
Transcriptomics and Proteomics Characterizing the Anticancer Mechanisms of Natural Rebeccamycin Analog Loonamycin in Breast Cancer Cells

Abstract
1. Introduction
2. Results
2.1. LM Targets TNBC In Vitro and In Vivo
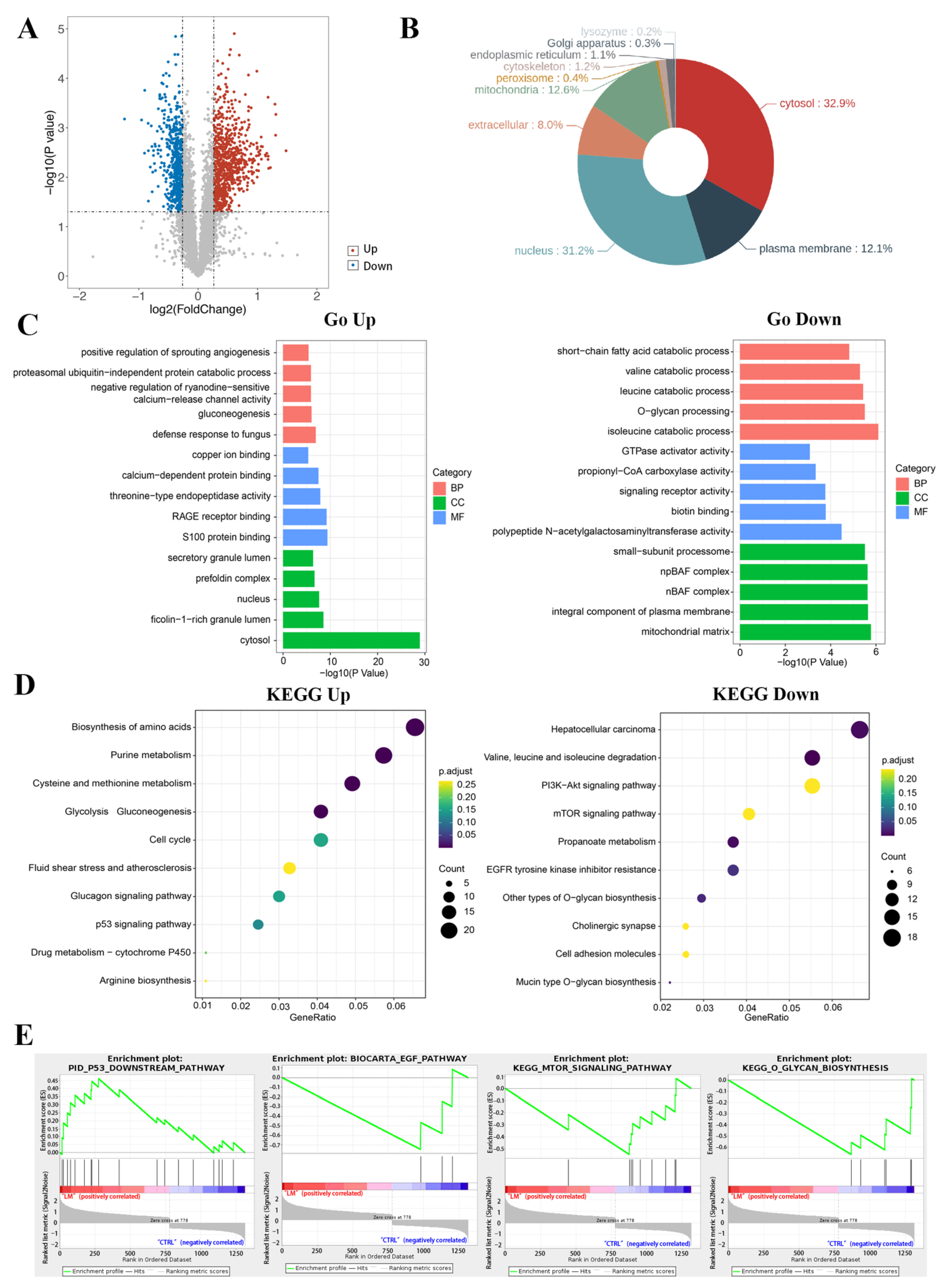
2.2. Functional Annotation Enrichment of LM-Regulated Genes
2.3. Proteomic Expression Profiling of LM-Treated TNBC Cells
2.4. Validation of Transcriptomic and Proteomic Results
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Growth Curve Measured by CCK-8 Method
4.4. Topoisomerase I-Mediated DNA Relaxation and Cleavage Assays
4.5. RNA-seq Analysis
4.6. TMT Labeling and LC-MS/MS Analysis
4.7. Molecular Docking
4.8. Western Blot Analysis
4.9. Xenograft Model
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernández, A.G.; Chabrera, C.; Font, M.G.; Fraile, M.; Lain, J.M.; Gónzalez, S.; Barco, I.; Torres, J.; Piqueras, M.; Cirera, L.; et al. Differential patterns of recurrence and specific survival between luminal A and luminal B breast cancer according to recent changes in the 2013 St Gallen immunohistochemical classification. Clin. Transl. Oncol. 2015, 17, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.L.; Zhang, B.; Xue, W.W.; Li, W.; Xu, Z.F.; Shi, J.; Shen, Y.; Jiao, R.H.; Tan, R.X.; Ge, H.M. Discovery, Biosynthesis, and Heterologous Production of Loonamycin, a Potent Anticancer Indolocarbazole Alkaloid. Org. Lett. 2020, 22, 4665–4669. [Google Scholar] [CrossRef]
- Bailly, C.; Colson, P.; Houssier, C.; Rodrigues-Pereira, E.; Prudhomme, M.; Waring, M.J. Recognition of Specific Sequences in DNA by a Topoisomerase I Inhibitor Derived from the Antitumor Drug Rebeccamycin. Mol. Pharmacol. 1998, 53, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Schwandt, A.; Mekhail, T.; Halmos, B.; O’Brien, T.; Ma, P.C.; Fu, P.; Ivy, P.; Dowlati, A. Phase-II Trial of Rebeccamycin Analog, a Dual Topoisomerase-I and -II Inhibitor, in Relapsed “Sensitive” Small Cell Lung Cancer. J. Thorac. Oncol. 2012, 7, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Sellers, S.; Diasio, R.B.; Douillard, J.-Y. A phase I dose-escalation study of edotecarin (J-107088) combined with infusional 5-fluorouracil and leucovorin in patients with advanced/metastatic solid tumors. Anti-Cancer Drugs 2010, 21, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Berger, J.M. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes 2019, 10, 859. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Perego, P.; Zunino, F. Targeting topoisomerase I: Molecular mechanisms and cellular determinants of response to topoisomerase I inhibitors. Expert Opin. Ther. Targets 2008, 12, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Manasrah, B.K.; McGregor, S.M.; Lera, R.F.; Norman, R.X.; Tucker, J.B.; Scribano, C.M.; Yan, R.E.; Humayun, M.; Wisinski, K.B.; et al. Paclitaxel Induces Micronucleation and Activates Pro-Inflammatory cGAS–STING Signaling in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2021, 20, 2553–2567. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Topoisomerase I Poisons and Suppressors as Anticancer Drugs. Curr. Med. Chem. 2000, 7, 39–58. [Google Scholar] [CrossRef]
- Nitiss, J.L. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Et Biophys. Acta Gene Struct. Expr. 1998, 1400, 63–81. [Google Scholar] [CrossRef]
- Wang, J.C. DNA TOPOISOMERASES. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef]
- Baglini, E.; Salerno, S.; Barresi, E.; Robello, M.; Da Settimo, F.; Taliani, S.; Marini, A.M. Multiple Topoisomerase I (TopoI), Topoisomerase II (TopoII) and Tyrosyl-DNA Phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci. 2021, 156, 105594. [Google Scholar] [CrossRef]
- Liu, J.; Geng, G.; Liang, G.; Wang, L.; Luo, K.; Yuan, J.; Zhao, S. A novel topoisomerase I inhibitor DIA-001 induces DNA damage mediated cell cycle arrest and apoptosis in cancer cell. Ann. Transl. Med. 2020, 8, 89. [Google Scholar] [CrossRef]
- Robey, R.W.; Obrzut, T.; Shukla, S.; Polgar, O.; Macalou, S.; Bahr, J.C.; Di Pietro, A.; Ambudkar, S.V.; Bates, S.E. Becatecarin (rebeccamycin analog, NSC 655649) is a transport substrate and induces expression of the ATP-binding cassette transporter, ABCG2, in lung carcinoma cells. Cancer Chemother. Pharmacol. 2009, 64, 575–583. [Google Scholar] [CrossRef]
- Saif, M.W.; Diasio, R.B. Edotecarin: A Novel Topoisomerase I Inhibitor. Clin. Color. Cancer 2005, 5, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Willenbrink, T.J.; Ruiz, E.S.; Cornejo, C.M.; Schmults, C.D.; Arron, S.T.; Jambusaria-Pahlajani, A. Field cancerization: Definition, epidemiology, risk factors, and outcomes. J. Am. Acad. Dermatol. 2020, 83, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Munisamy, M.; Mukherjee, N.; Thomas, L.; Pham, A.T.; Shakeri, A.; Zhao, Y.; Kolesar, J.; Rao, P.P.N.; Rangnekar, V.M.; Rao, M. Therapeutic opportunities in cancer therapy: Targeting the p53-MDM2/MDMX interactions. Am. J. Cancer Res. 2021, 11, 5762–5781. [Google Scholar]
- Yogosawa, S.; Yoshida, K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci. 2018, 109, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef]
- Berke, T.P.; Slight, S.H.; Hyder, S.M. Role of Reactivating Mutant p53 Protein in Suppressing Growth and Metastasis of Triple-Negative Breast Cancer. Onco Targets Ther. 2022, 15, 23–30. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function p53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef]
- Tutuska, K.; Parrilla-Monge, L.; Di Cesare, E.; Nemajerova, A.; Moll, U.M. Statin as anti-cancer therapy in autochthonous T-lymphomas expressing stabilized gain-of-function mutant p53 proteins. Cell Death Dis. 2020, 11, 274. [Google Scholar] [CrossRef]
- Sargolzaei, J.; Etemadi, T.; Alyasin, A. The P53/microRNA network: A potential tumor suppressor with a role in anticancer therapy. Pharmacol. Res. 2020, 160, 105179. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Xia, B.H.M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; To, K.-H.; et al. MDM2 Small-Molecule Antagonist RG7112 Activates p53 Signaling and Regresses Human Tumors in Preclinical Cancer Models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef]
- Bykov, V.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.-W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, A.E.; Rashed, H.E.; Abdelgawad, M.; Abdelhamid, M.I. Prognostic impact of EGFR and cytokeratin 5/6 immunohistochemical expression in triple-negative breast cancer. Ann. Diagn. Pathol. 2017, 28, 43–53. [Google Scholar] [CrossRef]
- Lev, S. Targeted therapy and drug resistance in triple-negative breast cancer: The EGFR axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Kaur, S.; Kumar, S.; Momi, N.; Sasson, A.R.; Batra, S.K. Mucins in pancreatic cancer and its microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Leon, F.; Rauth, S.; Batra, S.K.; Ponnusamy, M.P. A Systematic Review on the Implications of O-linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis. Cells 2020, 9, 446. [Google Scholar] [CrossRef]
- Cervoni, G.E.; Cheng, J.J.; Stackhouse, K.A.; Heimburg-Molinaro, J.; Cummings, R.D. O-glycan recognition and function in mice and human cancers. Biochem. J. 2020, 477, 1541–1564. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | IC50 (µM) | Cells | IC50 (µM) |
|---|---|---|---|
| L-02 | 1.022 ± 0.084 | L-02 | 1.02 ± 0.084 |
| MDA-MB-468 | 0.372 ± 0.201 | PANC-1 | 0.591 ± 0.091 |
| MDA-MB-231 | 0.197 ± 0.043 | SMMC-7721 | 0.623 ± 0.044 |
| A549 | 0.578 ± 0.096 | HeLa | 0.609 ± 0.037 |
| SH-SY5Y | 0.629 ± 0.028 | NCI-H446 | 0.664 ± 0.062 |
| PC-3 | 0.689 ± 0.102 | U251 | 0.935 ± 0.205 |
| MCF-7 | 0.517 ± 0.146 | SW1990 | 1.30 ± 0.022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Lu, Z.; Liang, Z.; Deng, B.; Zhu, Y.; Shi, J.; Lu, X. Transcriptomics and Proteomics Characterizing the Anticancer Mechanisms of Natural Rebeccamycin Analog Loonamycin in Breast Cancer Cells. Molecules 2022, 27, 6958. https://doi.org/10.3390/molecules27206958
Sun X, Lu Z, Liang Z, Deng B, Zhu Y, Shi J, Lu X. Transcriptomics and Proteomics Characterizing the Anticancer Mechanisms of Natural Rebeccamycin Analog Loonamycin in Breast Cancer Cells. Molecules. 2022; 27(20):6958. https://doi.org/10.3390/molecules27206958
Chicago/Turabian StyleSun, Xiao, Zhanying Lu, Zhenzhen Liang, Bowen Deng, Yuping Zhu, Jing Shi, and Xiaoling Lu. 2022. "Transcriptomics and Proteomics Characterizing the Anticancer Mechanisms of Natural Rebeccamycin Analog Loonamycin in Breast Cancer Cells" Molecules 27, no. 20: 6958. https://doi.org/10.3390/molecules27206958
APA StyleSun, X., Lu, Z., Liang, Z., Deng, B., Zhu, Y., Shi, J., & Lu, X. (2022). Transcriptomics and Proteomics Characterizing the Anticancer Mechanisms of Natural Rebeccamycin Analog Loonamycin in Breast Cancer Cells. Molecules, 27(20), 6958. https://doi.org/10.3390/molecules27206958