
Absolute Configuration and Biological Evaluation of Novel Triterpenes as Possible Anti-Inflammatory or Anti-Tumor Agents

Abstract
:1. Introduction
2. Results and Discussion
2.1. Phytochemical Identification
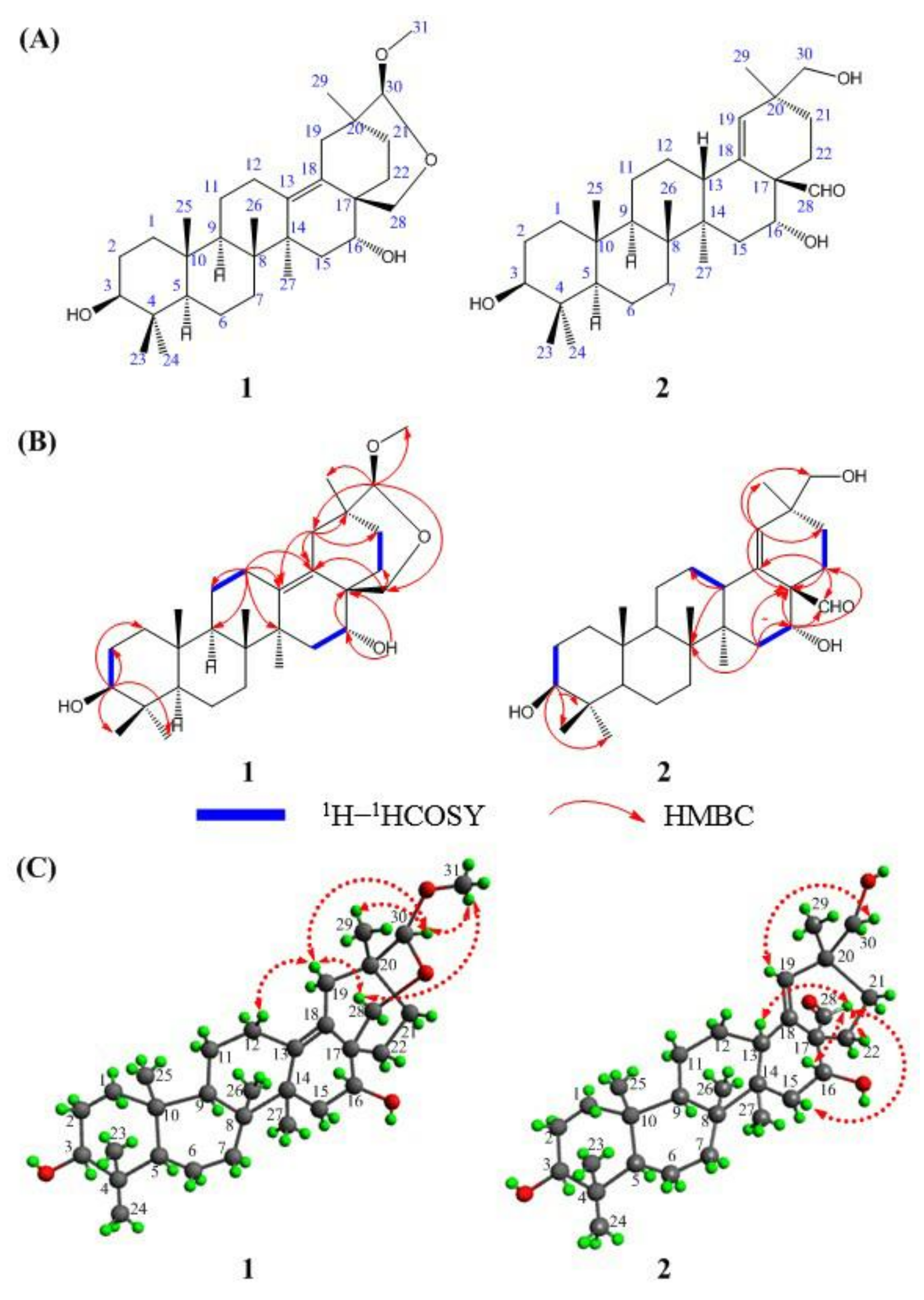
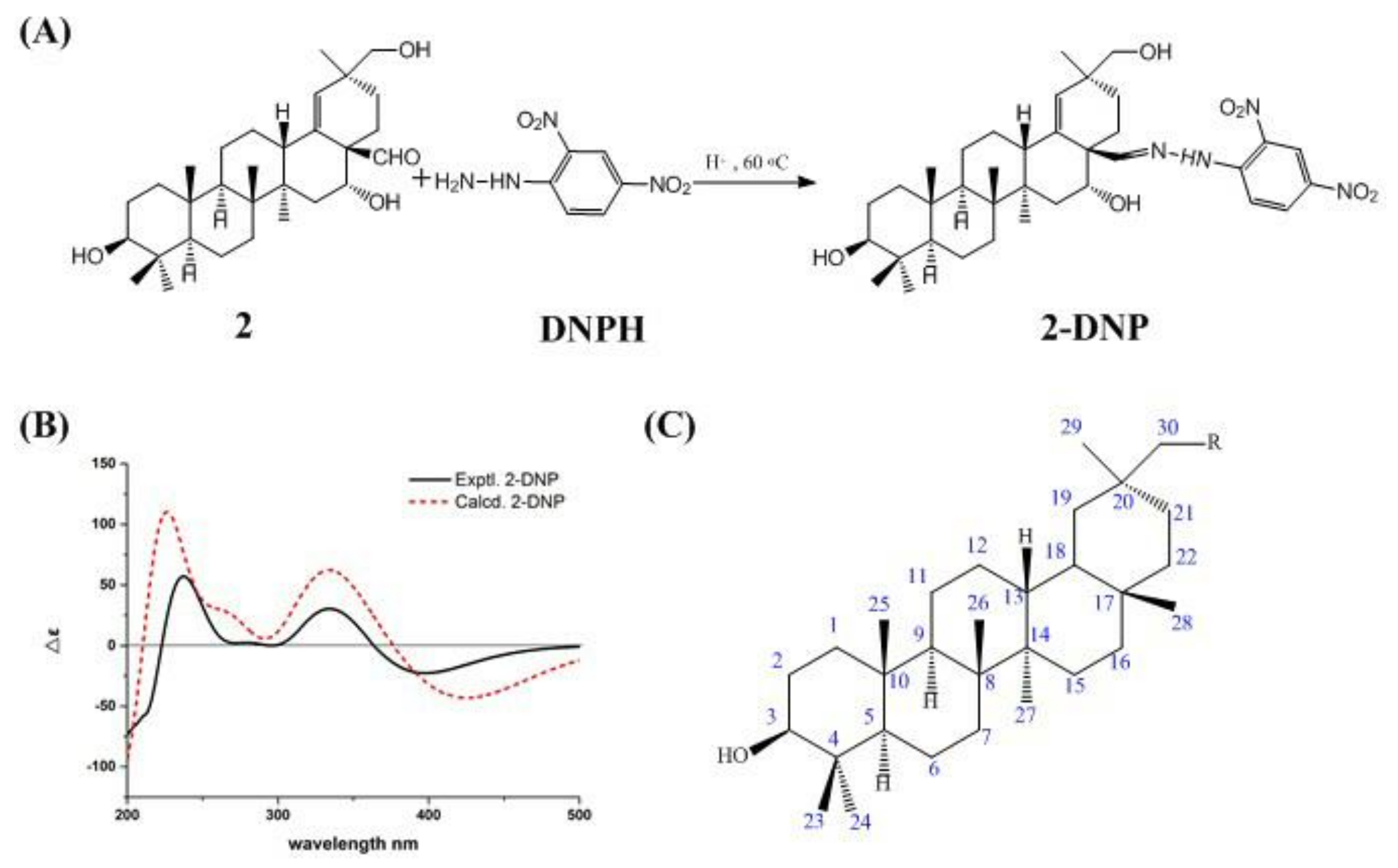
2.2. Structural Information of Compounds 1 and 2
2.3. X-Ray Crystallographic Analysis of Compound 1
2.4. Anti-Tumor Activity against Seven Cancer Cell Lines
2.4.1. Cytotoxic Activities
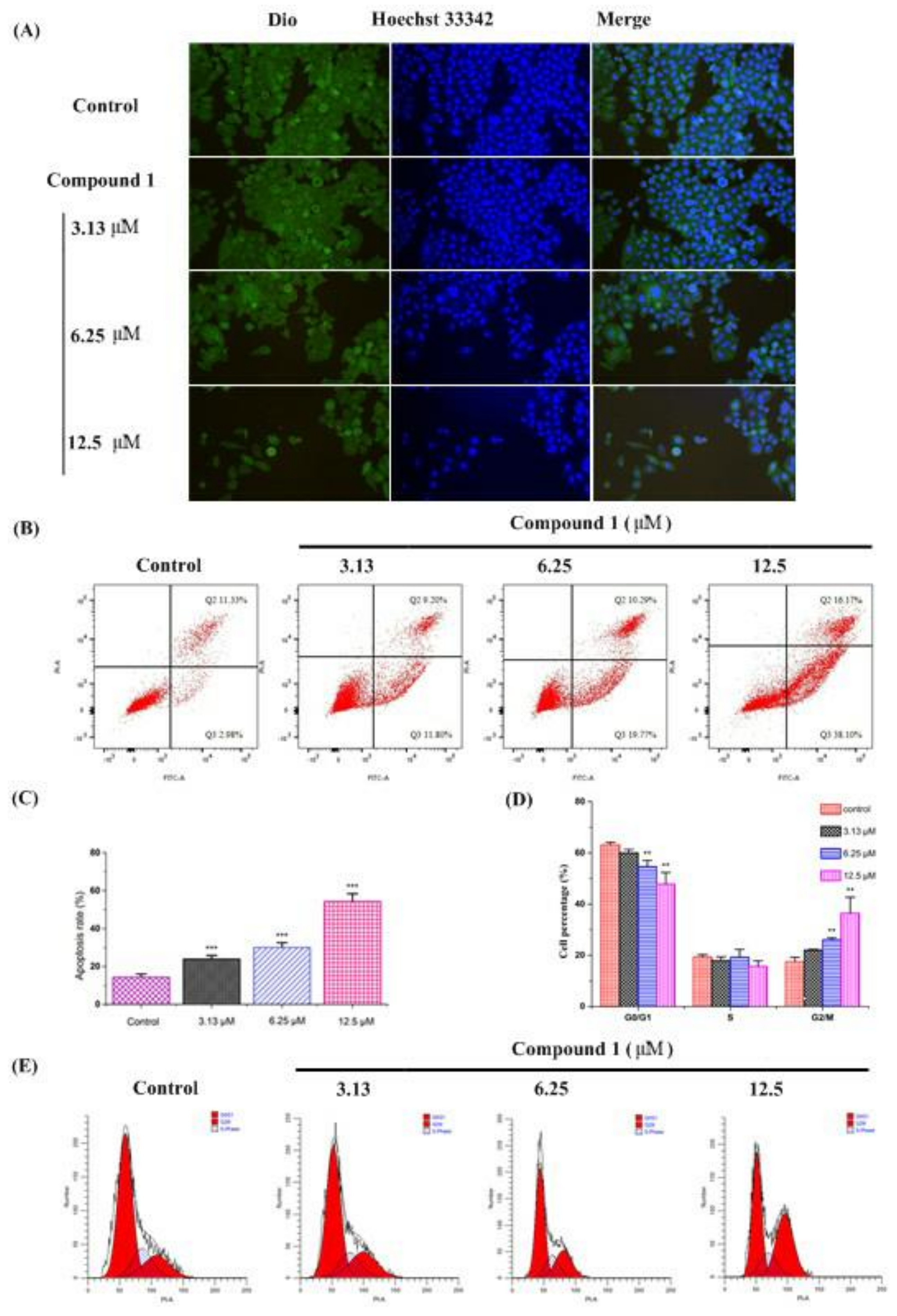
2.4.2. Cell Morphology
2.4.3. Cell Apoptosis and Cell Cycle
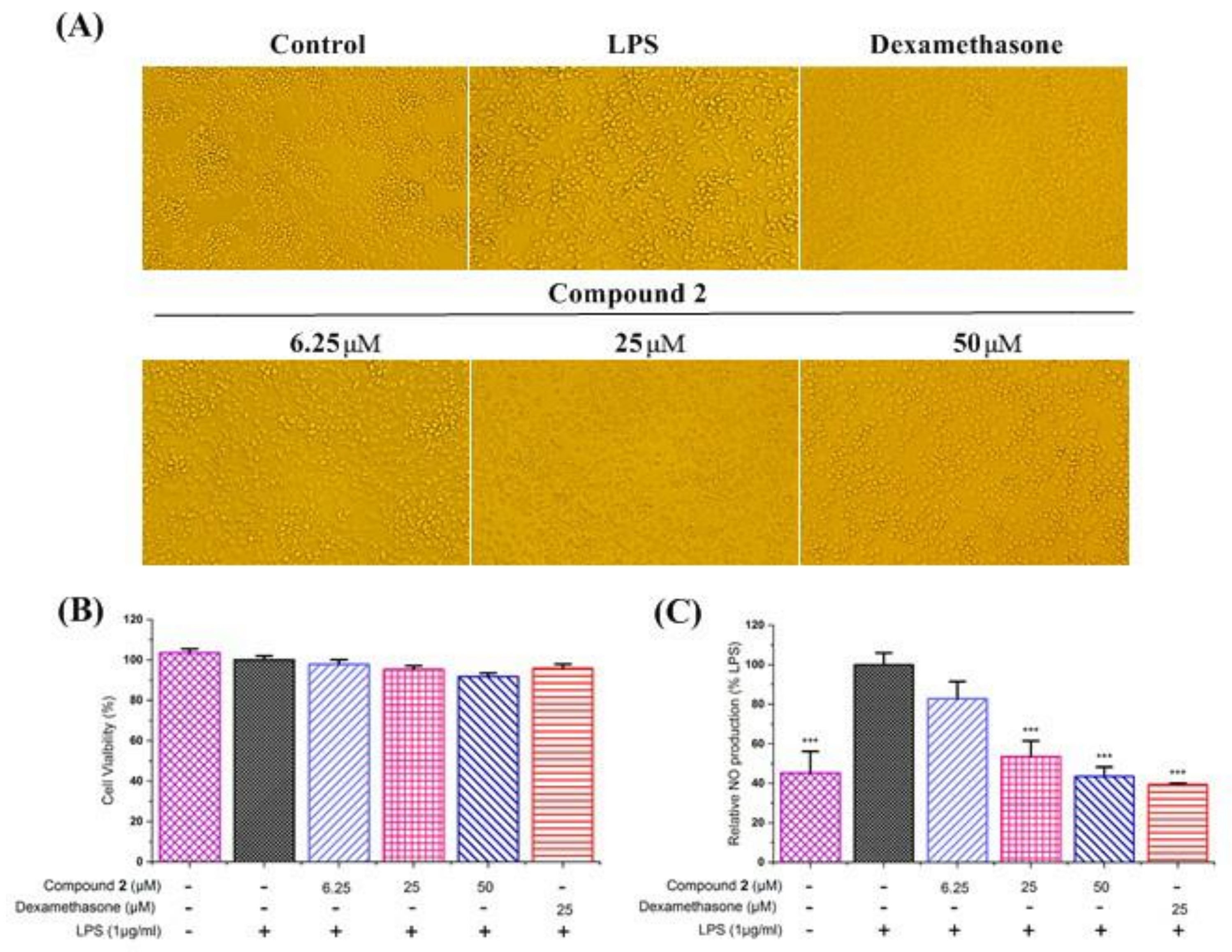
2.5. Anti–Inflammatory Activity against NO Production
3. Material and Methods
3.1. General Experimental Procedures
3.2. Plant Materials
3.3. Isolations of Compounds
3.4. Effect of Isolated Compounds on Cytotoxicity of Seven Cancer Cell Lines
3.5. Effect of Compound 1 on Cell Morphology, Cell Cycle, and Apoptosis of HepG2 Cells
3.5.1. Cell Morphology Assay
3.5.2. Cell Apoptosis and Cell Cycle Assay
3.6. Effect of Isolated Compounds on NO Production
3.6.1. Cell viability Assay on RAW264.7 Cells
3.6.2. Determination of the NO Content
3.6.3. Cell Morphology Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kobayashi, H.; De Mejía, E. The genus Ardisia: A novel source of health-promoting compounds and phytopharmaceuticals. J. Ethnopharmacol. 2005, 3, 47–354. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yue, D.K.; Bu, P.B.; Sun, Y.F. Chemical constituents from roots of Ardisia punctata. China J. Chin. Mater. Med. 2006, 7, 562–565. [Google Scholar]
- Ma, C.F.; Luo, M.; Lin, L.M.; Li, C.; Wang, Z.M.; Cheng, Y.Y. Chemical constituents of Ardisia punctata. China J. Chin. Mater. Med. 2012, 22, 3422–3425. [Google Scholar]
- Li, C.; Yue, D.K.; Bu, P.B.; Sun, Y.F. Three new belamcandaquinones from Ardisia punctata. Yao Xue Xue Bao 2006, 9, 830–834. [Google Scholar]
- Li, C.; Yue, D.K.; Bu, P.B.; Sun, Y.F. Two novel compounds from Ardisia punctata Lindl. Yao Xue Xue Bao 2007, 9, 959–963. [Google Scholar]
- De Mejía, E.G.; Ramírez-Mares, M.V. Ardisia: Health-promoting properties and toxicity of phytochemicals and extracts. Toxicol Mech. Methods 2011, 9, 667–674. [Google Scholar] [CrossRef] [PubMed]
- D’Yakonov, V.A.; Dzhemileva, L.U.; Dzhemilev, U.M. Advances in the Chemistry of Natural and Semisynthetic Topoisomerase I/II Inhibitors. Stud. Nat. Prod. Chem. 2017, 54, 21–86. [Google Scholar]
- Superchi, S.; Scafato, P.; Gorecki, M.; Pescitelli, G. Absolute Configuration Determination by Quantum Mechanical Calculation of Chiroptical Spectra: Basics and Applications to Fungal Metabolites. Curr. Med. Chem. 2018, 2, 287–320. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Di Bari, L.; Berova, N. Conformational aspects in the studies of organic compounds by electronic circular dichroism. Chem. Soc. Rev. 2011, 9, 4603–4625. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, S.; Inaba, Y.; Kunugita, N. Derivatization of carbonyl compounds with 2, 4-dinitrophenylhydrazine and their subsequent determination by high-performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 17–18, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.L.; Zhang, X.; Zhao, Y.M.; Wang, N.L.; Yao, X.S. Three new triterpenoid saponins from the roots of Ardisia crenata and their cytotoxic activities. Nat. Prod. Res. 2016, 23, 2694–2703. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Ruan, W.C.; Xu, Y.Z.; Wang, Y.J.; Pang, J.; Zhang, L.Y.; Liao, H.; Pang, T. The natural product 4,10-aromadendranediol induces neuritogenesis in neuronal cells in vitro through activation of the ERK pathway. Acta Pharmacol. Sin. 2007, 1, 29–40. [Google Scholar]
- Wu, S.; Romero-Ramírez, L.; Mey, J. Retinoic acid increases phagocytosis of myelin by macrophages. J. Cell. Physiol. 2021, 5, 3929–3945. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.S.; Sun, X.M.; Liu, X.Y.; Suo, X. Toxoplasma gondii: Effects of exogenous nitric oxide on egress of tachyzoites from infected macrophages. Exp. Parasitol. 2013, 1, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, H.; Radtke, O.A.; Kiderlen, A.F. Stimulus (polyphenol, IFN-gamma, LPS)-dependent nitric oxide production and antileishmanial effects in RAW264.7 macrophages. Phytochemistry 2008, 18, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.Y.; Cui, S.Y.; Wang, L.Y.; Fu, B.; Ding, R.; Yang, J.R.; Chen, X.M.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 5, 402–410. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO | 1 | 2 | 2-DNP | |||
|---|---|---|---|---|---|---|
| δC, Mult. | δH (J in Hz) | δC, Mult. | δH (J in Hz) | δC, Mult. | δH (J in Hz) | |
| 1 | 40.8, CH2 | overlapped | 39.1, CH2 | overlapped | 39.3, CH2 | overlapped |
| 2 | 29.6, CH2 | overlapped | 28.1, CH2 | overlapped | 28.1, CH2 | overlapped |
| 3 | 79.3, CH | 3.47, t (7.5) | 77.8, CH | 3.42, m | 77.8, CH | 3.44, t (7.5) |
| 4 | 40.9, C | 39.3, C | 39.3, C | |||
| 5 | 57.4, CH | overlapped | 55.8, CH | overlapped | 55.7, CH | overlapped |
| 6 | 20.2, CH2 | overlapped | 18.4, CH2 | overlapped | 18.4, CH2 | overlapped |
| 7 | 36.1, CH2 | overlapped | 34.8, CH2 | overlapped | 34.7, CH2 | overlapped |
| 8 | 43.2, C | 42.4, C | 42.4, C | |||
| 9 | 52.8, CH | overlapped | 50.6, CH | overlapped | 50.7, CH | overlapped |
| 10 | 38.9, C | 37.2, C | 37.2, C | |||
| 11 | 23.5, CH2 | overlapped | 20.9, CH2 | overlapped | 21.1, CH2 | overlapped |
| 12 | 27.1, CH2 | 2.57, d (13.7), Ha | 26.1, CH2 | overlapped, Ha | 26.2, CH2 | overlapped, Ha |
| overlapped, Hb | overlapped, Hb | overlapped, Hb | ||||
| 13 | 134.1, C | 40.7, CH | 2.67, d (9.4) | 40.3, CH | 2.71, d (9.6) | |
| 14 | 45.7, C | 40.8, C | 40.7, C | |||
| 15 | 38.1, CH2 | overlapped | 39.6, CH2 | overlapped | 39.5, CH2 | overlapped |
| 16 | 72.1, CH | overlapped | 75.5, CH | 4.12, dd (4.6, 11.9) | 75.8, CH | 4.12, d (11.4) |
| 17 | 45.8, C | 57.0, C | 50.0, C | |||
| 18 | 131.7, C | 137.4, C | 139.5, C | |||
| 19 | 36.9, CH2 | 3.16, d (16.3), Ha | 132.1, CH | 5.65, s | 131.5, CH | 5.70, s |
| 1.97, d (16.4), Hb | ||||||
| 20 | 40.7, C | 37.9, C | 38.2, C | |||
| 21 | 32.6, CH2 | overlapped | 28.5, CH2 | overlapped | 30.0, CH2 | overlapped |
| 22 | 25.9, CH2 | 2.24, m, Ha | 26.9, CH2 | 2.88, m, Ha | 27.9, CH2 | 2.80, m, Ha |
| overlapped, Hb | overlapped, Hb | overlapped, Hb | ||||
| 23 | 30.0, CH3 | 1.20, s | 28.4, CH3 | 1.19, s | 28.4, CH3 | 1.19, s |
| 24 | 17.7, CH3 | 1.02, s | 16.1, CH3 | 1.07, s | 16.1, CH3 | 1.01, s |
| 25 | 18.3, CH3 | 0.88, s | 16.7, CH3 | 0.85, s | 16.7, CH3 | 0.83, s |
| 26 | 20.2, CH3 | 1.00, s | 16.1, CH3 | 1.01, s | 16.1, CH3 | 1.01, s |
| 27 | 27.4, CH3 | 1.40, s | 16.0, CH3 | 0.95, s | 16.4, CH3 | 1.00, s |
| 28 | 72.2, CH2 | overlapped, Ha | 205.9, CH | 10.39, s | 157.4, CH | 8.65, s |
| 3.82, t (4.0), Hb | ||||||
| 29 | 29.9, CH3 | 0.98, s | 23.9, CH3 | 1.24, s | 23.9, CH3 | 1.30, s |
| 30 | 109.7, CH | 4.40, s | 71.5, CH2 | 3.68, m | 72.1, CH2 | 3.68, dd (10.2, 25.2) |
| 31 | 56.2, CH3 | 3.40, s | ||||
| 1′ | 145.6, C | |||||
| 2′ | 129.0, C | |||||
| 3′ | 122.7 (overlapped), CH | 9.04, d (2.5) | ||||
| 4′ | 137.2, C | |||||
| 5′ | 129.6, CH | 8.19, dd (2.5, 9.6) | ||||
| 6′ | 116.7, CH | 8.12, d (9.6) | ||||
| Parameter | Data |
|---|---|
| Empirical formula | C32H51Cl3O4 |
| Formula weight | 606.08 |
| Temperature / K | 115.85(10) |
| Crystal system | orthorhombic |
| Space group | P212121 |
| a / Å, b / Å, c / Å | 7.20942(19), 11.7688(3), 37.0363(10) |
| α/°, β/°, γ/° | 90.00, 90.00, 90.00 |
| Volume / Å3 | 3142.38(15) |
| Z | 4 |
| ρcalc / mg mm−3 | 1.281 |
| μ / mm−1 | 2.908 |
| F(000) | 1304 |
| Crystal size / mm3 | 0.34 × 0.33 × 0.13 |
| 2Θ range for data collection | 7.88 to 142.04° |
| Index ranges | −7 ≤ h ≤ 8, −14 ≤ k ≤ 10, −44 ≤ l ≤ 32 |
| Reflections collected | 10984 |
| Independent reflections | 5938[R(int) = 0.0324 (inf-0.9Å)] |
| Data/restraints/parameters | 5938/3/371 |
| Goodness-of-fit on F2 | 1.050 |
| Final R indexes (I > 2σ (I), i.e., Fo > 4σ (Fo)) | R1 = 0.0434, wR2 = 0.1161 |
| Final R indexes (all data) | R1 = 0.0461, wR2 = 0.1193 |
| Largest diff. peak/hole / e Å−3 | 0.497/−0.373 |
| Flack Parameters | −0.004(13) |
| Completeness | 0.9982 |
| Compound | IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| B16F10 a | A549 b | H460 c | MCF–7 d | HepG2 e | Hela f | U87 g | |
| 1 | 17.51 ± 3.55 | 29.86 ± 4.26 | 33.09 ± 0.58 | 21.73 ± 2.71 | 12.40 ± 3.76 | 24.96 ± 6.88 | 25.50 ± 3.45 |
| 2 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| cisplatin | 9.07 ± 0.66 | 8.45 ± 0.74 | 9.63 ± 0.49 | 8.23 ± 0.43 | 9.89 ± 0.61 | 8.26 ± 0.51 | 8.89 ± 0.63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Z.; Zhou, T.; Xia, Z.; Liu, S.; Li, M.; Zhang, G.; Tian, Y.; Li, B.; Wang, L.; Liu, S. Absolute Configuration and Biological Evaluation of Novel Triterpenes as Possible Anti-Inflammatory or Anti-Tumor Agents. Molecules 2022, 27, 6641. https://doi.org/10.3390/molecules27196641
Wei Z, Zhou T, Xia Z, Liu S, Li M, Zhang G, Tian Y, Li B, Wang L, Liu S. Absolute Configuration and Biological Evaluation of Novel Triterpenes as Possible Anti-Inflammatory or Anti-Tumor Agents. Molecules. 2022; 27(19):6641. https://doi.org/10.3390/molecules27196641
Chicago/Turabian StyleWei, Zhenzhen, Tiqiang Zhou, Ziming Xia, Sifan Liu, Min Li, Guangjie Zhang, Ying Tian, Bin Li, Lin Wang, and Shuchen Liu. 2022. "Absolute Configuration and Biological Evaluation of Novel Triterpenes as Possible Anti-Inflammatory or Anti-Tumor Agents" Molecules 27, no. 19: 6641. https://doi.org/10.3390/molecules27196641
APA StyleWei, Z., Zhou, T., Xia, Z., Liu, S., Li, M., Zhang, G., Tian, Y., Li, B., Wang, L., & Liu, S. (2022). Absolute Configuration and Biological Evaluation of Novel Triterpenes as Possible Anti-Inflammatory or Anti-Tumor Agents. Molecules, 27(19), 6641. https://doi.org/10.3390/molecules27196641