
AVA-NP-695 Selectively Inhibits ENPP1 to Activate STING Pathway and Abrogate Tumor Metastasis in 4T1 Breast Cancer Syngeneic Mouse Model

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. ENPP1 Exogenous Cell-Based Inhibition Assay
2.3. QUANTI-Luc™ Luciferase Assay
2.4. RNA Isolation and Real-Time PCR
2.5. Animal Studies
2.6. Lung Histopathology
2.7. KINOMEscan®® Assay
2.8. Bioinformatic Analysis
2.9. Statistical Analysis
3. Results
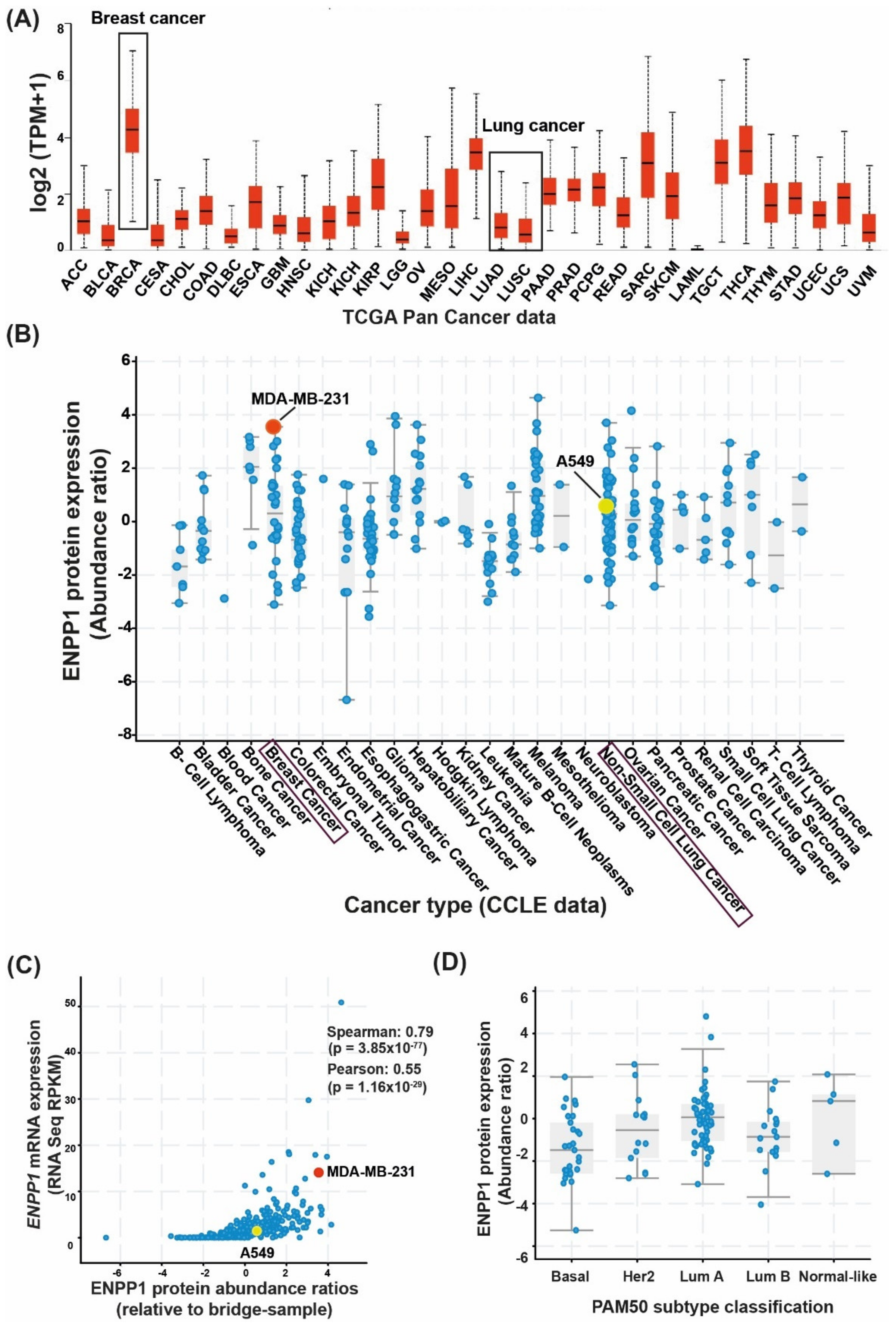
3.1. Differential Expression of ENPP1 across Cancer Types
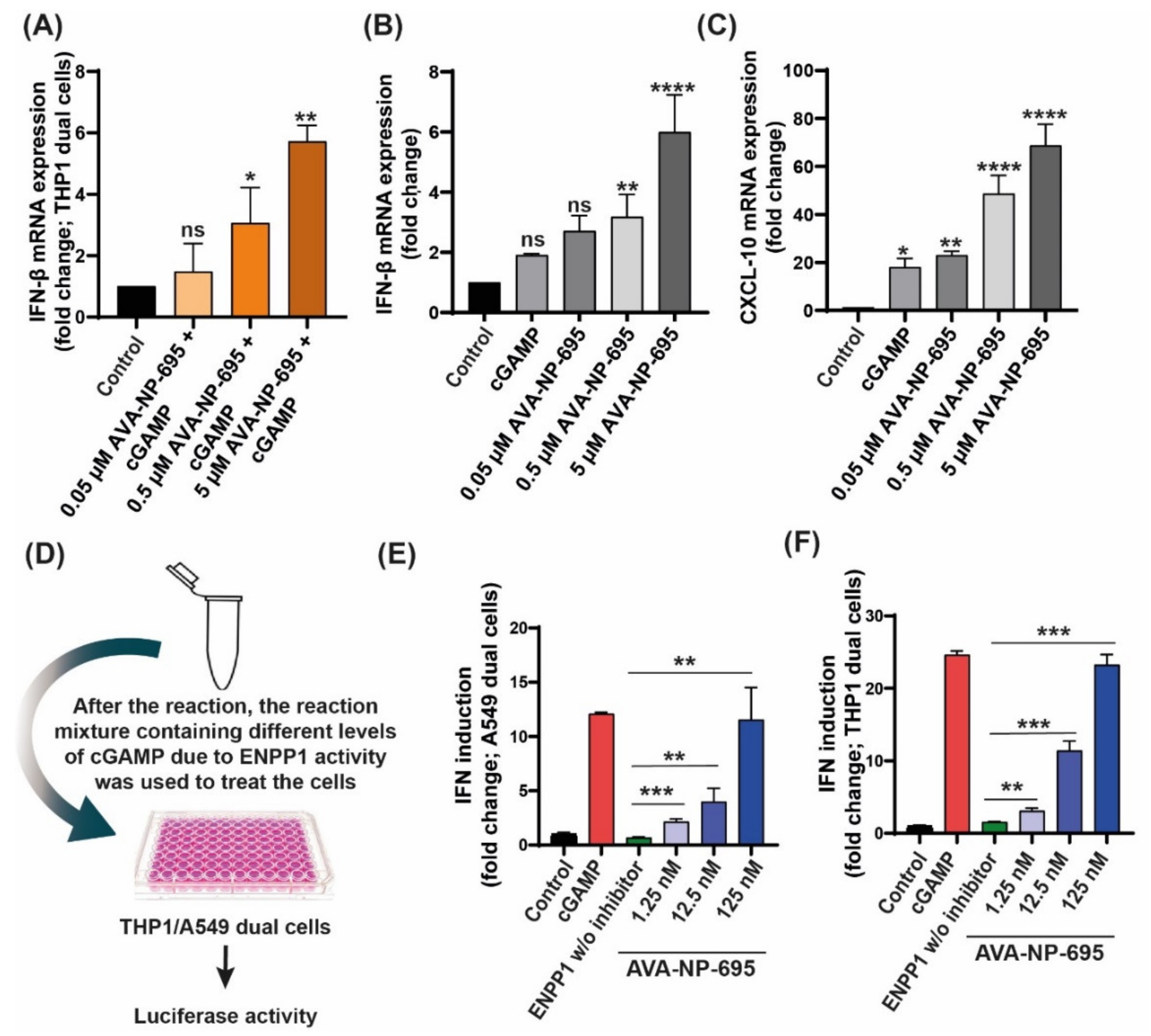
3.2. In Vitro Efficacy of AVA-NP-695 as a Potent Small ENPP1 Inhibitor
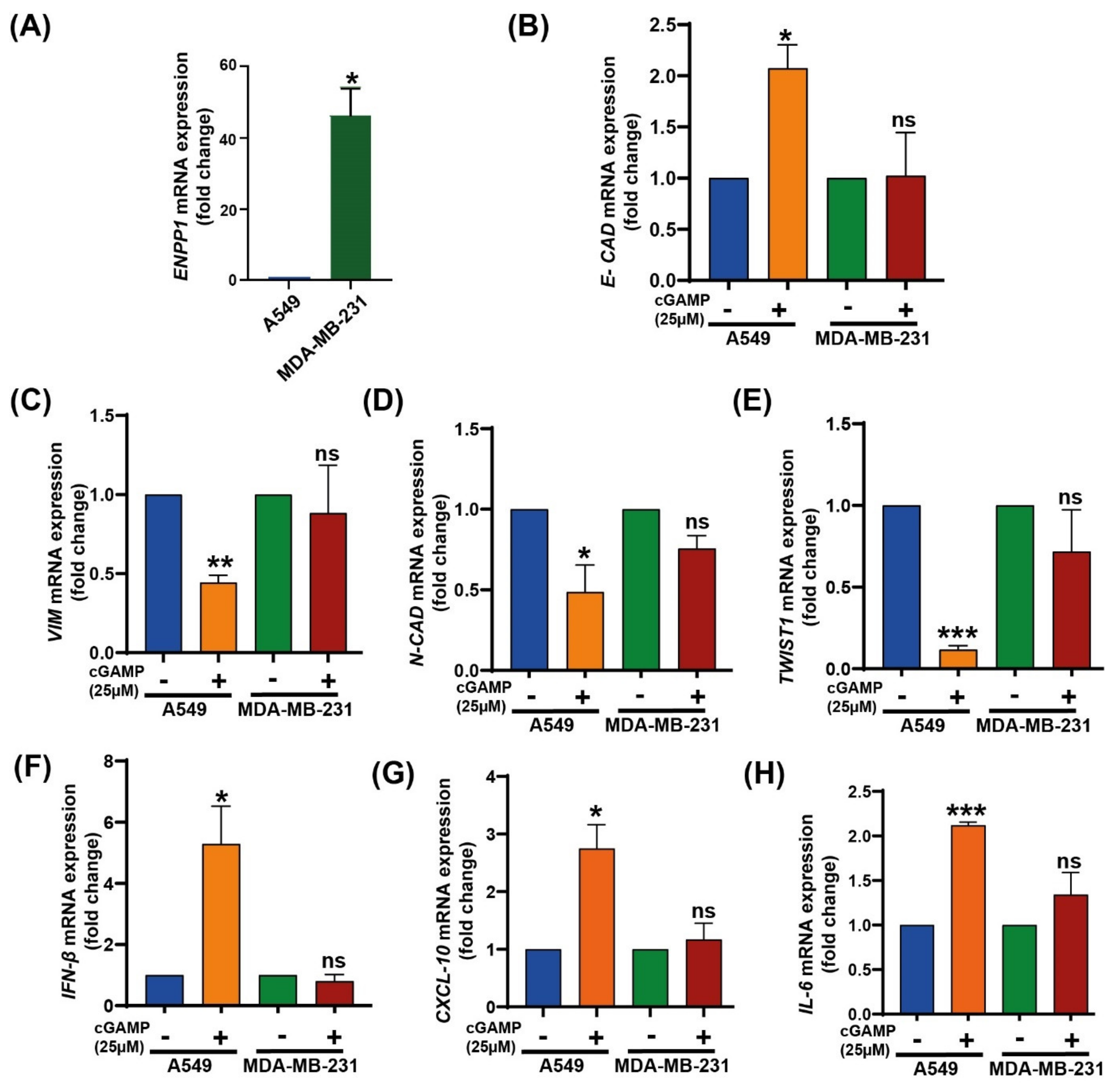
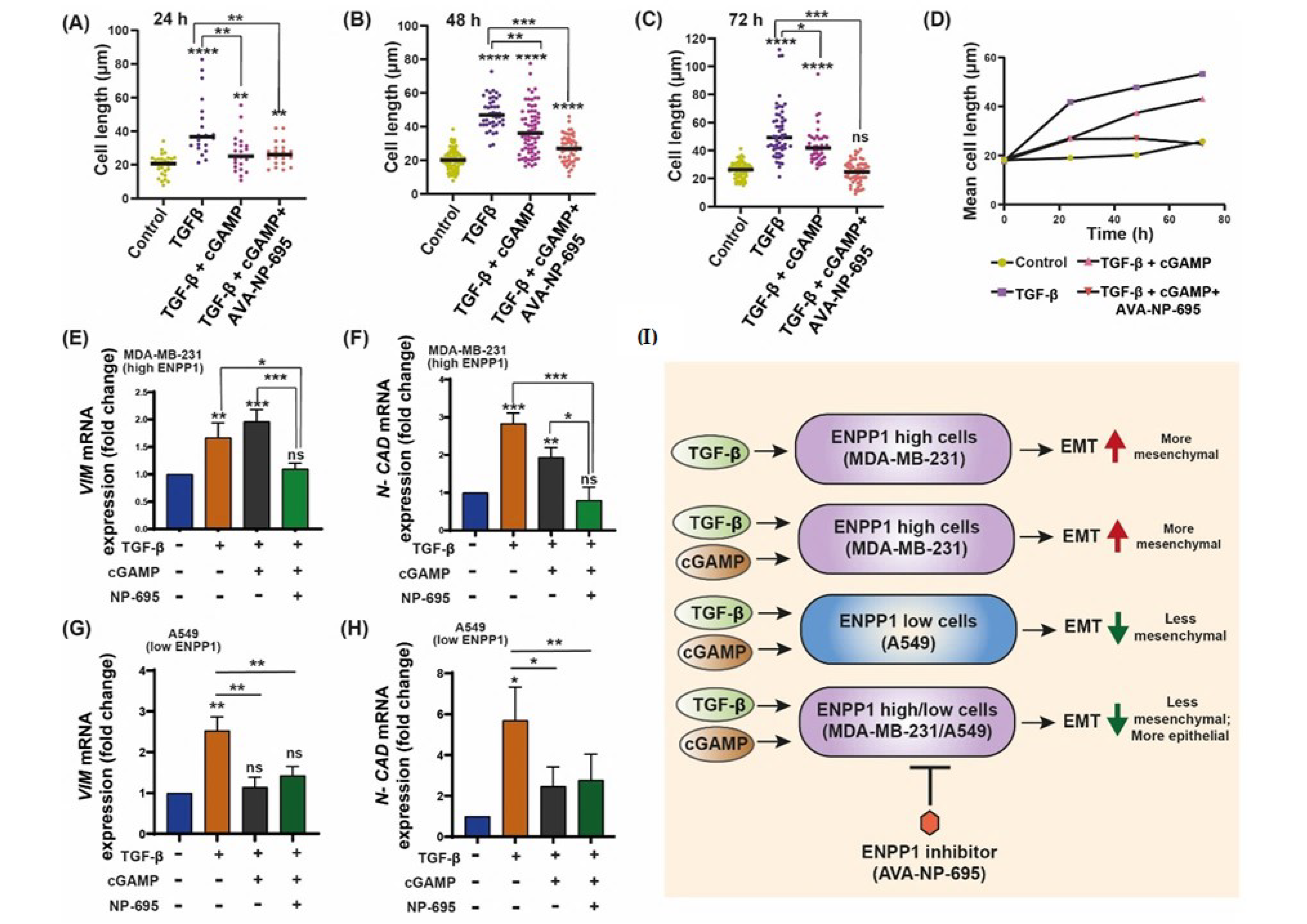
3.3. 2′3′-cGAMP Modulates EMT Markers Based on the Cellular ENPP1 Level
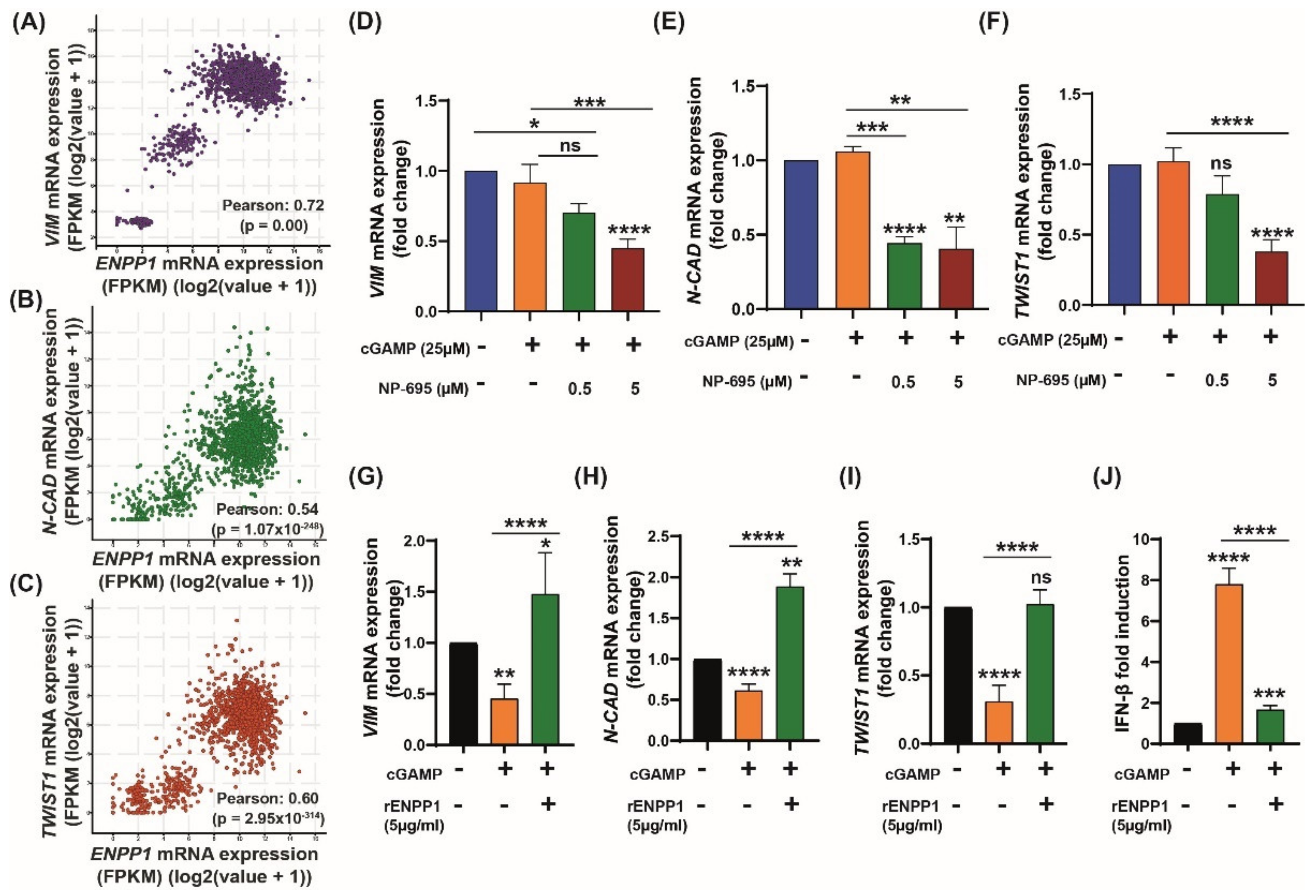
3.4. ENPP1 Determines 2′3′-cGAMP-Mediated Modulation of EMT Markers
3.5. 2′3′-cGAMP Blocks EMT Induced by TGF-β in Presence of ENPP1 Inhibitor, AVA-NP-695
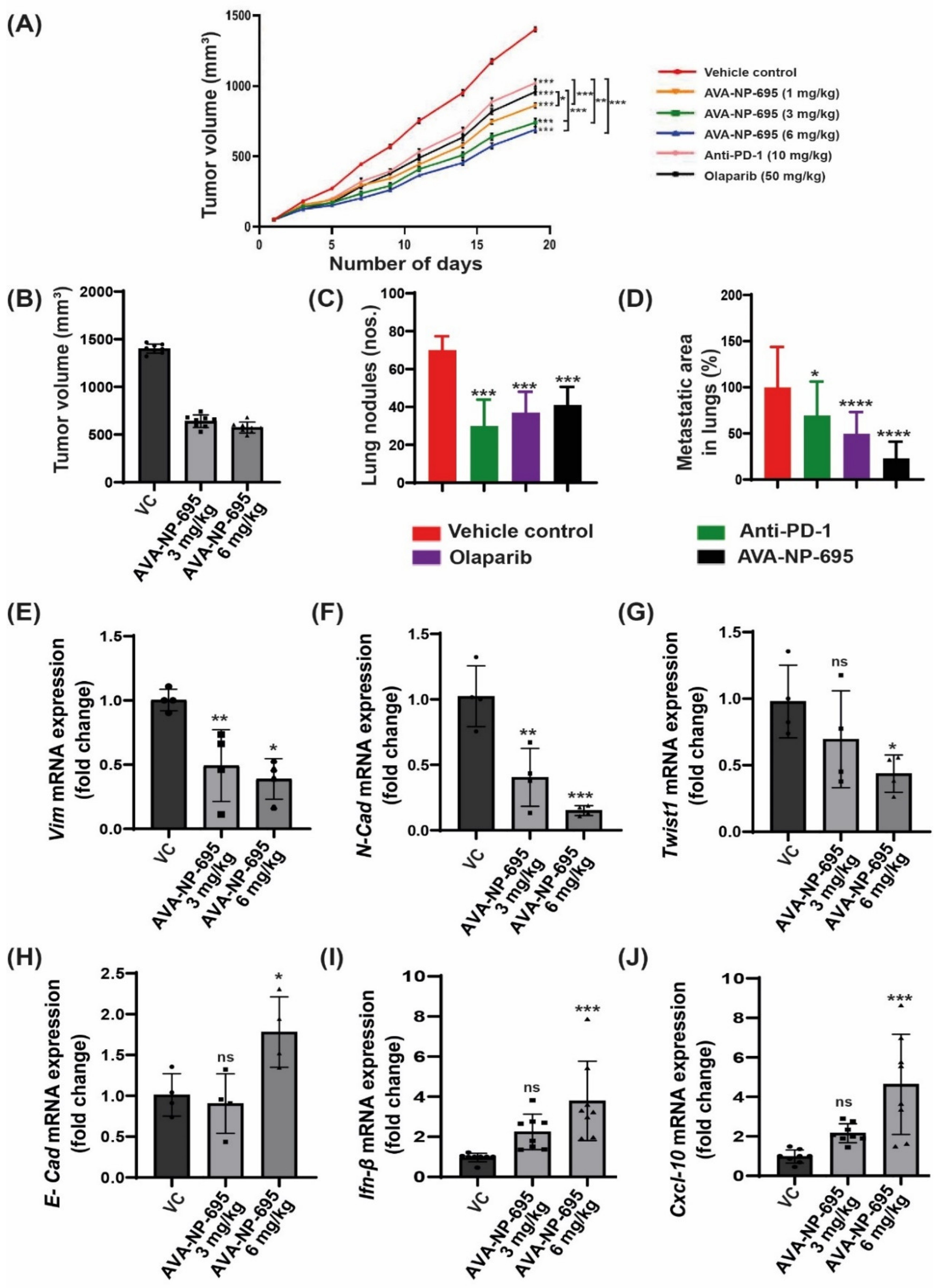
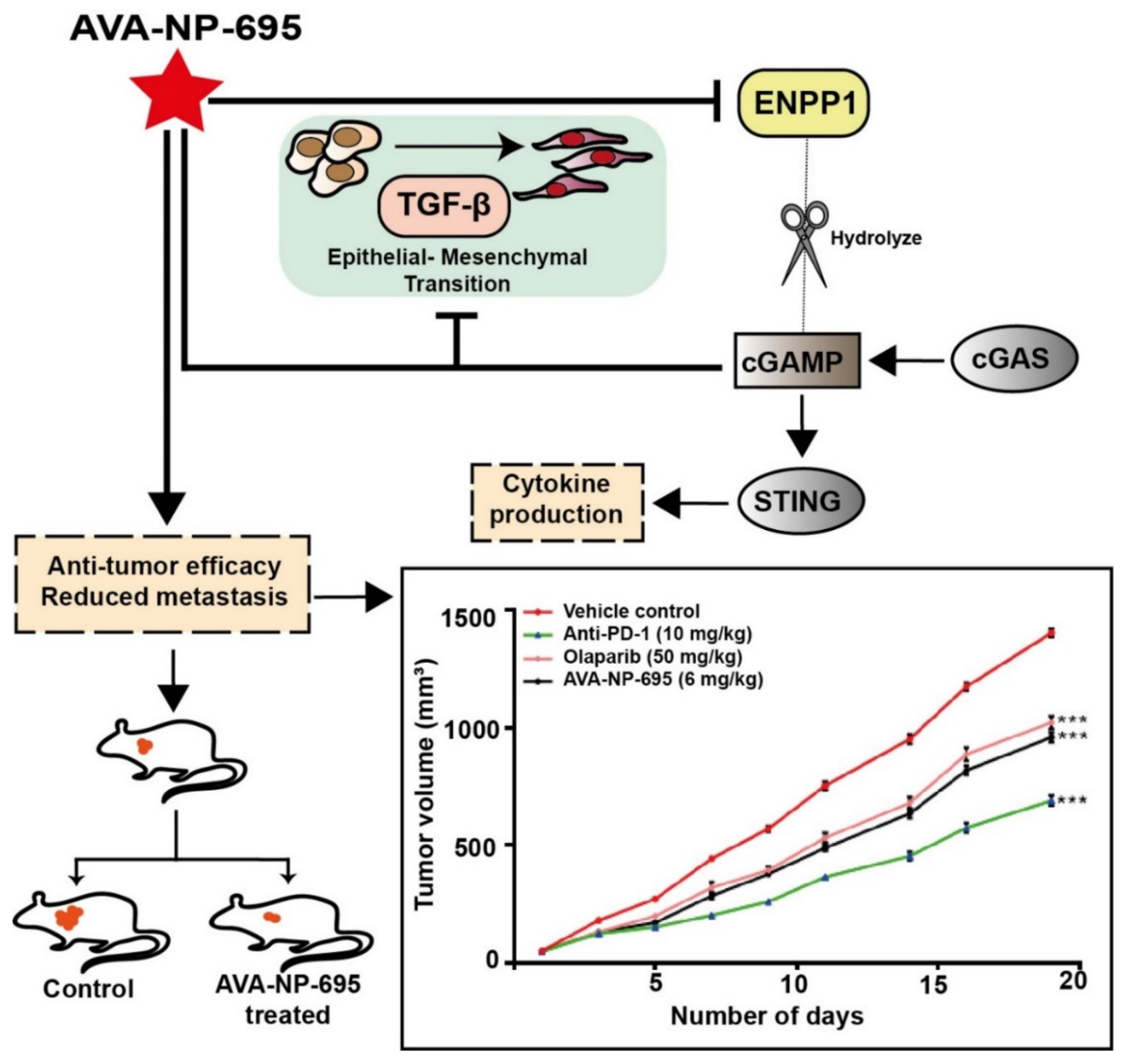
3.6. Pharmacological Inhibition of ENPP1 by AVA-NP-695 Abrogates 4T1 Tumor Growth and Metastasis in Syngeneic Mice Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef]
- Pépin, G.; Gantier, M.P. cGAS-STING Activation in the Tumor Microenvironment and its Role in Cancer Immunity. In Regulation of Inflammatory Signaling in Health and Disease; Xu, D., Ed.; Springer Singapore: Singapore, 2017; pp. 175–194. [Google Scholar]
- Baguley, B.C.; Ching, L.M. Immunomodulatory actions of xanthenone anticancer agents. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 1997, 8, 119–127. [Google Scholar] [CrossRef]
- Roberts, Z.J.; Goutagny, N.; Perera, P.Y.; Kato, H.; Kumar, H.; Kawai, T.; Akira, S.; Savan, R.; van Echo, D.; Fitzgerald, K.A.; et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J. Exp. Med. 2007, 204, 1559–1569. [Google Scholar] [CrossRef]
- Carozza, J.A.; Böhnert, V.; Nguyen, K.C.; Skariah, G.; Shaw, K.E.; Brown, J.A.; Rafat, M.; von Eyben, R.; Graves, E.E.; Glenn, J.S.; et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 2020, 1, 184–196. [Google Scholar]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e9. [Google Scholar] [CrossRef]
- Hoong, B.Y.D.; Gan, Y.H.; Liu, H.; Chen, E.S. cGAS-STING pathway in oncogenesis and cancer therapeutics. Oncotarget 2020, 11, 2930. [Google Scholar] [CrossRef]
- Motedayen Aval, L.; Pease, J.E.; Sharma, R.; Pinato, D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J. Clin. Med. 2020, 9, 3323. [Google Scholar] [CrossRef]
- Harrington, K.J.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.N.; Khilnani, A.D.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29 (Suppl. S8), viii712. [Google Scholar] [CrossRef]
- Onyedibe, K.I.; Wang, M.; Sintim, H.O. ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors-A STING in the Tale of ENPP1. Molecules 2019, 24, 4192. [Google Scholar] [CrossRef]
- Li, L.; Yin, Q.; Kuss, P.; Maliga, Z.; Millán, J.L.; Wu, H.; Mitchison, T.J. Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 2014, 10, 1043–1048. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Han, X.; Hisada, T.; Nishikawa, S.; Kano, K.; Ieda, N.; Aoki, J.; Toyama, T.; Nakagawa, H. Development of an ENPP1 Fluorescence Probe for Inhibitor Screening, Cellular Imaging, and Prognostic Assessment of Malignant Breast Cancer. J. Med. Chem. 2019, 62, 9254–9269. [Google Scholar] [CrossRef]
- Pérez-Valencia, J.A.; Prosdocimi, F.; Cesari, I.M.; da Costa, I.R.; Furtado, C.; Agostini, M.; Rumjanek, F.D. Angiogenesis and evading immune destruction are the main related transcriptomic characteristics to the invasive process of oral tongue cancer. Sci. Rep. 2018, 8, 2007. [Google Scholar] [CrossRef]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis, a Therapeutic Target in Chromosomally Unstable Tumors. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef]
- Lau, W.M.; Doucet, M.; Stadel, R.; Huang, D.; Weber, K.L.; Kominsky, S.L. Enpp1: A potential facilitator of breast cancer bone metastasis. PLoS ONE 2013, 8, e66752. [Google Scholar] [CrossRef]
- Wang, J.; Lu, S.F.; Wan, B.; Ming, S.L.; Li, G.L.; Su, B.Q.; Liu, J.Y.; Wei, Y.S.; Yang, G.Y.; Chu, B.B. Maintenance of cyclic GMP-AMP homeostasis by ENPP1 is involved in pseudorabies virus infection. Mol. Immunol. 2018, 95, 56–63. [Google Scholar] [CrossRef]
- Sautter, C.A.; Auray, G.; Python, S.; Liniger, M.; Summerfield, A. Phenotypic and functional modulations of porcine macrophages by interferons and interleukin-4. Dev. Comp. Immunol. 2018, 84, 181–192. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Eliahu, S.; Lecka, J.; Reiser, G.; Haas, M.; Bigonnesse, F.; Levesque, S.A.; Pelletier, J.; Sevigny, J.; Fischer, B. Diadenosine 5’,5’’-(boranated)polyphosphonate analogues as selective nucleotide pyrophosphatase/phosphodiesterase inhibitors. J. Med. Chem. 2010, 53, 8485–8497. [Google Scholar] [CrossRef]
- Nadel, Y.; Lecka, J.; Gilad, Y.; Ben-David, G.; Forster, D.; Reiser, G.; Kenigsberg, S.; Camden, J.; Weisman, G.A.; Senderowitz, H.; et al. Highly potent and selective ectonucleotide pyrophosphatase/phosphodiesterase I inhibitors based on an adenosine 5’-(alpha or gamma)-thio-(alpha, beta- or beta, gamma)-methylenetriphosphate scaffold. J. Med. Chem. 2014, 57, 4677–4691. [Google Scholar] [CrossRef]
- Forcellini, E.; Boutin, S.; Lefebvre, C.-A.; Shayhidin, E.E.; Boulanger, M.-C.; Rhéaume, G.; Barbeau, X.; Lagüe, P.; Mathieu, P.; Paquin, J.-F. Synthesis and biological evaluation of novel quinazoline-4-piperidinesulfamide derivatives as inhibitors of NPP1. Eur. J. Med. Chem. 2018, 147, 130–149. [Google Scholar] [CrossRef]
- Carozza, J.A.; Brown, J.A.; Böhnert, V.; Fernandez, D.; AlSaif, Y.; Mardjuki, R.E.; Smith, M.; Li, L. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem. Biol. 2020, 27, 1347–1358.e5. [Google Scholar] [CrossRef]
- Gangar, M.; Goyal, S.; Raykar, D.; Khurana, P.; Martis, A.M.; Goswami, A.; Ghoshal, I.; Patel, K.V.; Nagare, Y.; Raikar, S.; et al. Design, synthesis and biological evaluation studies of novel small molecule ENPP1 inhibitors for cancer immunotherapy. Bioorganic Chem. 2022, 119, 105549. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ye, F.; Zhou, C.; Cheng, Q.; Chen, H. High expression of ENPP1 in high-grade serous ovarian carcinoma predicts poor prognosis and as a molecular therapy target. PLoS ONE 2021, 16, e0245733. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Guo, W.; Liao, Y.; Xu, D.; Sun, B.; Song, H.; Wang, T.; Kuang, Y.; Jing, B.; Li, K.; et al. Dysregulated ENPP1 increases the malignancy of human lung cancer by inducing epithelial-mesenchymal transition phenotypes and stem cell features. Am. J. Cancer Res. 2019, 9, 134–144. [Google Scholar] [PubMed]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456.e31. [Google Scholar] [CrossRef]
- Kato, K.; Nishimasu, H.; Okudaira, S.; Mihara, E.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 16876–16881. [Google Scholar] [CrossRef]
- Cheng, H.; Xu, Q.; Lu, X.; Yuan, H.; Li, T.; Zhang, Y.; Tan, X. Activation of STING by cGAMP Regulates MDSCs to Suppress Tumor Metastasis via Reversing Epithelial-Mesenchymal Transition. Front. Oncol. 2020, 10, 896. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.-J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef]
- Edlund, S.; Landström, M.; Heldin, C.H.; Aspenström, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- O’Connor, J.W.; Gomez, E.W. Cell Adhesion and Shape Regulate TGF-Beta1-Induced Epithelial-Myofibroblast Transition via MRTF-A Signaling. PLoS ONE 2013, 8, e83188. [Google Scholar] [CrossRef]
- Wang, H.; Guo, S.; Kim, S.-J.; Shao, F.; Ho, J.W.K.; Wong, K.U.; Miao, Z.; Hao, D.; Zhao, M.; Xu, J.; et al. Cisplatin prevents breast cancer metastasis through blocking early EMT and retards cancer growth together with paclitaxel. Theranostics 2021, 11, 2442–2459. [Google Scholar] [CrossRef]
- Jalalirad, M.; Haddad, T.C.; Salisbury, J.L.; Radisky, D.; Zhang, M.; Schroeder, M.; Tuma, A.; Leof, E.; Carter, J.M.; Degnim, A.C.; et al. Aurora-A kinase oncogenic signaling mediates TGF-β-induced triple-negative breast cancer plasticity and chemoresistance. Oncogene 2021, 40, 2509–2523. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. United States Am. 2014, 111, 11774–11779. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Frankel, A.E.; Chen, Z.J. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 323–344. [Google Scholar] [CrossRef]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: A promising new anticancer strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Larkin, B.; Ilyukha, V.; Sorokin, M.; Buzdin, A.; Vannier, E.; Poltorak, A. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J. Immunol. 2017, 199, 397–402. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goswami, A.; Deb, B.; Goyal, S.; Gosavi, A.; Mali, M.; Martis, A.M.; Khurana, P.; Gangar, M.; Raykar, D.; Mohanty, A.; et al. AVA-NP-695 Selectively Inhibits ENPP1 to Activate STING Pathway and Abrogate Tumor Metastasis in 4T1 Breast Cancer Syngeneic Mouse Model. Molecules 2022, 27, 6721. https://doi.org/10.3390/molecules27196721
Goswami A, Deb B, Goyal S, Gosavi A, Mali M, Martis AM, Khurana P, Gangar M, Raykar D, Mohanty A, et al. AVA-NP-695 Selectively Inhibits ENPP1 to Activate STING Pathway and Abrogate Tumor Metastasis in 4T1 Breast Cancer Syngeneic Mouse Model. Molecules. 2022; 27(19):6721. https://doi.org/10.3390/molecules27196721
Chicago/Turabian StyleGoswami, Avijit, Barnali Deb, Sandeep Goyal, Abhishek Gosavi, Mukund Mali, Ashwita M. Martis, Princy Khurana, Mukesh Gangar, Digambar Raykar, Ankita Mohanty, and et al. 2022. "AVA-NP-695 Selectively Inhibits ENPP1 to Activate STING Pathway and Abrogate Tumor Metastasis in 4T1 Breast Cancer Syngeneic Mouse Model" Molecules 27, no. 19: 6721. https://doi.org/10.3390/molecules27196721
APA StyleGoswami, A., Deb, B., Goyal, S., Gosavi, A., Mali, M., Martis, A. M., Khurana, P., Gangar, M., Raykar, D., Mohanty, A., & Kulkarni, A. (2022). AVA-NP-695 Selectively Inhibits ENPP1 to Activate STING Pathway and Abrogate Tumor Metastasis in 4T1 Breast Cancer Syngeneic Mouse Model. Molecules, 27(19), 6721. https://doi.org/10.3390/molecules27196721