
New Bicyclic Azalide Macrolides Obtained by Tandem Palladium Catalyzed Allylic Alkylation/Conjugated Addition Reaction


Abstract
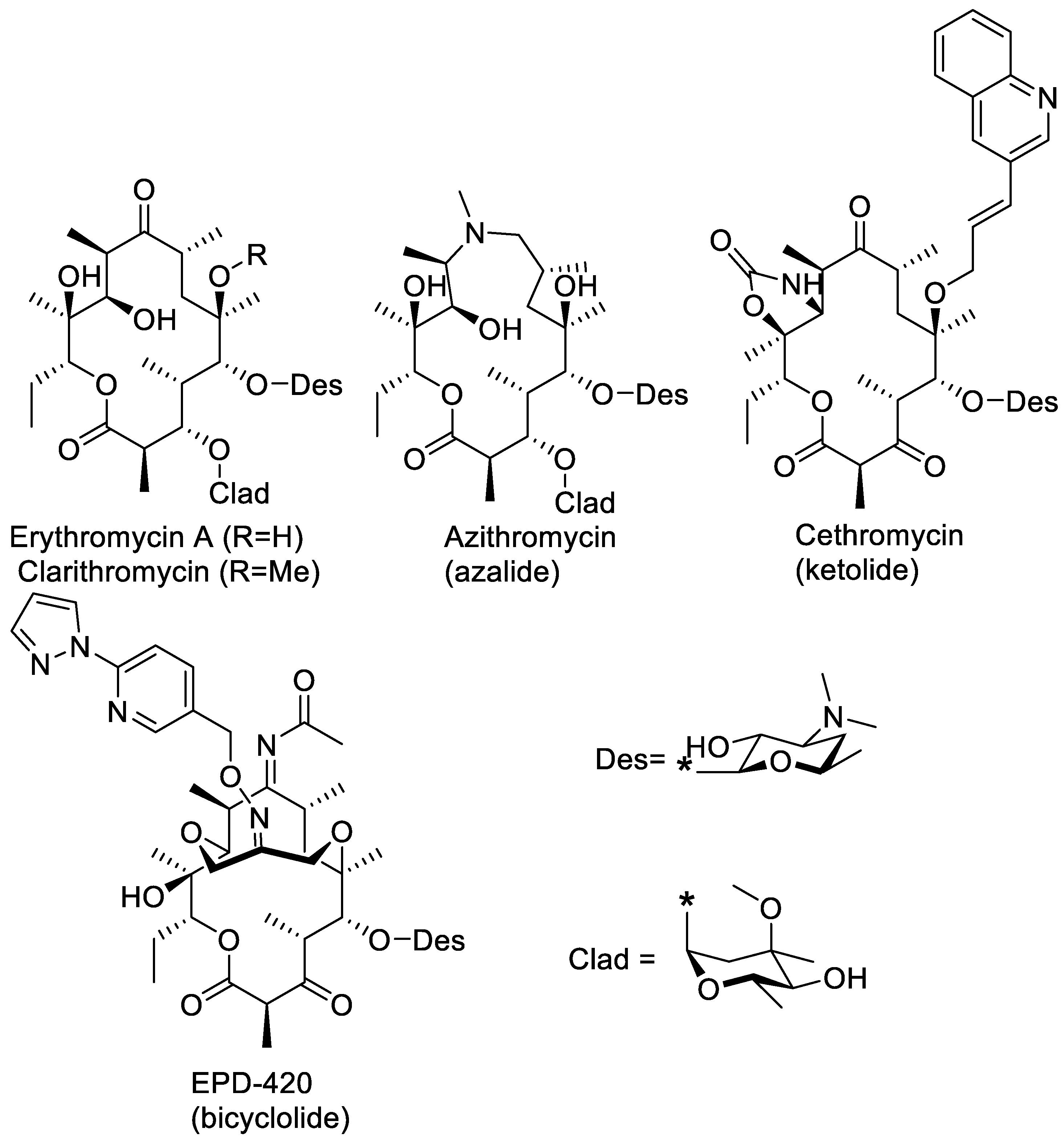
1. Introduction
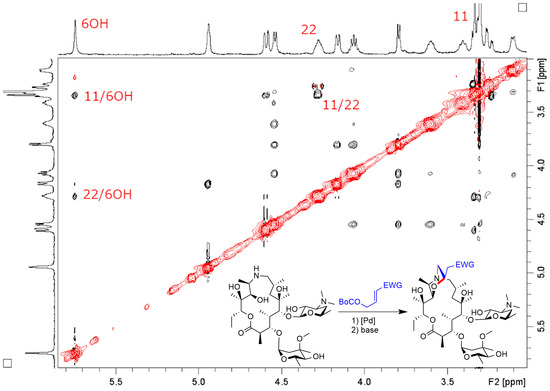
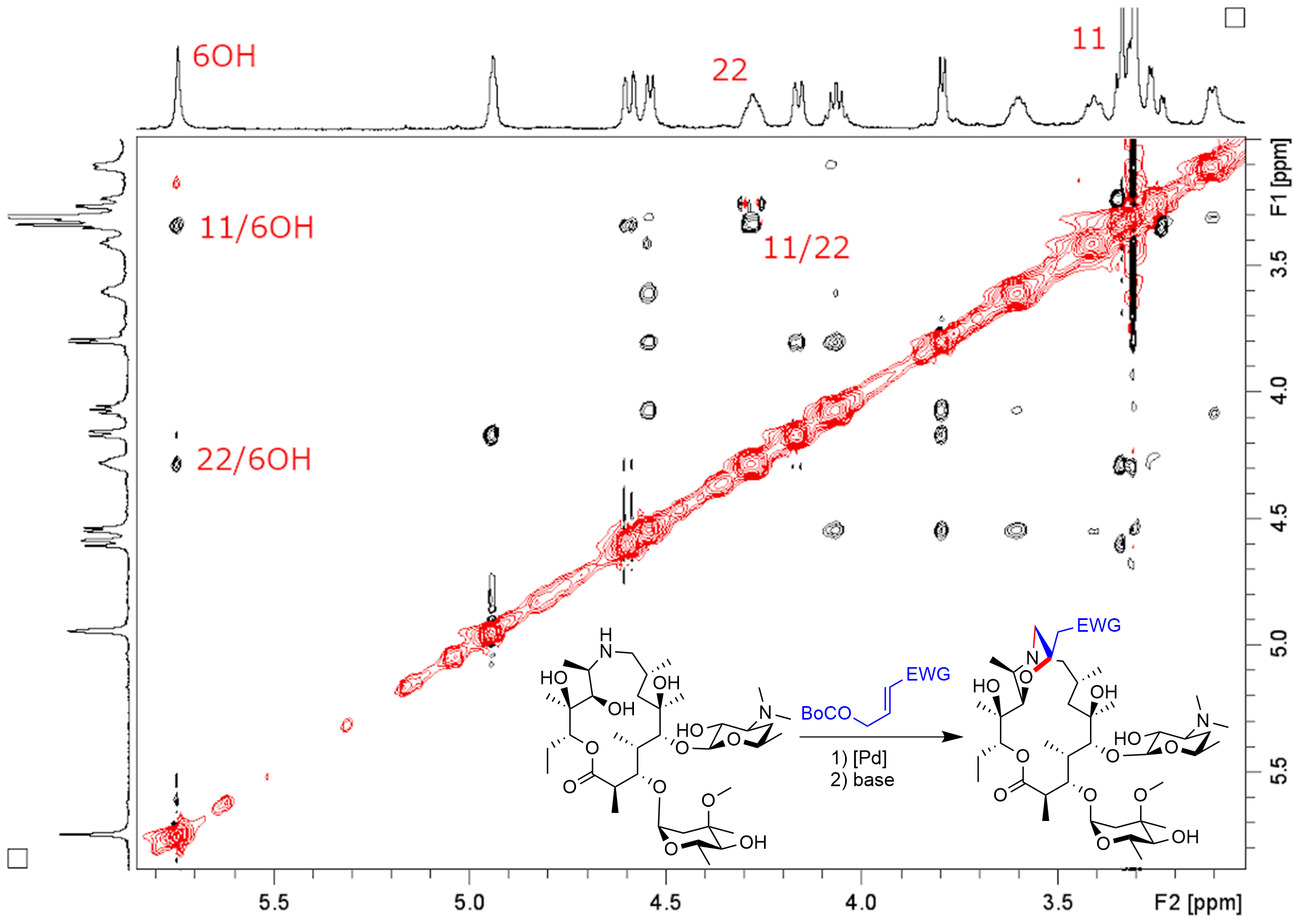
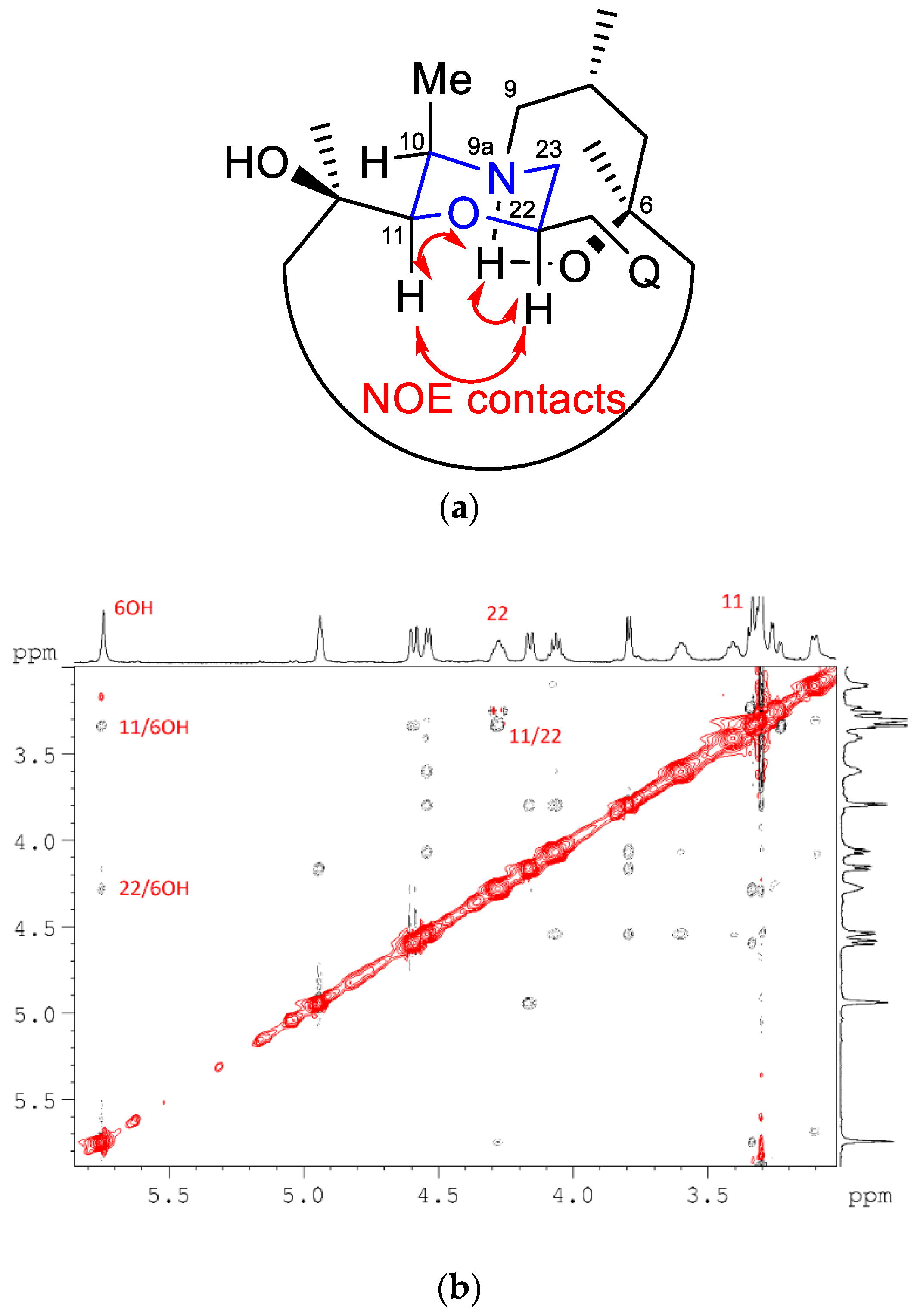
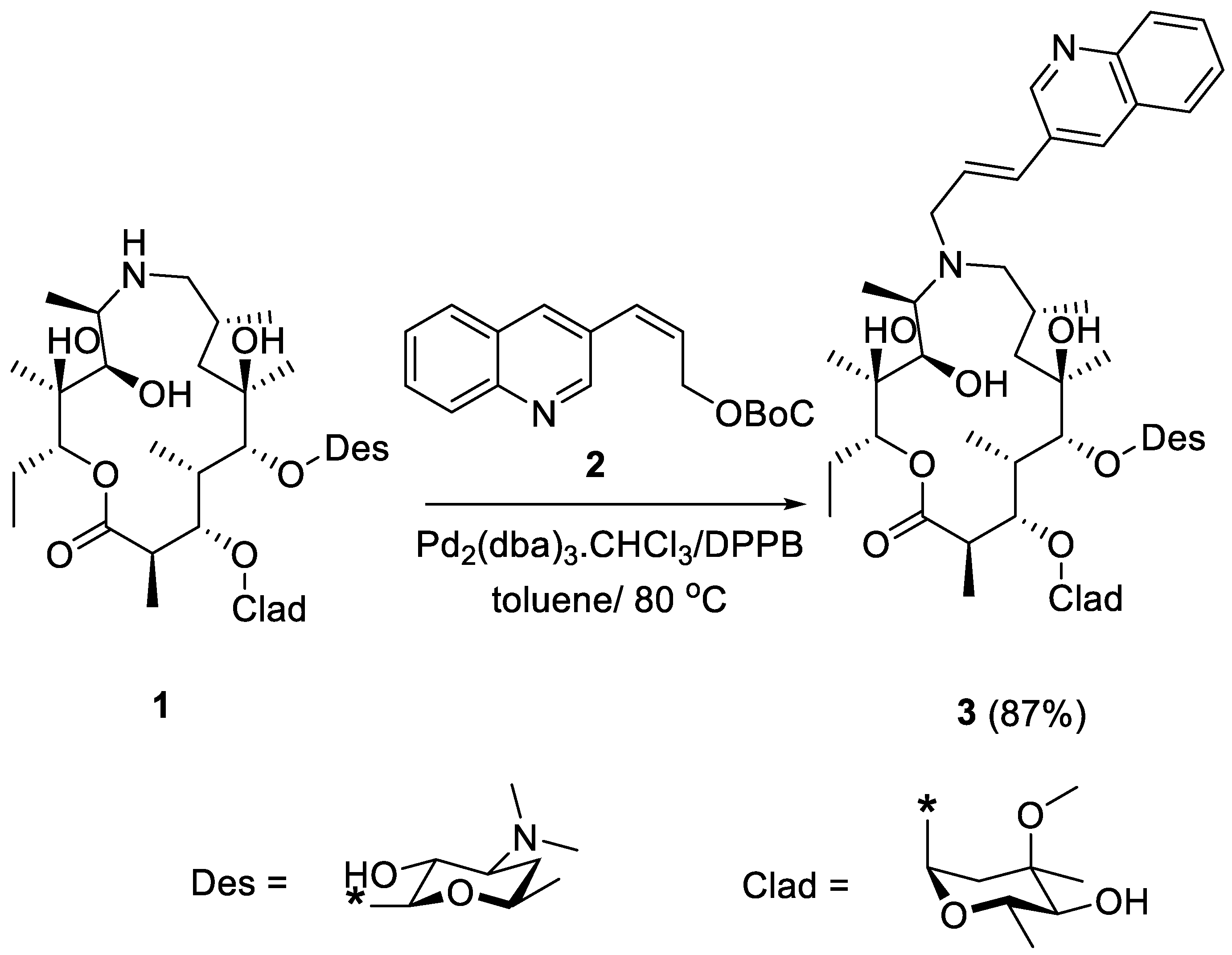
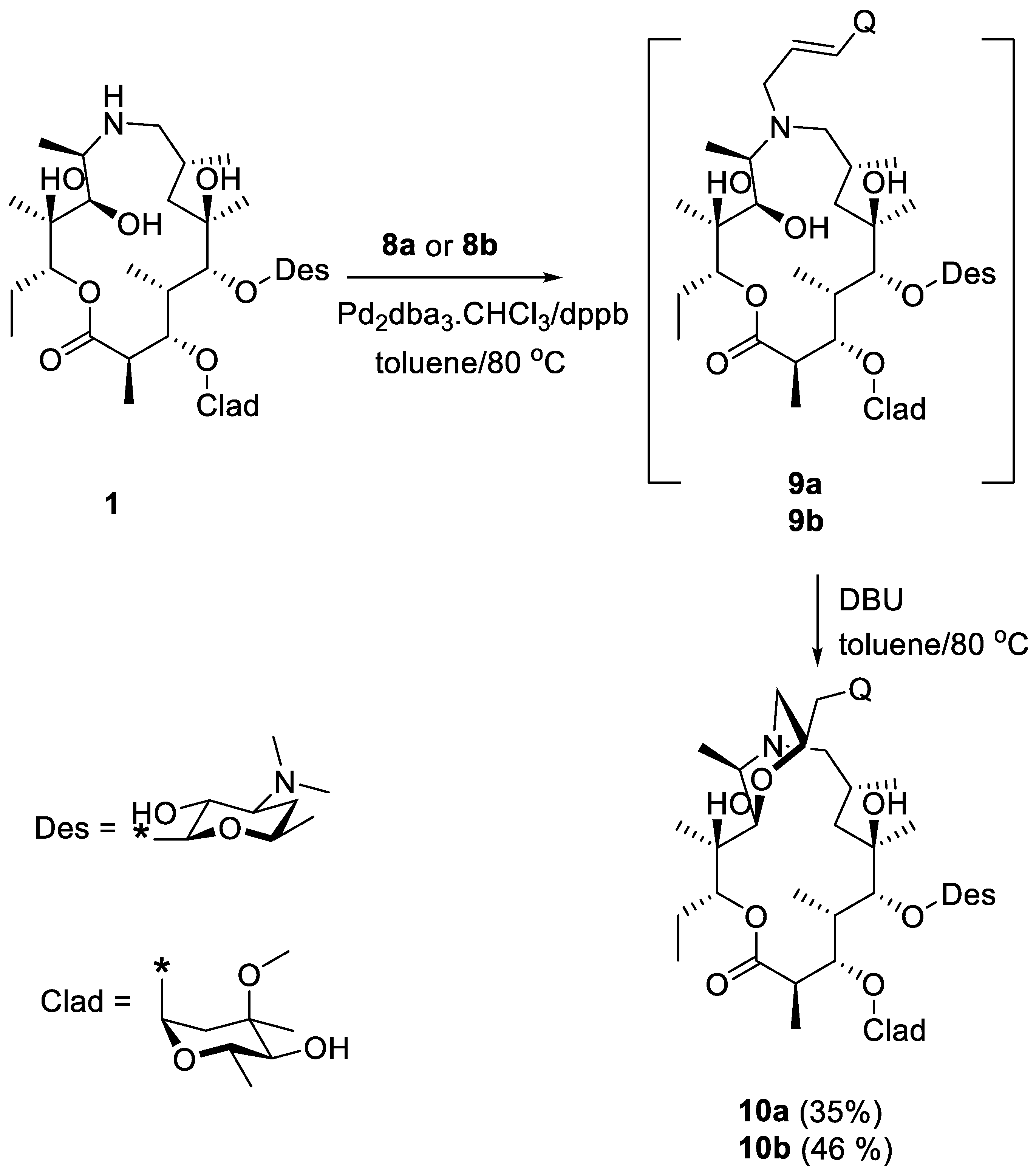
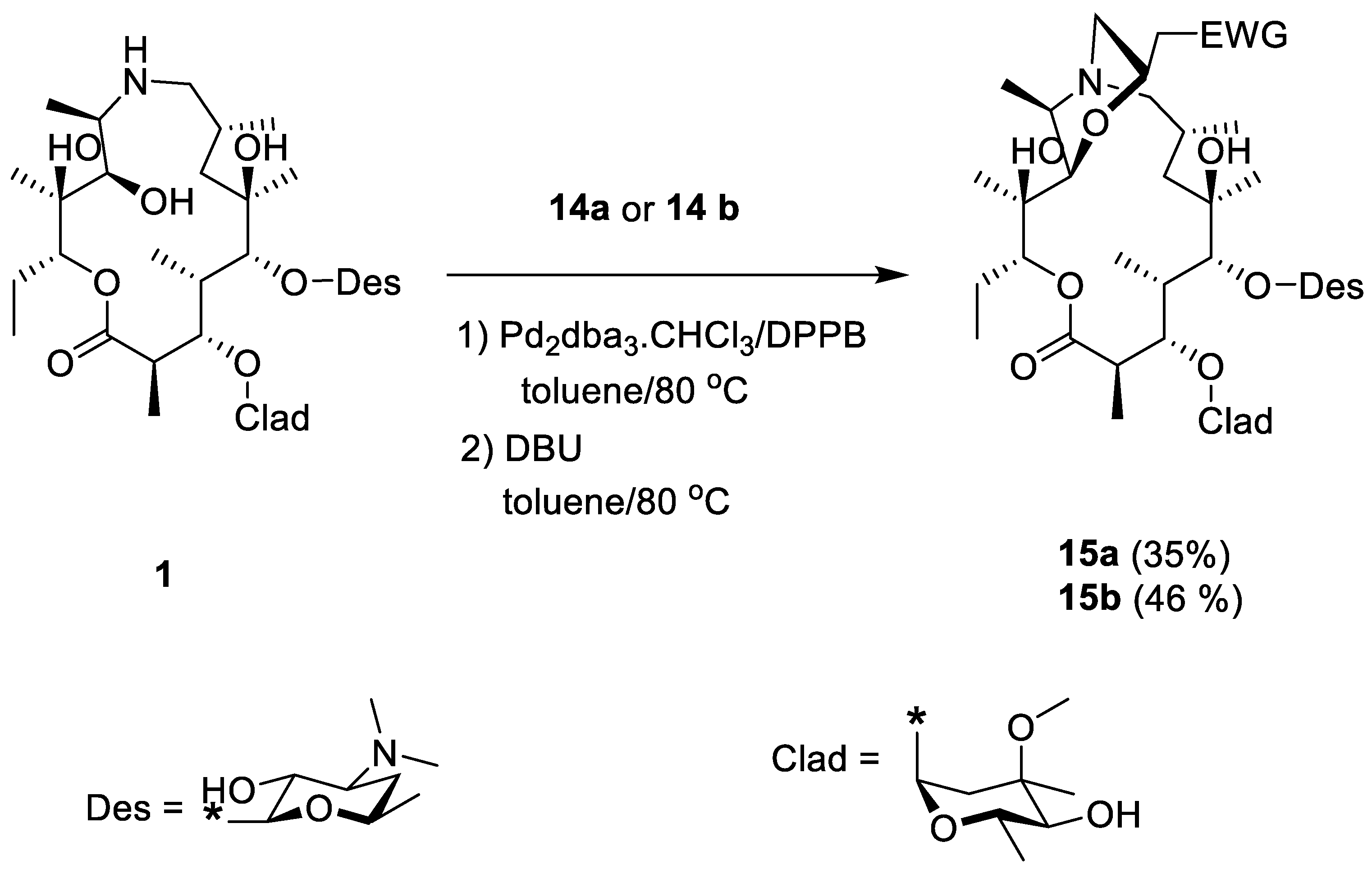
2. Results and Discussion
3. Experimental Section
3.1. General Methods
3.2. Synthetic Procedures
- 9-Deoxo-9a-(3-(3-Quinolyl)-2-propenyl)-9a-aza-9a-homoerythromycin A (3)
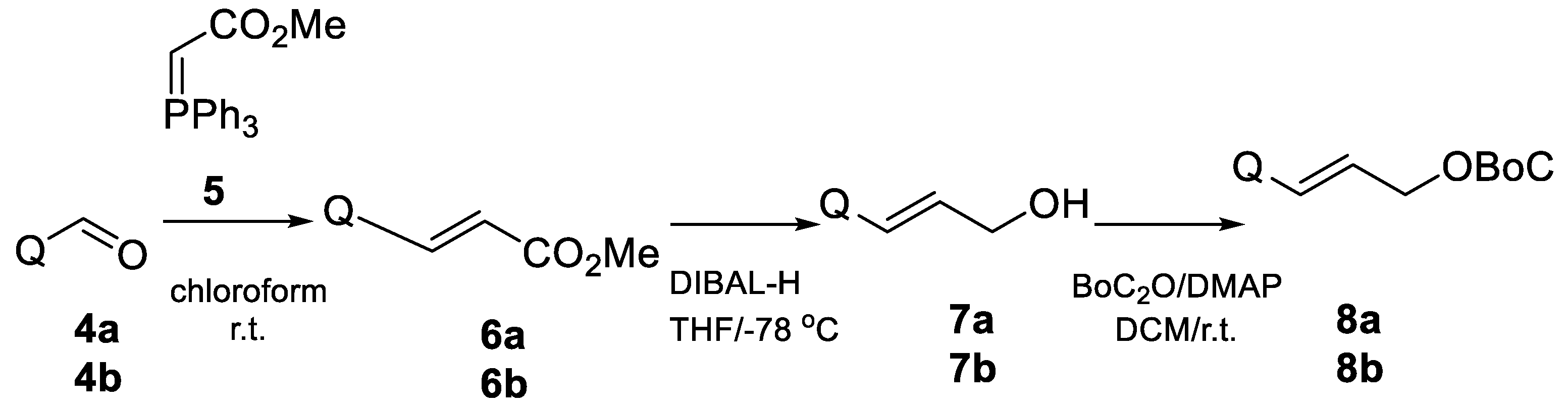
- Methyl 3-(2-quinolynyl)-Acrylate (6a)
- Methyl 3-(4-quinolynyl)-acrylate (6b)
- 3-(4-Quinolyl)-2-propene-1-ol (7b) [21]
- 3-(2-Quinolinyl)-2-propene-1-ol (7a) [22].
- 3-(4-Quinolyl)-allyl tert-butyl carbonate (8b).
- 3-(2-Quinolyl)-allyl tert-butyl carbonate (8a).
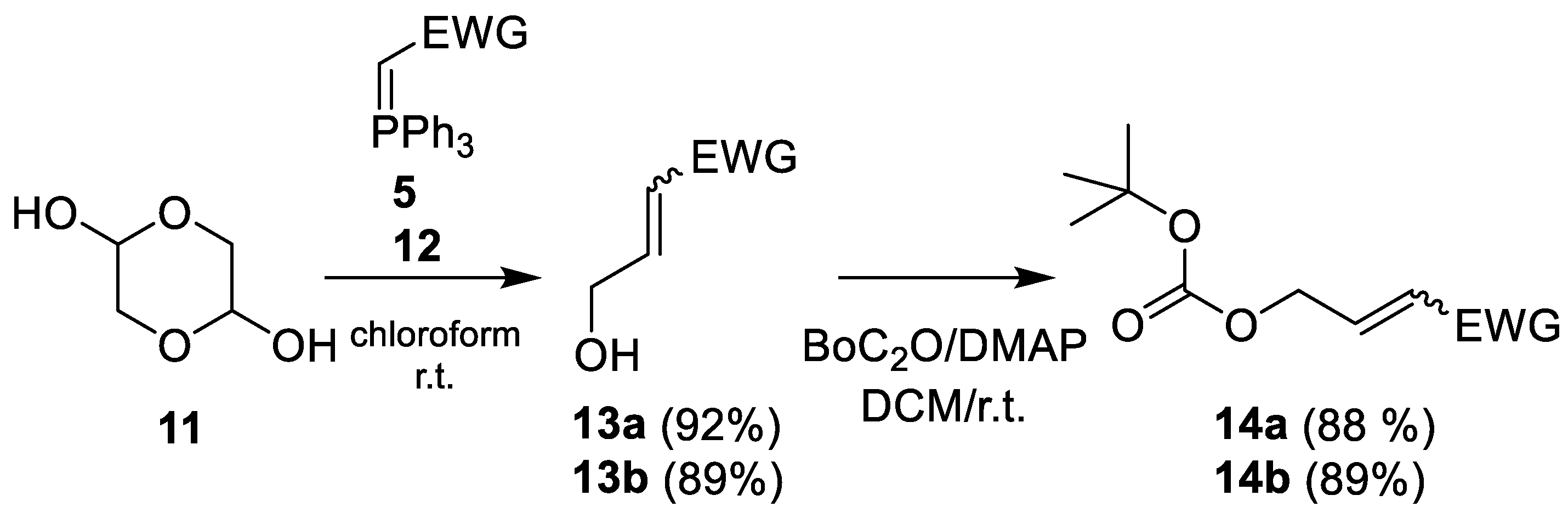
- Methyl 4-hydroxycrotonate (13a) [26]
- 4-Hydroxycrotononitrile (13b)
- Methyl-4-tert-butyloxycarbonyloxy-crotonate (14a)
- 4-tert-Butyloxycarbonyloxy-crotononitrile (14b)
- (22R)-9-Deoxo-11-deoxy-11-9a-(epoxyethano)-22-(2-quinolyl)methyl-9a-aza-9a-homoerythromycin A (10a)
- (22R)-9-Deoxo-11-deoxy-11-9a-(epoxyethano)-22-(4-quinolyl)methyl-9a-aza-9a-homoerythromycin A (10b)
- (22R)-9-Deoxo-11-deoxy-11-9a-(epoxyethano)-22-(methoxycarbonyl)methyl-9a-aza-9a-homoerythromycin A (15a)
- (22R)-9-Deoxo-11-deoxy-11-9a-(epoxyethano)-22-(cyano)methyl-9a-aza-9a-homoerythromycin A (15a)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pal, S. A journey across the sequential development of macrolides and ketolides related to erythromycin. Tetrahedron 2006, 62, 3171–3200. [Google Scholar] [CrossRef]
- Ma, X.; Ma, S. Significant breakthroughs in search for anti-infectious agents derived from erythromycin A. Curr. Med. Chem. 2011, 18, 1993–2015. [Google Scholar] [CrossRef]
- Janas, J.; Przybylski, P. 14- and 15-membered lactone macrolides and their analogues and hybrids: Structure, molecular mechanism of action and biological activity. Eur. J. Med. Chem. 2019, 182, 111662. [Google Scholar] [CrossRef]
- Morimoto, S.; Takahashi, Y.; Watanabe, Y.; Omura, S. Chemical modification of erythromycins. I. Synthesis and antibacterial activity of 6-0-methylerythromycins A. J. Antibiot. 1984, 37, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Djokić, S.; Kobrehel, G.; Lazarevski, G.; Lopotar, N.; Tamburašev, Z.; Kamenar, B.; Nagl, A.; Vicković, I. Erythromycin series. Part 11. Ring expansion of erythromycin A oxime by the Beckmann rearrangement. J. Chem. Soc. Perkin Trans. 1 1986, 1881–1890. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Walters, M.; Noreddin, A.; Vercaigne, L.M.; Wierzbowski, A.; Embil, J.M.; Gin, A.S.; Douthwaite, S.; Hoban, D.J. The Ketolides. Drugs 2002, 62, 1771–1804. [Google Scholar] [CrossRef]
- Wu, Y.-J. Highlights of Semi-synthetic Developments from Erythromycin, A. Curr. Pharm. Des. 2000, 6, 181–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Niu, D.; Qiu, Y.-L.; Phan, L.T.; Chen, Z.; Polemeropoulos, A.; Or, Y.S. Synthesis of Novel 6,11-O-Bridged Bicyclic Ketolides via a Palladium-Catalyzed Bis-allylation. Org. Lett. 2004, 6, 4455–4458. [Google Scholar] [CrossRef]
- Keyes, R.F.; Carter, J.J.; Zhang, X.; Ma, Z. Short and Efficient Synthesis of a Vinyl-Substituted Tricyclic Erythromycin Derivative. Org. Lett. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Gai, Y.; Tang, D.; Xu, G.; Chen, Z.; Polemeropoulos, A.; Wang, Z.; Or, Y.S. Synthesis of 3,6-bicyclolides: A novel class of macrolide antibiotics. Bioorg. Med. Chem. Lett. 2008, 18, 6315–6318. [Google Scholar] [CrossRef]
- Tang, D.; Gai, Y.; Polemeropoulos, A.; Chen, Z.; Wang, Z.; Or, Y.S. Design, synthesis, and antibacterial activities of novel 3,6-bicyclolide oximes: Length optimization and zero carbon linker oximes. Bioorg. Med. Chem. Lett. 2008, 18, 5078–5082. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, S.; Rubin, B.K. Mechanisms of Action and Clinical Application of Macrolides as Immunomodulatory Medications. Clin. Mibrobiol. Rev. 2010, 23, 590–615. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, V.; Striepen, B. Teaching old drugs new tricks to stop malaria invasion in its tracks. BMC Biol. 2015, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.J.; Haber, V.E.; Giamarellos-Bourboulis, E.J.; Perletti, G.; Verleden, G.M.; Vos, R. Azithromycin: Mechanisms of action and their relevance for clinical applications. Pharmacol. Therapeut. 2014, 143, 225–245. [Google Scholar] [CrossRef]
- Echeverría-Esnal, D.; Martin-Ontiyuelo, C.; Navarrete-Rouco, M.E.; De-Antonio Cuscó, M.; Ferrández, O.; Horcajada, J.P.; Grau, S. Azithromycin in the treatment of COVID-19: A review. Exp. Rev. Antiinfect. Ther. 2020, 19, 147–163. [Google Scholar] [CrossRef]
- Shapovalova, E.N.; Fedorova, I.A.; Anan’eva, I.A.; Shpigun, O.A.J. Macrocyclic Antibiotics as Chiral Selectors in High-Performance Liquid Chromatography and Capillary Electrophoresis. Anal. Chem. 2018, 73, 1064–1075. [Google Scholar]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem. 2005, 117, 4516–4563, Angew. Chem. Int. Ed.2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Trost, B.M.; Crawley, M.L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2943. [Google Scholar] [CrossRef]
- Stoner, E.J.; Peterson, M.J.; Allen, M.S.; DeMattei, J.A.; Haight, A.R.; Leanna, M.R.; Patel, S.R.; Plata, D.J.; Premchandran, R.H.; Rasmussen, M. Allylation of Erythromycin Derivatives: Introduction of Allyl Substituents into Highly Hindered Alcohols. J. Org. Chem. 2003, 68, 8847–8852. [Google Scholar] [CrossRef]
- Trost, B.M.; Machacek, M.R.; Aponick, A. Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model. Acc. Chem. Res. 2006, 39, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Henninger, T.C.; Macielag, M.J.; Tennakoon, M.A.; Xu, X. Preparation of 6-O-acyl Ketolide Derivatives of Erythromycin Useful as Antibacterials. WO2003050132A1, 19 June 2003. [Google Scholar]
- Fakhfakh, M.A.; Fournet, A.; Prina, E.; Mouscadet, J.-F.; Franck, X.; Hocquemiller, R.; Figaderea, B. Synthesis and Biological Evaluation of Substituted Quinolines: Potential Treatment of Protozoal and Retroviral Co-infections. Bioorg. Med. Chem. 2003, 11, 5013–5023. [Google Scholar] [CrossRef]
- Wilkening, R.R.; Ratcliffe, R.W.; Doss, G.A.; Mosley, R.T.; Ball, R.G. Novel Transannular Rearrangements of Azalide Iminoethers. Tetrahedron 1997, 53, 16923–16944. [Google Scholar] [CrossRef]
- Everett, J.R.; Tyler, J.W. An Analysis of the 1H and 13C n.m.r. spectra of erythromycin a using two-dimentional methods. J. Chem. Soc. Perkin Trans. 1 1985, 2599–2603. [Google Scholar] [CrossRef]
- Lazarevski, G.; Vinković, M.; Kobrehel, G.; Đokić, S.; Metelko, B.; Vikić-Topić, D. Conformational Analysis of Azithromycin by Nuclear Magnetic Resonance Spectroscopy and Molecular Modelling. Tetrahedron 1993, 49, 721–730. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chanthamath, S.; Shibatomi, K.; Iwasa, S. Ru(II)–Pheox-Catalyzed Asymmetric Intramolecular Cyclopropanation of Electron-Deficient Olefins. Org. Lett. 2015, 17, 2792–2795. [Google Scholar] [CrossRef]
- Or, Y.S.; Phan, T.; Wang, G.; Wang, Y.; Peng, Y. Preparation of 9A,11-2C-bicyclic 9a-azalide Erythromycin Derivatives as Prodrugs and Antibacterial Agents. U.S. Patent US20060154881A1, 13 July 2006. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance standards for Antimicrobial Susceptibility Testing; 15th Informational Supplement; CLSI document M100-S15; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2005. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | MIC (µg/mL) | ||||||
|---|---|---|---|---|---|---|---|
| Azithromycin | 3 | 10a | 10b | 15a | 15b | ||
| S. pneumoniae SP030 | ery S | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 |
| S. pyogenes 3565 | ery S | ≤0.125 | ≤0.125 | ≤0.125 | ≤0.125 | 0.25 | 0.25 |
| S. aureus ATCC13709 | ery S | 0.5 | 0.25 | ≤0.125 | 0.25 | 4 | 8 |
| S. pneumoniae Ci137 | M | 8 | 8 | 4 | 16 | 32 | 32 |
| S. pyogenes Finland 2 | M | 8 | 4 | 2 | 4 | 16 | 8 |
| S. aureus PK1 | M | >64 | >64 | >64 | >64 | >64 | >64 |
| S. pneumoniae 134 GR-M | iMcLS | >64 | >64 | >64 | >64 | >64 | >64 |
| S. pyogenes Finland 11 | iMLS | 16 | 2 | 0.5 | 4 | >64 | >64 |
| S. aureus PK2 | iMLS | >64 | >64 | >64 | >64 | >64 | >64 |
| M. catarrhalis ATCC 49247 | ≤0.125 | 1 | 4 | 4 | 0.5 | 1.25 | |
| H. influenzae ATCC 23246 | 1 | 2 | 4 | 4 | 4 | 4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alihodžić, S.; Čipčić Paljetak, H.; Čikoš, A.; Elenkov, I.J. New Bicyclic Azalide Macrolides Obtained by Tandem Palladium Catalyzed Allylic Alkylation/Conjugated Addition Reaction. Molecules 2022, 27, 432. https://doi.org/10.3390/molecules27020432
Alihodžić S, Čipčić Paljetak H, Čikoš A, Elenkov IJ. New Bicyclic Azalide Macrolides Obtained by Tandem Palladium Catalyzed Allylic Alkylation/Conjugated Addition Reaction. Molecules. 2022; 27(2):432. https://doi.org/10.3390/molecules27020432
Chicago/Turabian StyleAlihodžić, Sulejman, Hana Čipčić Paljetak, Ana Čikoš, and Ivaylo Jivkov Elenkov. 2022. "New Bicyclic Azalide Macrolides Obtained by Tandem Palladium Catalyzed Allylic Alkylation/Conjugated Addition Reaction" Molecules 27, no. 2: 432. https://doi.org/10.3390/molecules27020432
APA StyleAlihodžić, S., Čipčić Paljetak, H., Čikoš, A., & Elenkov, I. J. (2022). New Bicyclic Azalide Macrolides Obtained by Tandem Palladium Catalyzed Allylic Alkylation/Conjugated Addition Reaction. Molecules, 27(2), 432. https://doi.org/10.3390/molecules27020432