cassis, R.; Valderrama, J.A. Studies on Quinones. XI. Synthesis of Quinones from Hydroquinones by Using Manganese Dioxide and Acid-Impregnated Manganese Dioxide. Synth. Commun. 1983, 13, 347–356. [Google Scholar] [CrossRef]
- Yamaguchi, K.S.; Sawyer, D.T. The Redox Chemistry of Manganese(III) and -(IV) Complexes. Israel J. Chem. 1985, 25, 164–176. [Google Scholar] [CrossRef]
- Moir, M.; Lane, S.; Montgomery, A.P.; Hibbs, D.; Connor, M.; Kassiou, M. The discovery of a potent and selective pyrazolo-[2,3-e]-[1,2,4]-triazine cannabinoid type 2 receptor agonist. Eur. J. Med. Chem. 2021, 210, 113087. [Google Scholar] [CrossRef]
- Zhu, Z.; Glinkerman, C.M.; Boger, D.L. Selective N1/N4 1,4-Cycloaddition of 1,2,4,5-Tetrazines Enabled by Solvent Hydrogen Bonding. J. Am. Chem. Soc. 2020, 142, 20778–20787. [Google Scholar] [CrossRef] [PubMed]
- Branowska, D.; Olender, E.; Świętochowska, M.; Karczmarzyk, Z.; Wysocki, W.; Cichosz, I.; Woźna, A.; Urbańczyk-Lipkowska, Z.; Kalicki, P.; Gil, M. Synthesis and optical properties of some 3,4-(ethylenedioxythiophen-2-yl)-1,2,4-triazine derivatives. Tetrahedron 2017, 73, 411–417. [Google Scholar] [CrossRef]
- Fatykhov, R.F.; Chupakhin, O.N.; Rusinov, V.L.; Khalymbadzha, I.A. Copper catalysis for triazines. In Copper in N-Heterocyclic Chemistry; Srivastava, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 161–220. [Google Scholar] [CrossRef]
- Finlay, M.R.V.; Anderton, M.; Bailey, A.; Boyd, S.; Brookfield, J.; Cairnduff, C.; Charles, M.; Cheasty, A.; Critchlow, S.E.; Culshaw, J.; et al. Discovery of a Thiadiazole–Pyridazine-Based Allosteric Glutaminase 1 Inhibitor Series That Demonstrates Oral Bioavailability and Activity in Tumor Xenograft Models. J. Med. Chem. 2019, 62, 14, 6540–6560. [Google Scholar] [CrossRef]
- Sahin, Z.; Biltekin, S.N.; Ozansoy, M.; Hemiş, B.; Ozansoy, M.B.; Yurttaş, L.; Berk, B.; Demirayak, Ş. Synthesis and in vitro antitumor activities of novel thioamide substituted piperazinyl-1,2,4-triazines. J. Heterocycl. Chem. 2022, 59, 1333–1340. [Google Scholar] [CrossRef]
- Bielawska, A.; Bielawski, K.; Czarnomysy, R.; Gornowicz, A.; Mojzych, M.; Szymanowska, A. The anticancer action of a novel 1,2,4-triazine sulfonamide derivative in colon cancer cells. Molecules 2021, 26, 2045. [Google Scholar] [CrossRef]
- Bashir, M.; Bano, A.; Ijaz, A.S.; Chaudhary, B.A. Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A Review. Molecules 2015, 20, 13496–13517. [Google Scholar] [CrossRef]
- Majid, A.; Ashid, M.; Nasir, H.; Joshi, A. A Convenient Synthesis and Reactions of some Substituted 1,2,4-Triazine, and Their Derivatives with Carbazole, Sulfonamide and Trityl Chloride Moiety of Biological Interest. Eur. J. Mol. Clin. Med. 2020, 7, 994–1002. [Google Scholar]
- Zassowski, P.; Ledwon, P.; Kurowska, A.; Herman, A.P.; Lapkowski, M.; Cherpak, V.; Hotra, Z.; Turyk, P.; Ivaniuk, K.; Stakhira, P.; et al. 1,3,5-Triazine and carbazole derivatives for OLED applications. Dyes Pigment. 2018, 149, 804–811. [Google Scholar] [CrossRef]
- Fatiadi, A.J. Active manganese dioxide oxidation in organic chemistry-part I. Synthesis 1976, 1976, 65–104. [Google Scholar] [CrossRef]
- Miri, R.; Firuzi, O.; Peymani, P.; Zamani, M.; Mehdipour, A.R.; Heydari, Z.; Farahani, M.M.; Shafiee, A. Synthesis, Cytotoxicity, and QSAR Study of New Aza-cyclopenta[b]fluorene-1,9-dione Derivatives. Chem. Biol. Drug Des. 2012, 79, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Lubinu, M.C. Microwave-assisted oxidative aromatization of Hantzsch 1, 4-dihydropyridines using manganese dioxide. Synthesis 2006, 2006, 1283–1288. [Google Scholar] [CrossRef]
- Sengoku, T.; Murata, Y.; Suzuki, C.; Takahashi, M.; Yoda, H. Synthesis of new chiral lactam-fused pyridine derivatives. RSC Adv. 2015, 5, 73562–73565. [Google Scholar] [CrossRef]
- Quinonero, O.; Jean, M.; Vanthuyne, N.; Roussel, C.; Bonne, D.; Constantieux, T.; Bressy, C.; Bugaut, X.; Rodriguez, J. Combining organocatalysis with central-to-axial chirality conversion: Atroposelective hantzsch-type synthesis of 4-arylpyridines. Angew. Chem. Int. Ed. 2016, 55, 1401–1405. [Google Scholar] [CrossRef]
- Shabunina, O.V.; Starnovskaya, E.S.; Shaitz, Y.K.; Kopchuk, D.S.; Sadieva, L.K.; Kim, G.A.; Taniya, O.S.; Nikonov, I.L.; Santra, S.; Zyryanov, G.V.; et al. Asymmetrically substituted 5,5′′-diaryl-2,2′:6′,2′′-terpyridines as efficient fluorescence “turn-on” probes for Zn2+ in food/cosmetic samples and human urine. J. Photochem. Photobiol. Chem. 2021, 408, 113101. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Ban, K.; Duffy, S.; Khakham, Y.; Avery, V.M.; Hughes, A.; Montagnat, O.; Katneni, K.; Ryan, E.; Baell, J.B. 3-Alkylthio-1,2,4-triazine dimers with potent antimalarial activity. Bioorg. Med. Chem. Lett. 2010, 20, 6024–6029. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.C.; Macor, J.E.; Pont, J.L. Intramolecular diels-alder reactions of 1, 2, 4-triazines.: A general synthesis of furo [2,3-b]pyridines, 2,3-dihydropyrano [2,3-b]pyridines, and pyrrolo [2,3-b]pyridines. Tetrahedron 1987, 43, 5145–5158. [Google Scholar] [CrossRef]
- Von Neunhoeffer, H.; Hennig, H.; Frühauf, H.-W.; Mutterer, M. Zur synthese von 1,2,4-triazinen. Tetrahedron Lett. 1969, 10, 3147–3150. [Google Scholar] [CrossRef]
- Shkurko, O.P.; Gogin, L.L.; Baram, S.G.; Mamaev, V.P. Electronic spectra of asym-triazinyl groups. Chem. Heterocycl. Compd. 1987, 23, 216–221. [Google Scholar] [CrossRef]
- Courcot, B.; Tran, D.N.; Fraisse, B.; Bonhomme, F.; Marsura, A.; Ghermani, N.E. Electronic Properties of 3,3′-Dimethyl-5,5′-bis(1,2,4-triazine): Towards Design of Supramolecular Arrangements of N-Heterocyclic CuI Complexes. Chem. Eur. J. 2007, 13, 3414–3423. [Google Scholar] [CrossRef]
- Alekseev, S.G.; Charushin, V.N.; Chupakhin, O.N.; Shorshnev, S.V.; Chernyshev, A.I.; Klyuev, N.A. Reactions of azinium cations. 6. N(1)-alkyl-1,2,4-triazinium salts. Reactions with indoles—The first case of the double addition of nucleophiles to a triazine ring. Chem. Heterocycl. Compd. 1986, 22, 1242–1249. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
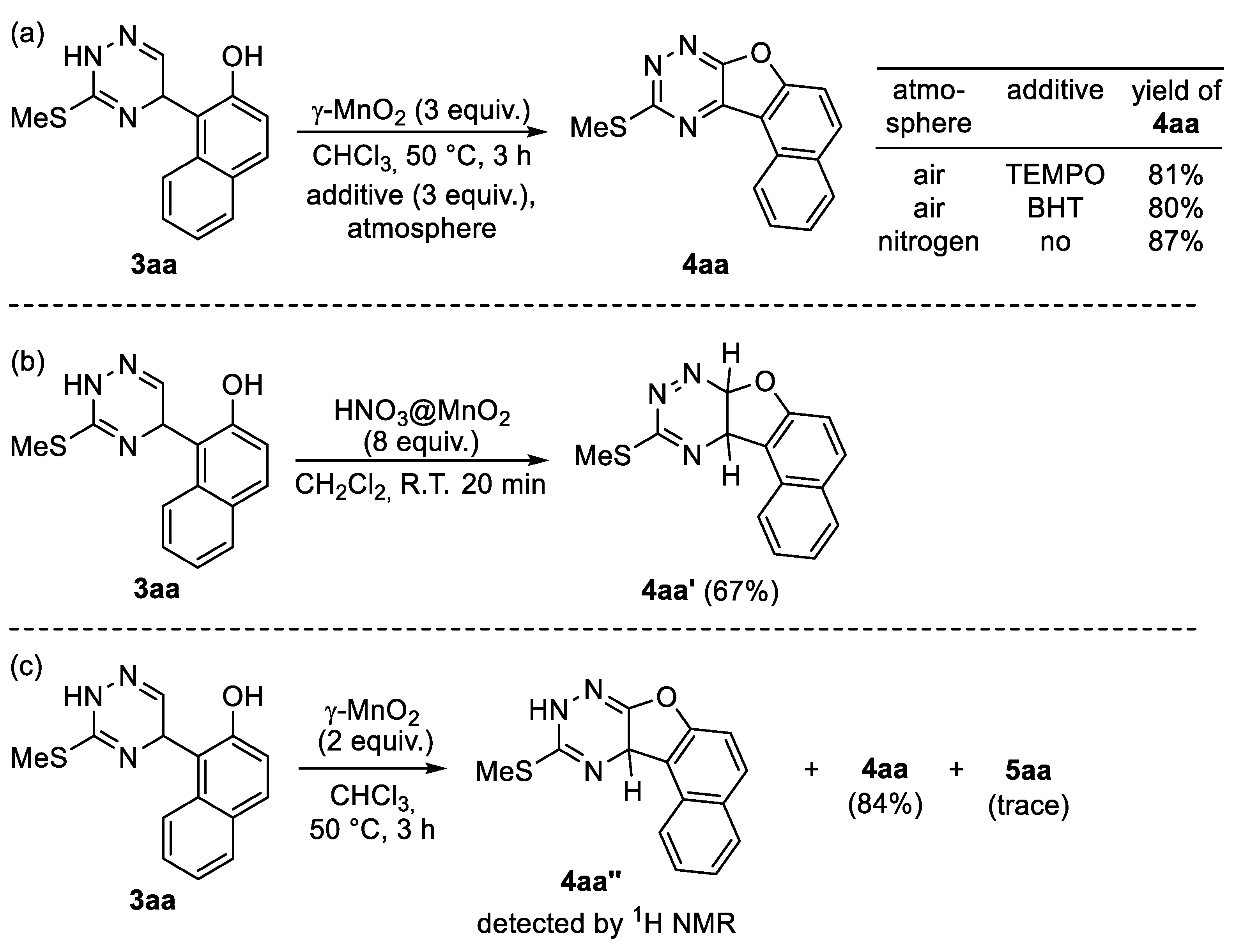
| Entry | Conditions | 4aa (%) b | 5aa (%) b |
|---|---|---|---|
| 1 | Using 3 equiv. of γ-MnO2 | >99 | trace |
| 2 | HNO3@γ-MnO2 instead of γ-MnO2 | 75 | trace |
| 3 | Mn(OAc)2.4H2O instead of γ-MnO2 | - | - |
| 4 | MnCl2 instead of γ-MnO2 | - | - |
| 5 | Mn(acac)2 instead of γ-MnO2 | - | - |
| 6 | Mn(OAc)3.2H2O instead of γ-MnO2 | 29 | 5 |
| 7 | Ag2O instead of γ-MnO2 | 60 | - |
| 8 | DTBP instead of γ-MnO2 | 35 | 15 |
| 9 | DDQ instead of γ-MnO2 | 39 | 43 |
| 10 | p-Chloranil instead of γ-MnO2 | - | 89 |
| 11 | HFIP instead of CHCl3 | 89 | trace |
| 12 | EtOH instead of CHCl3 | 66 | trace |
| 13 | DCE instead of CHCl3 | 85 | trace |
| 14 | Benzene instead of CHCl3 | 90 | trace |
| 15 | Performed at 60 °C | 94 | 4 |
| 16 | Performed at 25 °C | 91 | 3 |
| 17 | Using 2 equiv. of γ-MnO2 | 85 | 3 |
| 18 | Using 5 equiv. of γ-MnO2 | 90 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatykhov, R.F.; Khalymbadzha, I.A.; Sharapov, A.D.; Potapova, A.P.; Mochulskaya, N.N.; Tsmokalyuk, A.N.; Ivoilova, A.V.; Mozharovskaia, P.N.; Santra, S.; Chupakhin, O.N. MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines. Molecules 2022, 27, 7105. https://doi.org/10.3390/molecules27207105
Fatykhov RF, Khalymbadzha IA, Sharapov AD, Potapova AP, Mochulskaya NN, Tsmokalyuk AN, Ivoilova AV, Mozharovskaia PN, Santra S, Chupakhin ON. MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines. Molecules. 2022; 27(20):7105. https://doi.org/10.3390/molecules27207105
Chicago/Turabian StyleFatykhov, Ramil F., Igor A. Khalymbadzha, Ainur D. Sharapov, Anastasia P. Potapova, Nataliya N. Mochulskaya, Anton N. Tsmokalyuk, Alexandra V. Ivoilova, Polina N. Mozharovskaia, Sougata Santra, and Oleg N. Chupakhin. 2022. "MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines" Molecules 27, no. 20: 7105. https://doi.org/10.3390/molecules27207105
APA StyleFatykhov, R. F., Khalymbadzha, I. A., Sharapov, A. D., Potapova, A. P., Mochulskaya, N. N., Tsmokalyuk, A. N., Ivoilova, A. V., Mozharovskaia, P. N., Santra, S., & Chupakhin, O. N. (2022). MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines. Molecules, 27(20), 7105. https://doi.org/10.3390/molecules27207105