
Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
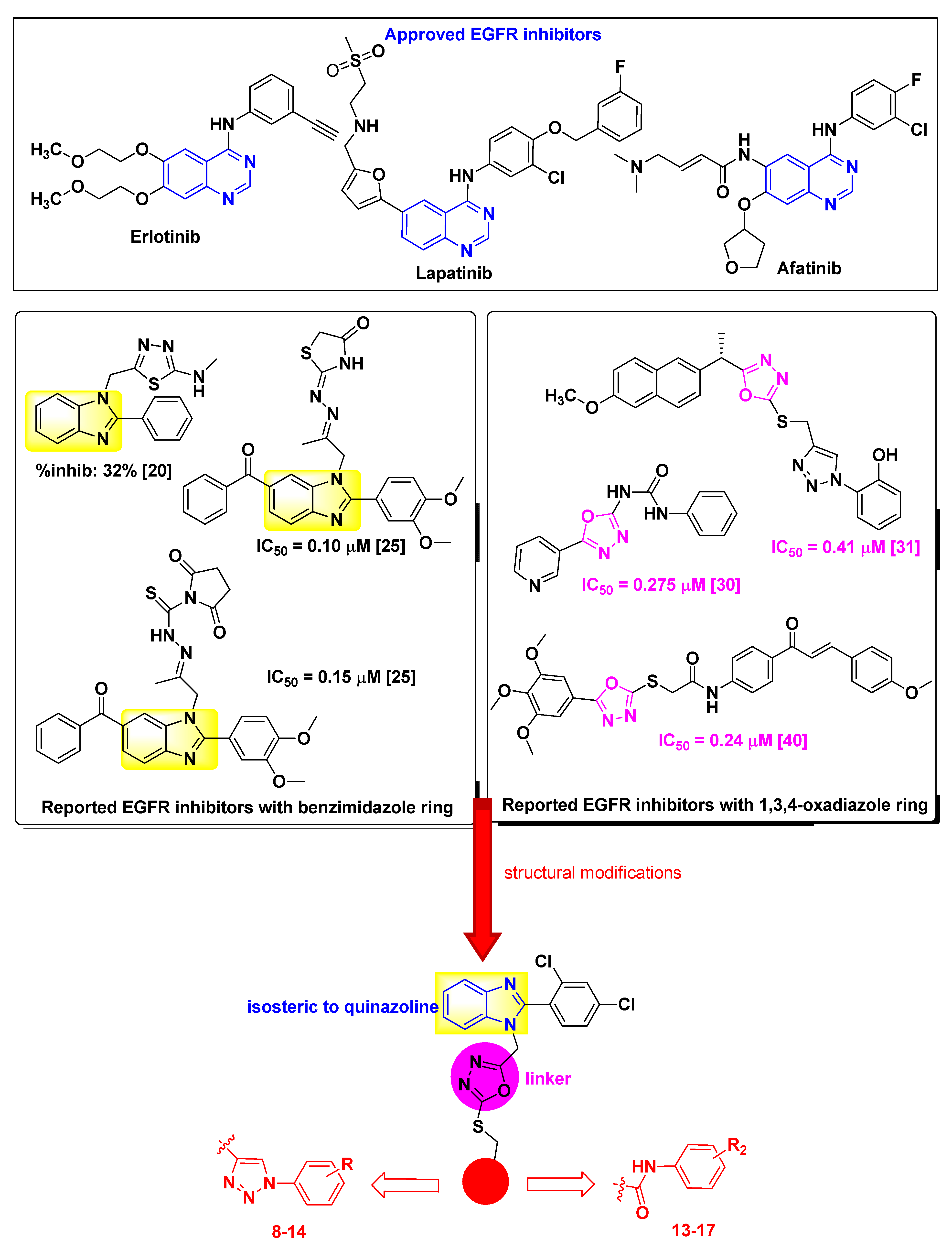
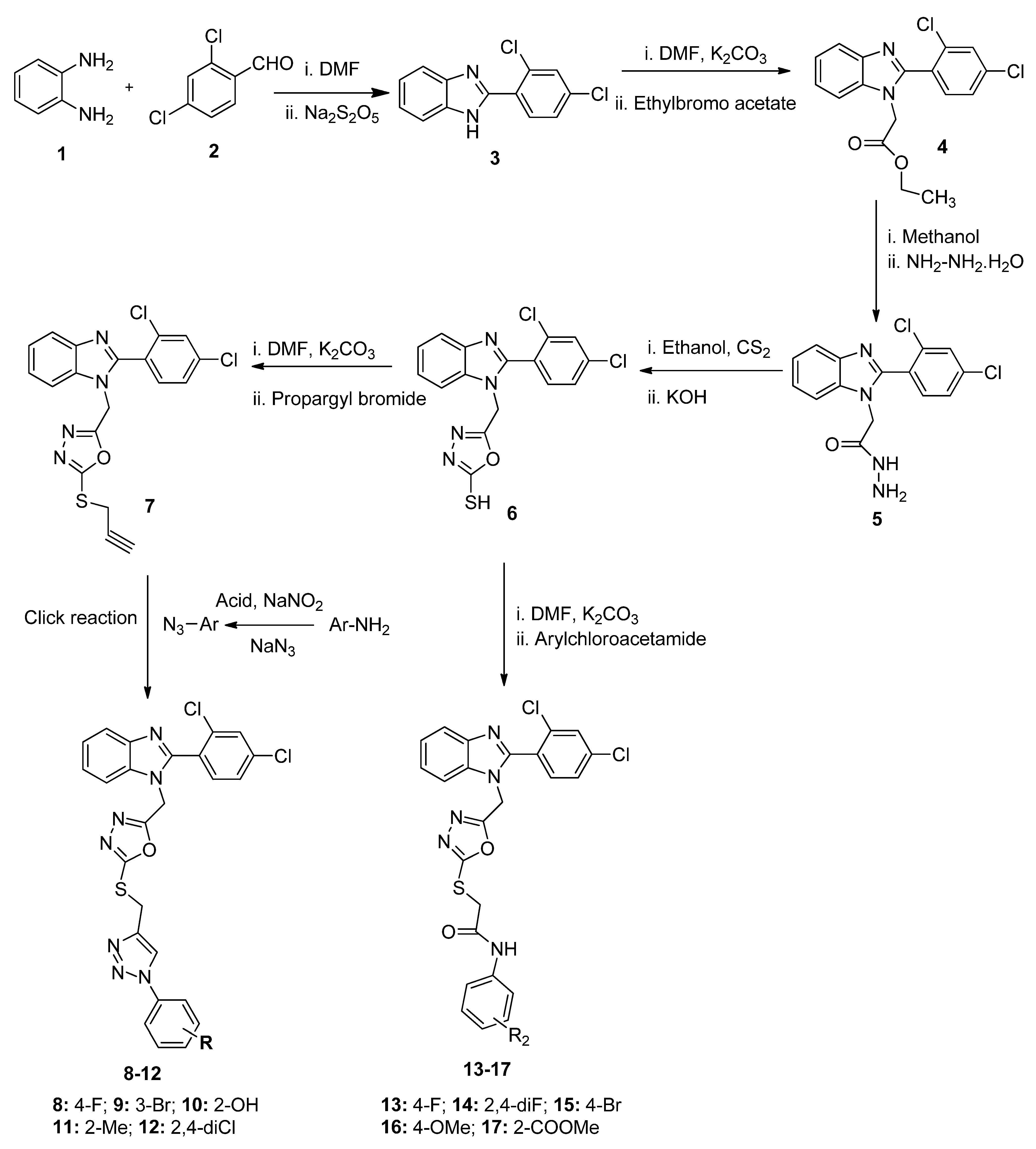
2.1. Chemistry
2.2. Biological Activity
2.2.1. Cytotoxicity
2.2.2. In Vitro EGFR Activity
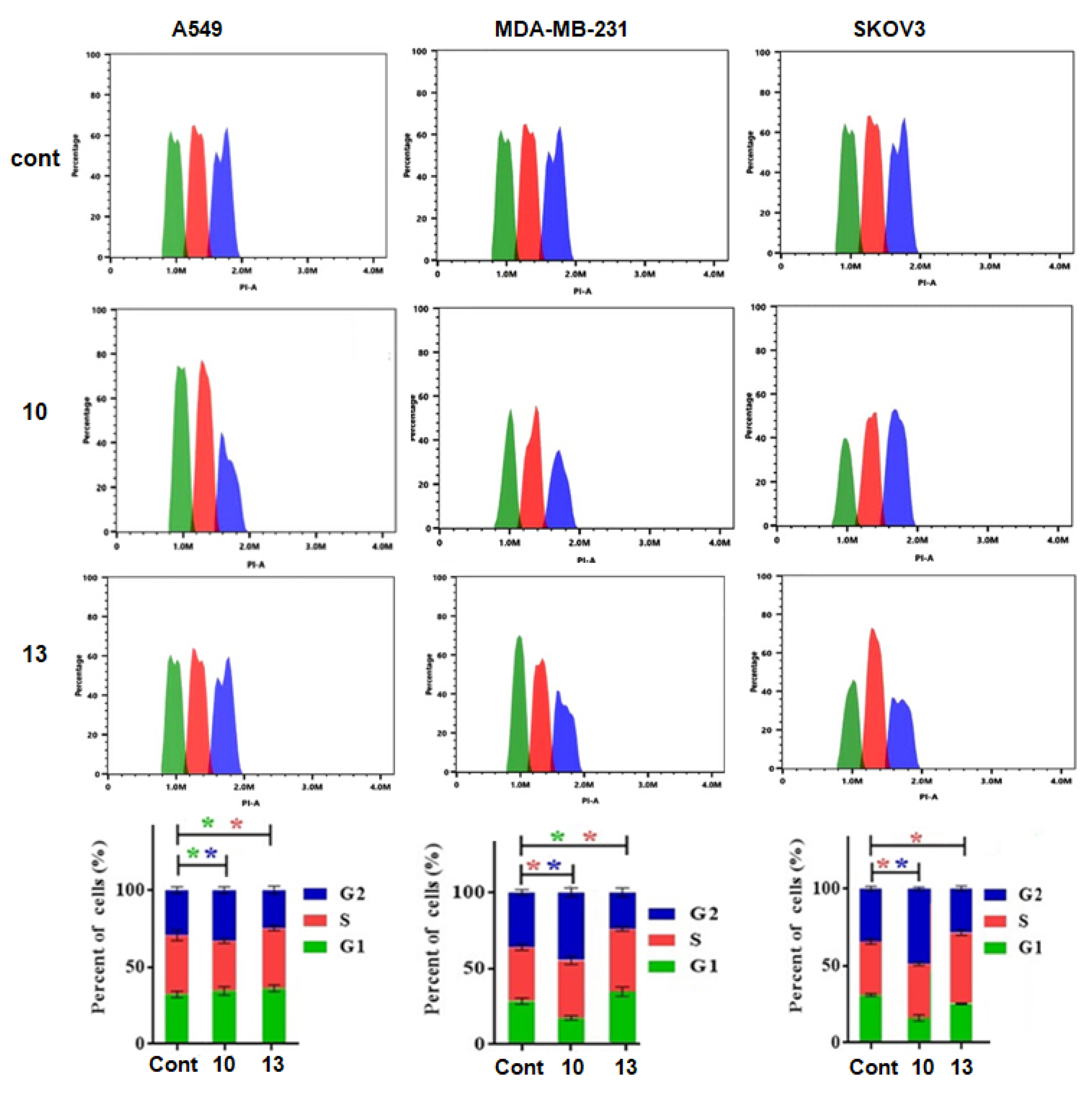
2.2.3. Cell Cycle Studies
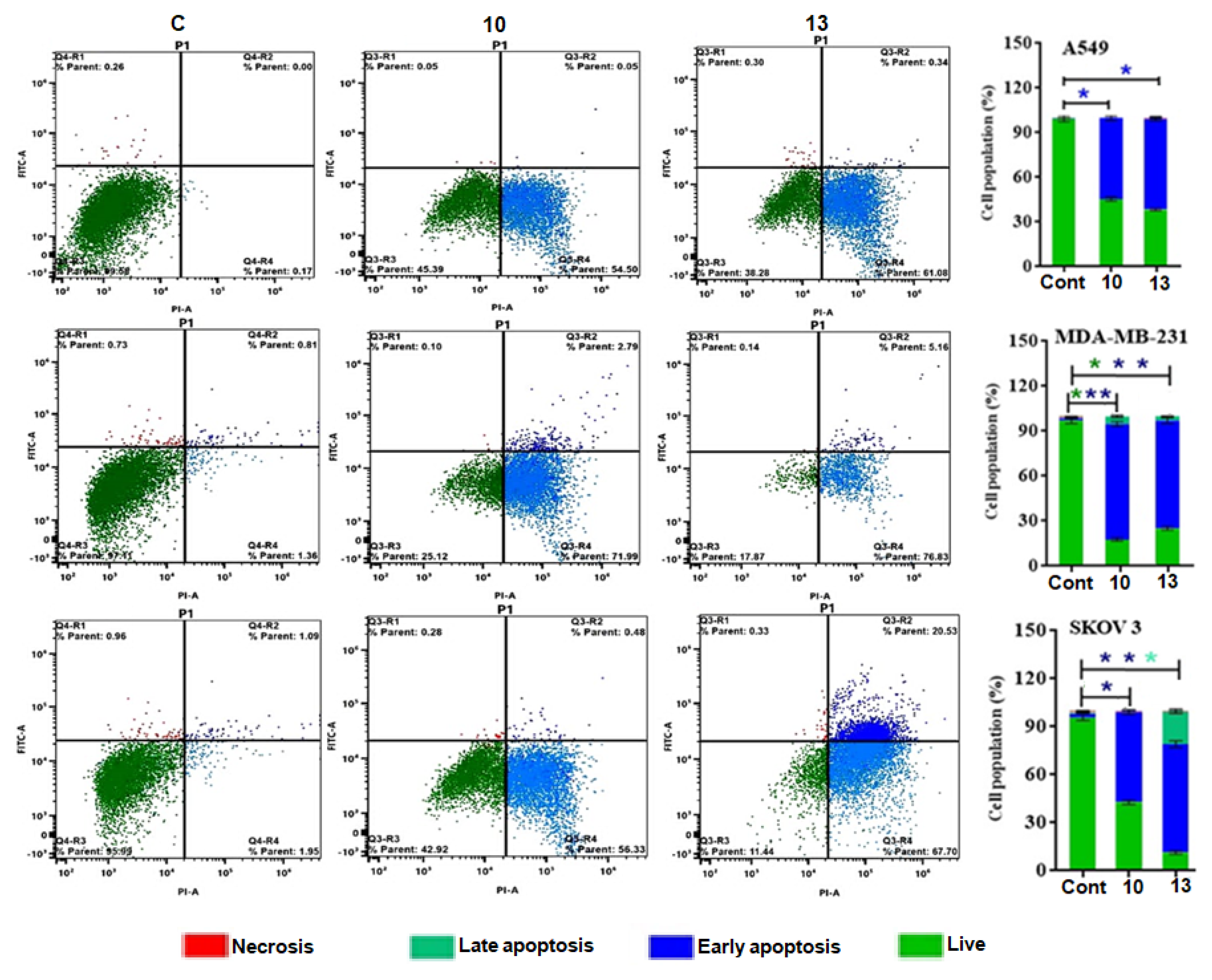
2.2.4. Apoptosis Studies
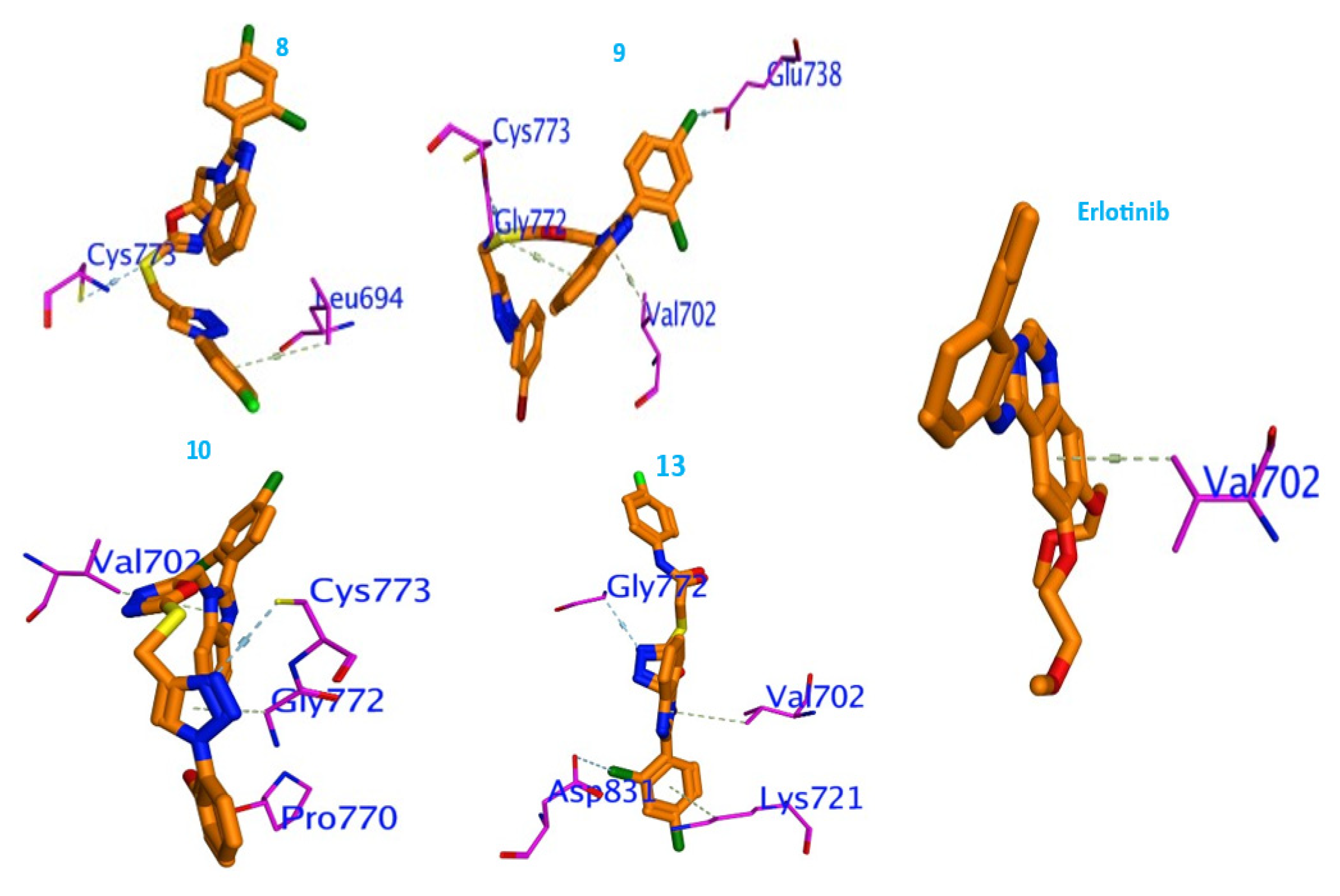
2.3. Molecular Docking Studies
2.4. In Silico Toxicity Studies
3. Experimental
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis of Final Compounds 8–12
3.1.3. Synthesis of Final Compounds 13–17
3.2. Biological Activity
3.2.1. Cytotoxicity
3.2.2. In Vitro EGFR Activity
3.2.3. Cell Cycle Analysis
3.2.4. Apoptosis Analysis
3.2.5. Statistical Analysis
3.3. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warnault, P.; Yasri, A.; Coisy-Quivy, M.; Cheve, G.; Bories, C.; Fauvel, B.; Benhida, R. Recent Advances in Drug Design of Epidermal Growth Factor Receptor Inhibitors. Curr. Med. Chem. 2013, 20, 2043–2067. [Google Scholar] [CrossRef] [PubMed]
- Allama, H.A.; Aly, E.E.; Farouka, A.K.B.A.W.; El Kerdawya, A.M.; Rashwan, E.; Abbass, S.E.S. Design and Synthesis of some new 2,4,6-trisubstituted quinazoline EGFR inhibitors as targeted anticancer agents. Bioorg. Chem. 2020, 98, 103726. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Marzouk, M.; Ghabbour, H.; AL-Salahi, R. Synthesis and anticancer activity of new quinazoline derivatives. Saudi Pharm. J. 2017, 25, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Abd El Hadi, S.R.; Lasheen, D.S.; Hassan, M.A.; Abouzid, K.A.M. Design and Synthesis of 4-Anilinothieno[2,3-d]pyrimidine-Based Compounds as Dual EGFR/HER-2 Inhibitors. Arch. Pharm. 2016, 349, 827–847. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.R.; Ferguson, K.M. Interaction of antibodies with ErbB receptor extracellular regions. Exp. Cell Res. 2009, 315, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.R.; Bhagavanraju, M.; Reddy, G.D.; Peters, G.J.; Rajendra Prasad, V.V.S. Design and synthesis of quinazolinone tagged acridones as cytotoxic agents and their effects on EGFR tyrosine kinase. Arch. Pharm. 2014, 347, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Gan, Y.; Fu, Y.; Liu, J.; Li, W.; Zou, X.; Zhou, Z.; Wang, Z.; Ouyang, G.; Yan, L. Design, synthesis and in vitro biological evaluation of quinazolinone derivatives as EGFR inhibitors for antitumor treatment. J. Enzyme Inhib. Med. Chem. 2020, 35, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef]
- Zhang, D.; Yan, Y.; Jin, G.; Liu, B.; Ma, X.; Han, D.; Jia, X. Synthesis and antitumor evaluation of novel 4-anilino-7,8-dihydropyrido[4,3-d]pyrimidine-6(5H)-carboxylate derivatives as potential EGFR inhibitors. Arch. Pharm. Chem. Life Sci. 2018, 351, e1800110. [Google Scholar] [CrossRef]
- Patel, H.M.; Pawara, R.; Ansari, A.; Noolvi, M.; Surana, S. Design and synthesis of quinazolinones as EGFR inhibitors to overcome EGFR resistance obstacle. Bioorg. Med. Chem. 2017, 25, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Hu, D.Y.; Jin, L.H.; Song, B.A.; Yang, S.; Liu, P.S.; Bhadury, P.S.; Ma, Y.; Luo, H.; Zhou, X. Synthesis and bioactivities of 6,7,8-trimethoxy-N-aryl-4-aminoquinazoline derivatives. Bioorg. Med. Chem. 2007, 15, 6608–6617. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer. 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zhou, Z.; Chu, C.; Zhang, Q.; Zhou, L.; Yang, Z.; Li, X.; Yu, L.; Zheng, P.; Xu, S.; et al. Design, synthesis and antitumor activity of novel thiophene-pyrimidine derivatives as EGFR inhibitors overcoming T790M and L858R/T790M mutations. Eur. J. Med. Chem. 2020, 203, 112511. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chu, C.; Zhou, L.; Zhou, Z.; Zhang, Q.; Yang, F.; Yang, Z.; Zheng, P.; Xu, S.; Zhun, W. Discovery of thiapyran-pyrimidine derivatives as potential EGFR inhibitors. Bioorg. Med. Chem. 2020, 28, 115669. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, F.; Alam, O.; Perwez, A.; Rizvi, M.A.; Naim, M.J.; Siddiqui, N.; Pottoo, F.H.; Jha, M. 3’-(4-(Benzyloxy)phenyl)-1’-phenyl-5-(heteroaryl/aryl)-3,4-dihydro-1’H,2H-[3,4’-bipyrazole]-2-carboxamides as EGFR kinase inhibitors: Synthesis, anticancer evaluation, and molecular docking studies. Arch. Pharm. Chem. Life Sci. 2020, 353, e1900262. [Google Scholar] [CrossRef] [PubMed]
- Khattab, R.R.; Alshamari, A.K.; Hassan, A.A.; Elganzory, H.H.; El-Sayed, W.A.; Awad, H.M.; Nossier, E.S.; Hassan, N.A. Click chemistry based synthesis, cytotoxic activity and molecular docking of novel triazole-thienopyrimidine hybrid glycosides targeting EGFR. J. Enzyme Inhib. Med. Chem. 2021, 36, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Ihmaid, S.K.; Yahya Alraqa, S.; Aouad, M.R.; Aljuhani, A.; Elbadawy, H.M.; Salama, S.A.; Rezki, N.; Ahmed, H.E.A. Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: Synthesis, EGFR inhibitory effect, and metabolic stability. Bioorg. Chem. 2021, 111, 104835. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.A.A.; El-Husseiny, W.M.; Abdel-Aziz, N.I.; El-Azab, A.S.; Abuelizz, H.A.; Abdel-Aziz, A.A.M. Synthesis and biological evaluation of 2-styrylquinolines as antitumour agents and EGFR kinase inhibitors: Molecular docking study. J. Enzyme Inhib. Med. Chem. 2018, 33, 199–209. [Google Scholar] [CrossRef]
- Rezki, N.; Almehmadi, M.A.; Ihmaid, S.; Shehata, A.M.; Omar, A.M.; Ahmed, H.E.A.; Aouad, M.R. Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1,2,3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses. Bioorg. Chem. 2020, 103, 104133. [Google Scholar] [CrossRef]
- Celik, I.; Ayhan-Kılcıgil, G.; Guven, B.; Kara, Z.; Gurkan-Alp, S.; Karayel, A.; Onay-Besikci, A. Design, synthesis and docking studies of benzimidazole derivatives as potential EGFR inhibitors. Eur. J. Med. Chem. 2019, 173, 240–249. [Google Scholar] [CrossRef]
- Abdullah, M.N.; Ali, Y.; Hamid, S.A. Insights into the structure and drug design of benzimidazole derivatives targeting the epidermal growth factor receptor (EGFR). Chem. Biol. Drug Des. 2021. [Google Scholar] [CrossRef]
- Elrayess, R.; Abdel Aziz, Y.M.; Elgawish, M.S.; Elewa, M.; Elshihawy, H.A.; Said, M.M. Pharmacophore modeling, 3D-QSAR, synthesis, and anti-lung cancer evaluation of novel thieno[2,3-d][1,2,3]triazines targeting EGFR. Arch. Pharm. 2020, 353, e1900108. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, Z.M.M.; Alam, M.M.; Nazreen, S. Recent Advancements on Benzimidazole: A Versatile Scaffold in Medicinal Chemistry. Mini. Rev. Med. Chem. 2022, 22, 365–386. [Google Scholar] [CrossRef]
- Shrivastava, N.; Naim, M.J.; Alam, M.J.; Nawaz, F.; Ahmed, S.; Alam, O. Benzimidazole Scaffold as Anticancer Agent: Synthetic Approaches and Structure-Activity Relationship. Arch. Pharm. 2017, 350, 1. [Google Scholar] [CrossRef] [PubMed]
- El-Meguid, E.A.; El-Deen, E.M.; Nael, M.A.; Anwar, M.M. Novel Benizmidazole derivatives as anti cervical cancer agents for potential multi targetiing kinase inhibitory activity. Arab. J. Chem. 2020, 13, 9179–9195. [Google Scholar]
- Cui, J.; Xiao, Z.; Zhang, L. Clinical efficacy and safety of nazartinib for epidermal growth factor receptor mutated non-small cell lung cancer: Study protocol for a prospective, multicenter, open-label. Medicine 2021, 100, e25992. [Google Scholar] [CrossRef] [PubMed]
- Siwach, A.; Verma, P.K. Therapeutic potential of oxadiazole or furadiazole containing compounds. BMC Chem. 2020, 14, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.R.W.; Boger, D.L.; Jorgensen, W.L. Elucidation of fatty acid amide hydrolase inhibition by potent alpha-ketoheterocycle derivatives from Monte Carlo simulations. J. Am. Chem. Soc. 2005, 127, 17377–17384. [Google Scholar] [CrossRef]
- Abou-Seri, S.M. Synthesis and biological evaluation of novel 2,4’-bis substituted diphenylamines as anticancer agents and potential epidermal growth factor receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 4113–4121. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.A.; Nour, M.S.; Alaraby Salem, M.; Arafa, R.K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019, 183, 111693. [Google Scholar] [CrossRef] [PubMed]
- Fathi, M.A.A.; Abd-El-Hafeez, A.A.; Abdelhamid, D.; Abbas, S.H.; Montano, M.M.; Abdel-Aziz, M. 1,3,4-oxadiazole/chalcone hybrids: Design, synthesis, and inhibition of leukemia cell growth and EGFR, Src, IL-6 and STAT3 activities. Bioorg. Chem. 2019, 84, 150–163. [Google Scholar] [CrossRef]
- Nazreen, S. Design, synthesis, and molecular docking studies of thiazolidinediones as PPAR-γ agonists and thymidylate synthase inhibitors. Arch. Pharm. 2021, 354, 2100021. [Google Scholar] [CrossRef] [PubMed]
- Elbehairi, S.E.I.; Ahmed, A.E.; Alshati, A.A.; ALkahtani, M.A.; Alfaifi, M.Y.; Alsyaad, K.M.; Alalmie, A.Y.A.; Ahamed, M.M.E.; Moustafa, M.F.; Alhag, S.K.; et al. Prosopis juliflora leave extracts induce cell death of MCF-7, HepG2, and LS-174T cancer cell lines. EXCLI J. 2020, 19, 1282–1294. [Google Scholar] [PubMed]
- Ling, Y.H.; Li, T.; Yuan, Z.; Haigentz Jr, M.; Weber, T.K.; Perez-Soler, R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol. Pharmacol. 2007, 72, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Huether, A.; Hopfner, M.; Sutter, A.P.; Schuppan, D.; Scherubl, H. Erlotinib induces cell cycle arrests and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostattics. J. Heptaol. 2005, 43, 661–669. [Google Scholar] [CrossRef]
- Sutter, A.P.; Heopfner, M.; Huether, A.; Maaser, K.; Scherubl, H. Targeting the epidermal growth factor receptor by erlotinib (Tarceva) for the treatment of esophageal cancer. Int. J. Cancer 2006, 118, 1814–1822. [Google Scholar] [CrossRef]
- Bashmail, H.A.; Alamoudi, A.A.; Noorwali, A.; Hegazy, G.A.; Ajabnoor, G.; Choudhry, H.; Al-Abd, A.M. Thymoquinone synergizes gemcitabine anti-breast cancer activity via modulating its apoptotic and autophagic activities. Sci. Rep. 2018, 8, 11674–11685. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, Maestro, Schrödinger. 2018. Available online: https://www.schrodinger.com/products/maestro (accessed on 10 September 2022).
- McGuffin, L.J.; Street, S.; Sørensen, S.A.; Jones, D.T. The genomic threading database. Bioinformatics 2004, 20, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Fouda, A.M.; El-Nassag, M.A.; Elhenawy, A.A.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Alam, M.M.; El-Agrody, A.M. Synthesis of 1,4-dihydropyrano[2,3-c]pyrazole derivatives and exploring molecular and cytotoxic properties based on DFT and molecular docking studies. J. Mol. Struct. 2022, 1249, 131555. [Google Scholar] [CrossRef]
- ALmalki, A.S.A.; Nazreen, S.; Elbehairi, S.E.I.; Asad, M.; Shati, A.A.; ALfaifi, M.Y.; Alhadhrami, A.; Elhenawy, A.A.; Alorai, A.Q.; Alam, M.M. Design, synthesis, anticancer activity and molecular docking studies of new benzimidazole derivatives bearing 1,3,4-oxadiazole moieties as potential thymidylate synthase inhibitors. New J. Chem. 2022, 46, 14967. [Google Scholar] [CrossRef]
- Alzahrani, H.A.; Alam, M.M.; Elhenawy, A.A.; Malebari, A.M.; Nazreen, S. Synthesis, antiproliferative, docking and DFT studies of benzimidazole derivatives as EGFR inhibitors. J. Mol. Struct. 2022, 1253, 132265. [Google Scholar] [CrossRef]
- Alam, M.M.; Nazreen, S.; Almalki, A.S.A.; Elhenawy, A.A.; Alsenani, N.I.; Elbehairi, S.E.I.; Malebari, A.M.; Alfaifi, M.Y.; Alsharif, M.A.; Alfaifi, S.Y.M. Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies. Pharmaceuticals 2021, 14, 870. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| A549 | SKOV3 | MDA-MB231 | |
| 8 | 26.65 ± 1.26 | 17.68 ± 0.68 | 15.49 ± 1.64 |
| 9 | 17.41 ± 0.54 | 13.60 ± 0.71 | 10.88 ± 0.58 |
| 10 | 3.31 ± 0.14 | 6.98 ± 0.28 | 1.18 ± 0.68 |
| 11 | 43.80 ± 0.87 | 25.69 ± 1.16 | 36.74 ± 0.75 |
| 12 | 37.15 ± 0.67 | 33.60 ± 0.91 | 25.18 ± 1.82 |
| 13 | 5.30 ± 0.44 | 4.35 ± 0.38 | 2.90 ± 0.53 |
| 14 | 11.64 ± 0.29 | 12.84 ± 0.53 | 20.85 ± 0.24 |
| 15 | 20.59 ± 1.41 | 13.71 ± 0.45 | 16.38 ± 0.44 |
| 16 | 7.83 ± 0.35 | 8.13 ± 0.28 | 10.49 ± 0.88 |
| 17 | 17.97 ± 0.87 | 12.18 ± 0.35 | 11.87 ± 0.91 |
| doxorubicin | 5.85 ± 0.61 | 8.65 ± 0.37 | 4.76 ± 0.44 |
| Compounds | IC50 (µM) |
|---|---|
| 9 | 0.95 ± 0.023 |
| 10 | 0.33 ± 0.051 |
| 13 | 0.38 ± 0.022 |
| 14 | 1.21 ± 0.047 |
| 16 | 1.09 ± 0.011 |
| 17 | 1.54 ± 0.026 |
| Erlotinib | 0.39 ± 0.034 |
| Compd | ΔΕ | RMSD | Econf | Eplace | E-Int. | LE | Ki | Fit Quality |
|---|---|---|---|---|---|---|---|---|
| 8 | −8.01 | 2.05 | −7.45 | −19.93 | −9.63 | −4.42 | 1.49 | −1.80 |
| 9 | −8.16 | 2.28 | 53.09 | −29.59 | −9.09 | −4.67 | 1.54 | −1.63 |
| 10 | −8.27 | 1.30 | 19.01 | −26.40 | −9.21 | −6.93 | 1.94 | −3.91 |
| 11 | −7.33 | 2.79 | 57.17 | −24.50 | −9.22 | −8.65 | 2.16 | −5.91 |
| 12 | −7.83 | 1.61 | 34.88 | −24.49 | −10.29 | −5.66 | 1.73 | −2.82 |
| 13 | −8.31 | 1.34 | 23.09 | −16.95 | −10.68 | −6.14 | 1.81 | −3.14 |
| 14 | −7.23 | 1.99 | 39.33 | −26.71 | −9.58 | −7.24 | 1.98 | −4.59 |
| 15 | −7.25 | 4.15 | −3.82 | −24.83 | −10.81 | −1.17 | 0.16 | 1.96 |
| 16 | −6.06 | 3.36 | −11.40 | −30.67 | −9.46 | −4.77 | 1.56 | −1.79 |
| 17 | −7.87 | 1.16 | 41.09 | −41.70 | −2.56 | −2.56 | 0.94 | 0.55 |
| Erlotinib | −7.90 | 1.94 | −30.17 | −15.64 | −14.74 | −8.39 | 2.47 | 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazreen, S.; Almalki, A.S.A.; Elbehairi, S.E.I.; Shati, A.A.; Alfaifi, M.Y.; Elhenawy, A.A.; Alsenani, N.I.; Alfarsi, A.; Alhadhrami, A.; Alqurashi, E.A.; et al. Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation. Molecules 2022, 27, 6899. https://doi.org/10.3390/molecules27206899
Nazreen S, Almalki ASA, Elbehairi SEI, Shati AA, Alfaifi MY, Elhenawy AA, Alsenani NI, Alfarsi A, Alhadhrami A, Alqurashi EA, et al. Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation. Molecules. 2022; 27(20):6899. https://doi.org/10.3390/molecules27206899
Chicago/Turabian StyleNazreen, Syed, Abdulraheem S. A. Almalki, Serag Eldin I. Elbehairi, Ali A. Shati, Mohammad Y. Alfaifi, Ahmed A. Elhenawy, Nawaf I. Alsenani, Anas Alfarsi, Abdulrahman Alhadhrami, Esam A. Alqurashi, and et al. 2022. "Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation" Molecules 27, no. 20: 6899. https://doi.org/10.3390/molecules27206899
APA StyleNazreen, S., Almalki, A. S. A., Elbehairi, S. E. I., Shati, A. A., Alfaifi, M. Y., Elhenawy, A. A., Alsenani, N. I., Alfarsi, A., Alhadhrami, A., Alqurashi, E. A., & Alam, M. M. (2022). Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation. Molecules, 27(20), 6899. https://doi.org/10.3390/molecules27206899