
Indane Based Molecular Motors: UV-Switching Increases Number of Isomers
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
- 6-Nitroindan-1-one (2) and 4-nitroindan-1-one (3)
- Ethyl-2-[6-nitroindan-1-ylidene]acetate (4) and ethyl 2-[4-nitroindan-1-ylidene]acetate (5)
- 4-Aphin and 6-Aphin
2.1. Absorption Spectra and UV-Isomerization
2.2. Quantum Chemical Calculations
3. Results
3.1. Synthesis
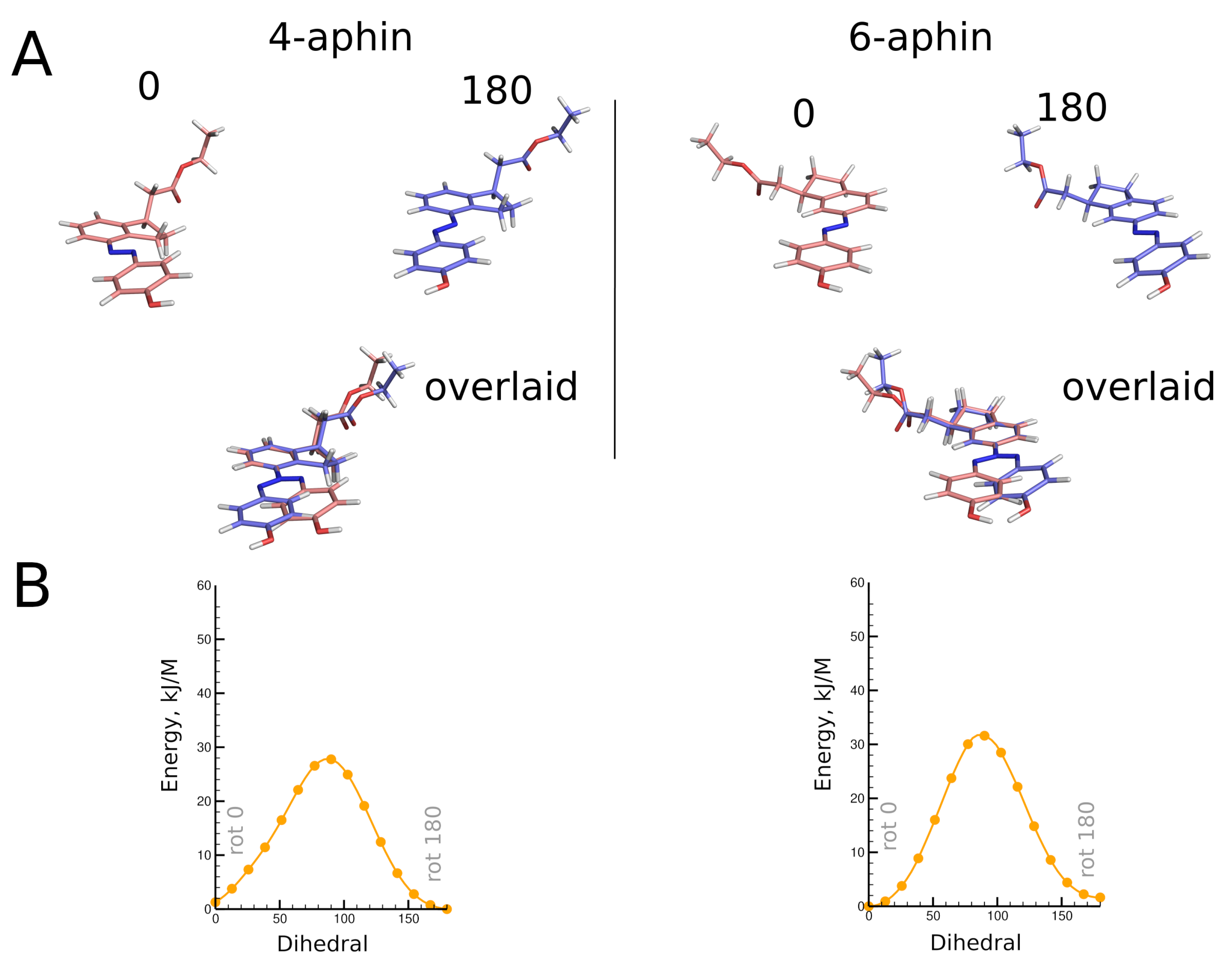
3.2. Molecular Geometry
3.2.1. Trans Isomers
3.2.2. Cis Isomers
3.3. UV-Spectra
3.4. Photoisomerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jerca, F.A.; Jerca, V.V.; Hoogenboom, R. Advances and opportunities in the exciting world of azobenzenes. Nat. Rev. Chem. 2022, 6, 51–69. [Google Scholar] [CrossRef]
- Villarón, D.; Wezenberg, S.J. Stiff-Stilbene Photoswitches: From Fundamental Studies to Emergent Applications. Angew. Chem. Int. Ed. 2020, 59, 13192. [Google Scholar] [CrossRef]
- Feringa, B.L. The Art of Building Small: From Molecular Switches to Molecular Motors. J. Org. Chem. 2007, 72, 6635–6652. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Marazzi, M.; Frutos, L.M.; Sampedro, D. E/Z Photochemical switches: Syntheses, properties and applications. RSC Adv. 2013, 3, 6241–6266. [Google Scholar] [CrossRef]
- Lerch, M.M.; Szymanski, W.; Feringa, B.L. The (photo)chemistry of Stenhouse photoswitches: Guiding principles and system design. Chem. Soc. Rev. 2018, 47, 1910. [Google Scholar] [CrossRef]
- Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Photochromism of diarylethene molecules and crystals: Memories, switches, and actuators. Chem. Rev. 2014, 114, 12174–12277. [Google Scholar] [CrossRef]
- Li, Z.; He, C.; Li, P.; Zhu, Y.-P. Recent progress in all-visible-light-triggered diarylethenes. Dyes Pigments 2020, 182, 108623. [Google Scholar] [CrossRef]
- Bercovic, G.; Krongauz, V.; Weiss, V. Spiropyrans and spirooxazines for memories and switches. Chem. Rev. 2000, 100, 1741–1754. [Google Scholar] [CrossRef]
- Leith, G.A.; Martin, C.R.; Mathur, A.; Kittikhunnatham, P.; Park, K.C.; Shustova, N.R. Dynamically controlled electronic behavior of stimuli-responsive materials: Exploring dimensionality and connectivity. Adv. Energy Mater. 2022, 12, 2100441. [Google Scholar] [CrossRef]
- Sahoo, D.; Benny, R.; Ks, N.K.; De, S. Stimuli-Responsive Chiroptical Switching. ChemPlusChem 2022, 87, e202100322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Q. Light-Driven Chiral Molecular Switches or Motors in Liquid Crystals. Adv. Mater. 2012, 24, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Moulin, E.; Giuseppone, N. Extraction of mechanical work from stimuli-responsive molecular systems and materials. Trends Chem. 2021, 3, 926–942. [Google Scholar] [CrossRef]
- Wang, H.; Bisoyi, H.K.; Zhang, X.; Hassan, F.; Li, Q. Visible Light-Driven Molecular Switches and Motors: Recent Developments and Applications. Chem. Eur. J. 2022, 28, e202103906. [Google Scholar] [PubMed]
- Fujiwara, H.; Yonezawa, Y. Photoelectric response of a black lipid membrane containing an amphiphilic azobenzene derivative. Nature 1991, 351, 724–726. [Google Scholar] [CrossRef]
- Song, X.; Perlstein, J.; Whitten, D. Supramolecular aggregates of azobenzene phospholipids and related compounds in bilayer assemblies and other microheterogeneous media: Structure, properties, and photoreactivity. J. Am. Chem. Soc. 1997, 119, 9144–9159. [Google Scholar] [CrossRef]
- Hamada, T.; Sugimoto, R.; Nagasaki, T.; Takagi, M. Photochemical control of membrane raft organization. Soft Matter 2011, 7, 220–224. [Google Scholar] [CrossRef]
- Frank, J.; Franquelim, H.; Schwille, P.; Trauner, D. Optical Control of Lipid Rafts with Photoswitchable Ceramides. J. Am. Chem. Soc. 2016, 138, 12981–12986. [Google Scholar] [CrossRef]
- Saitov, A.; Akimov, S.; Galimzyanov, T.; Glasnov, T.; Pohl, P. Ordered Lipid Domains Assemble via Concerted Recruitment of Constituents from Both Membrane Leaflets. Phys. Rev. Lett. 2020, 124, 108102. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Yushchenko, D.; Hodson, D.; Lipstein, N.; Nagpal, J.; Rutter, G.; Rhee, J.; Gottschalk, A.; Brose, N.; Schultz, C.; et al. Photoswitchable diacylglycerols enable optical control of protein kinase C. Nat. Chem. Biol. 2016, 12, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Moroni, M.; Moshourab, R.; Sumser, M.; Lewin, G.; Trauner, D. Photoswitchable fatty acids enable optical control of TRPV1. Nat. Commun. 2015, 6, 7118. [Google Scholar] [CrossRef]
- Pfeffermann, J.; Eicher, B.; Boytsov, D.; Hannesschlaeger, C.; Galimzyanov, T.; Glasnov, T.; Pabst, G.; Akimov, S.; Pohl, P. Photoswitching of model ion channels in lipid bilayers. J. Photochem. Photobiol. B 2021, 224, 112320. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, V.; Grafmüller, A.; Bléger, D.; Hecht, S.; Kunstmann, S.; Barbirz, S.; Lipowsky, R.; Dimova, R. Area Increase and Budding in Giant Vesicles Triggered by Light: Behind the Scene. Adv. Sci. 2018, 5, 1800432. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, I. Optical Switches for Lipid Membranes: Computed Molecular Projection Area as a Switch Selection Criterion. Colloids Interfaces 2022, 6, 30. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Leong, M.K.; Mastryukov, V.S.; Boggs, J.E. Structure and conformation of cyclopentene, cycloheptene and trans-cyclooctene. J. Mol. Struct. 1998, 445, 149–160. [Google Scholar] [CrossRef]
- Caputto, M.E.; Fabian, L.E.; Benítez, D.; Merlino, A.; Ríos, N.; Cerecetto, H.; Moltrasio, G.Y.; Moglioni, A.G.; González, M.; Finkielsztein, L.M. Thiosemicarbazones derived from 1-indanones as new anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. 2011, 19, 6818–6826. [Google Scholar] [CrossRef]
- Keeffe, J.R.; Kresge, A.J.; Yin, Y. 2-Indanone and its enol. The effect of a conjugated phenyl group on enol and enolate stability. J. Am. Chem. Soc. 1988, 110, 8201–8206. [Google Scholar] [CrossRef]
- Alekseeva, A.S.; Korotaeva, A.A.; Samoilova, E.V.; Volynsky, P.E.; Vodovozova, E.L.; Boldyrev, I.A. Secretory phospholipase A2 activity in blood serum: The challenge to sense. Biochem. Biophys. Res. Commun. 2014, 454, 178–182. [Google Scholar] [CrossRef]
- Erickson, L.E.; Morris, K.F. The Energy Profile for Rotation about the C-C Bond in Substituted Ethanes: A Multi-Part Experimental Computational Project for the Physical Chemistry Laboratory. J. Chem. Educ. 1998, 75, 900. [Google Scholar] [CrossRef]
- Mahmoud, M.R.; Ibrahim, S.A.; Hamed, M.A. Low excitation energy band of 4-hydroxyazobenzene derivatives. Spectrochim. Acta Part A Mol. Spectrosc. 1983, 39, 729–733. [Google Scholar] [CrossRef]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Terentjev, E.M. Effects of solvent viscosity and polarity on the isomerization of azobenzene. Macromolecules 2008, 41, 981–986. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| trans rot 180 | trans rot 0 | cis anti rot 180 | cis anti rot 0 | cis syn rot 180 | cis syn rot 0 | |
|---|---|---|---|---|---|---|
| OH–COOEt distance, Å | 12.8 | 12.6 | 12.2 | 10.0 | 9.5 | 5.5 |
| OH–C1 distance, Å | 10.8 | 10.5 | 8.6 | 6.8 | 8.6 | 6.5 |
| C5-C4N=N dihedral | 5.3 | −165.8 | 58.6 | 123.0 | −57.7 | −128.6 |
| Dipole moment, D | 3.77 | 4.34 | 3.09 | 3.55 | 6.48 | 7.02 |
| G, Ha | −1070.80480 | −1070.80387 | −1070.78583 | −1070.78943 | −1070.78556 | −1070.78775 |
| S, Ha | 0.06865 | 0.06839 | 0.06585 | 0.07048 | 0.06586 | 0.06979 |
| , kJ/mol | 0 | 2.4 | 49.7 | 40.3 | 50.5 | 44.7 |
| Fraction, % | 73 | 27 | 2 | 83 | 1 | 14 |
| trans rot 180 | trans rot 0 | cis anti rot 180 | cis anti rot 0 | cis syn rot 180 | cis syn rot 0 | |
|---|---|---|---|---|---|---|
| OH–COOEt distance, Å | 13.7 | 12.7 | 12.3 | 9.7 | 9.7 | 5.2 |
| OH–C1 distance, Å | 11.1 | 10.7 | 8.7 | 6.7 | 8.7 | 6.3 |
| C5-C4N=N dihedral | 4.6 | 176.4 | −54.0 | −134.2 | 53.8 | 135.3 |
| Dipole moment, D | 2.08 | 3.19 | 4.40 | 4.18 | 7.13 | 6.66 |
| G, Ha | −1070.80445 | −1070.80692 | −1070.78706 | −1070.78663 | −1070.78638 | −1070.785292 |
| S, Ha | 0.06845 | 0.07097 | 0.06549 | 0.06845 | 0.06839 | 0.06577503 |
| , kJ/mol | 6.5 | 0 | 52.1 | 53.3 | 53.9 | 56.8 |
| Fraction, % | 7 | 93 | 44 | 28 | 21 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shendrikov, V.P.; Alekseeva, A.S.; Kot, E.F.; Mineev, K.S.; Tretiakova, D.S.; Ece, A.; Boldyrev, I.A. Indane Based Molecular Motors: UV-Switching Increases Number of Isomers. Molecules 2022, 27, 6716. https://doi.org/10.3390/molecules27196716
Shendrikov VP, Alekseeva AS, Kot EF, Mineev KS, Tretiakova DS, Ece A, Boldyrev IA. Indane Based Molecular Motors: UV-Switching Increases Number of Isomers. Molecules. 2022; 27(19):6716. https://doi.org/10.3390/molecules27196716
Chicago/Turabian StyleShendrikov, Valeriy P., Anna S. Alekseeva, Erik F. Kot, Konstantin S. Mineev, Daria S. Tretiakova, Abdulilah Ece, and Ivan A. Boldyrev. 2022. "Indane Based Molecular Motors: UV-Switching Increases Number of Isomers" Molecules 27, no. 19: 6716. https://doi.org/10.3390/molecules27196716
APA StyleShendrikov, V. P., Alekseeva, A. S., Kot, E. F., Mineev, K. S., Tretiakova, D. S., Ece, A., & Boldyrev, I. A. (2022). Indane Based Molecular Motors: UV-Switching Increases Number of Isomers. Molecules, 27(19), 6716. https://doi.org/10.3390/molecules27196716