
Synthesis of Novel Lipophilic Polyamines via Ugi Reaction and Evaluation of Their Anticancer Activity

 , , ,
, , ,  , , , ,
, , , ,  and
and
Abstract
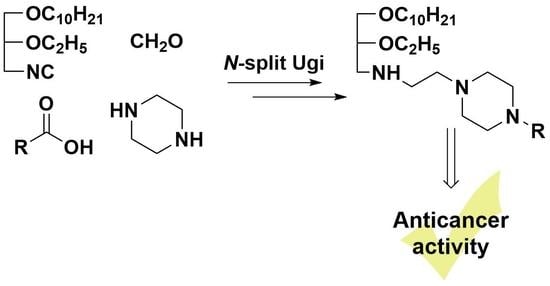
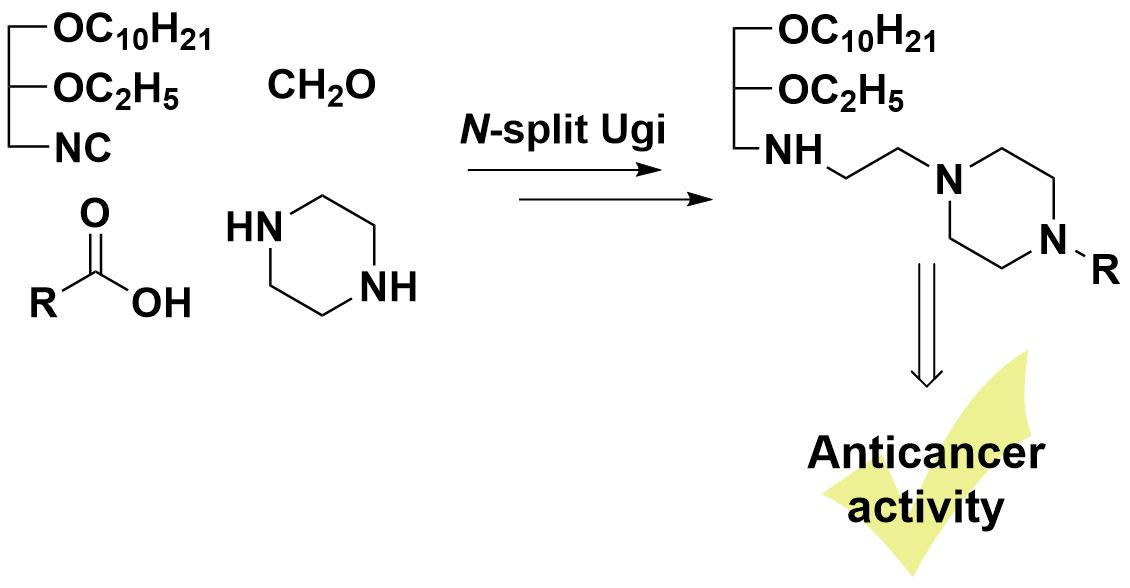
1. Introduction
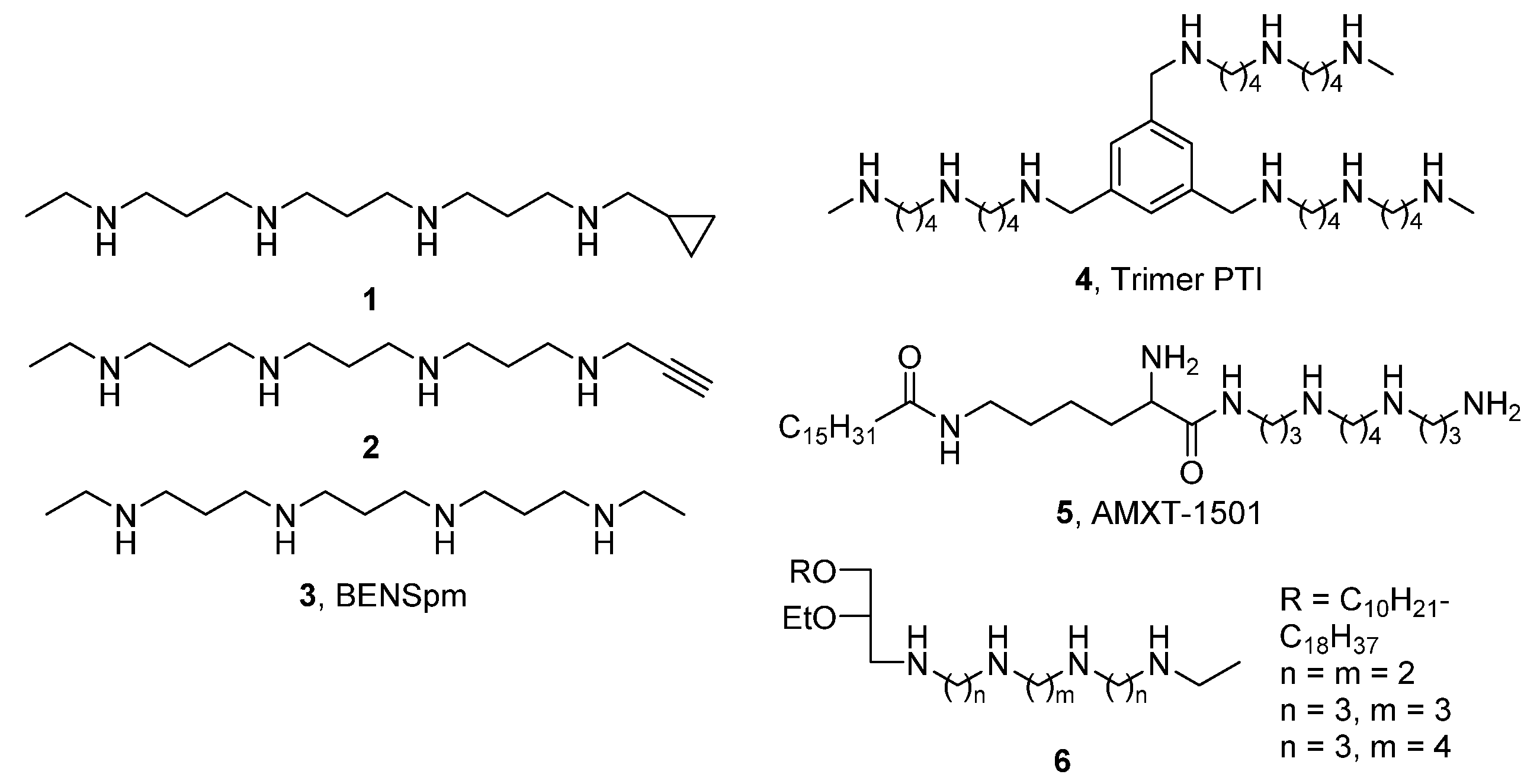
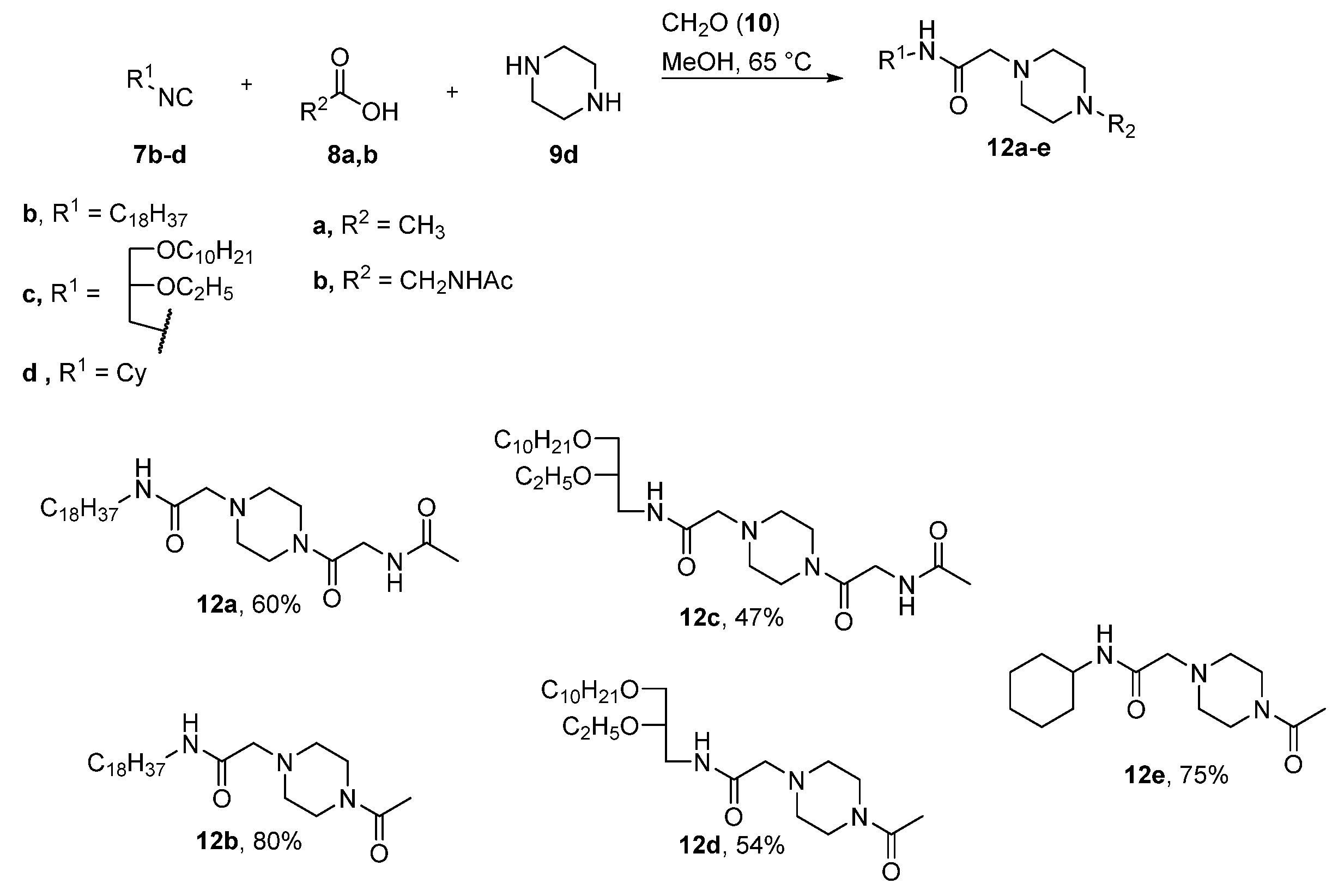
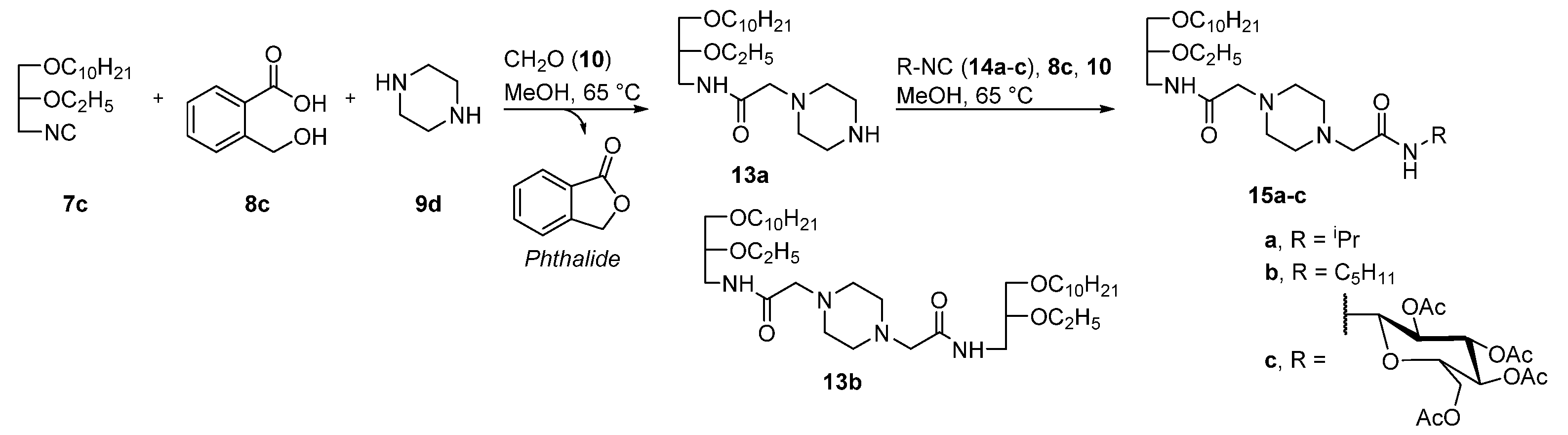
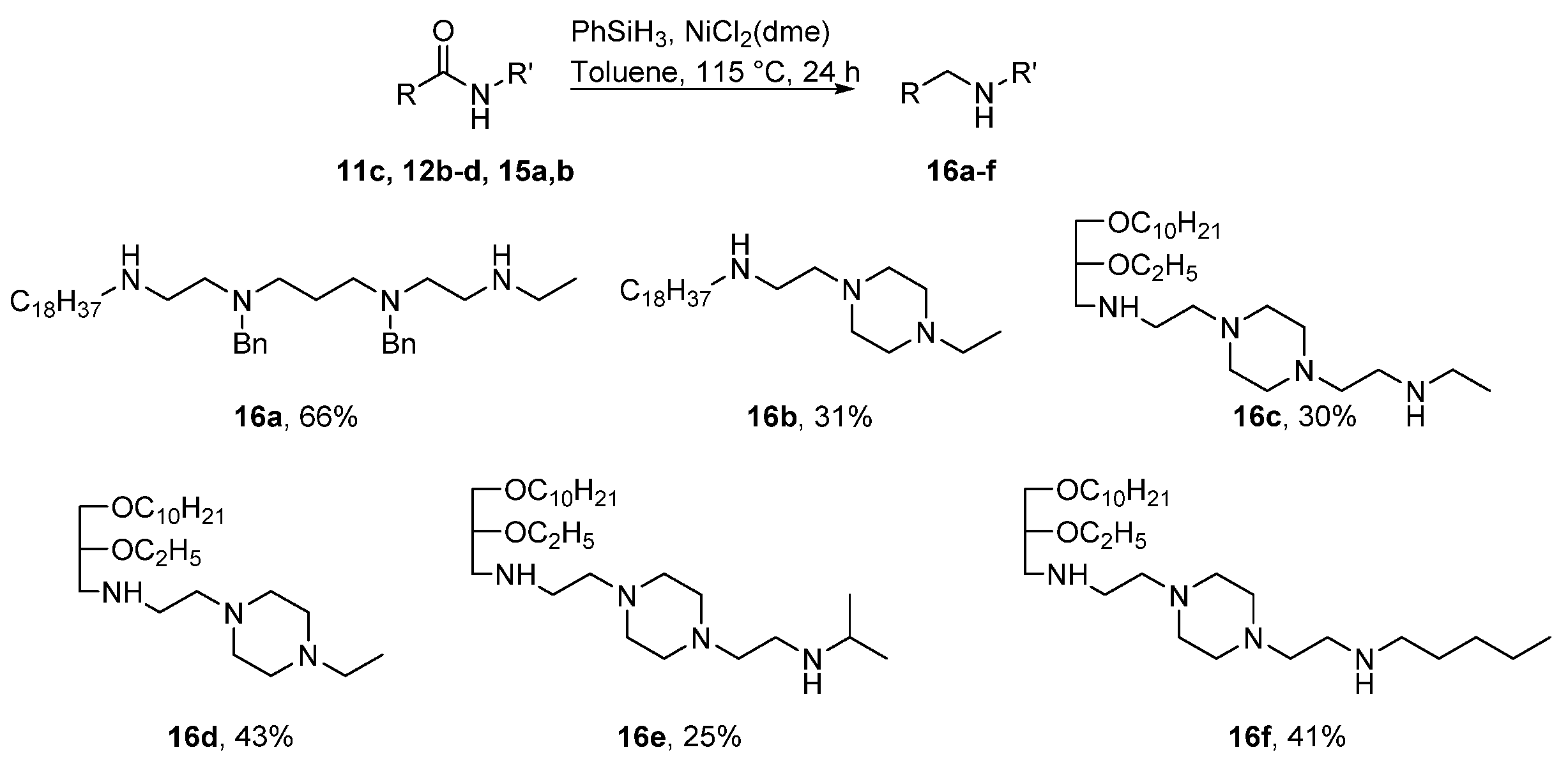
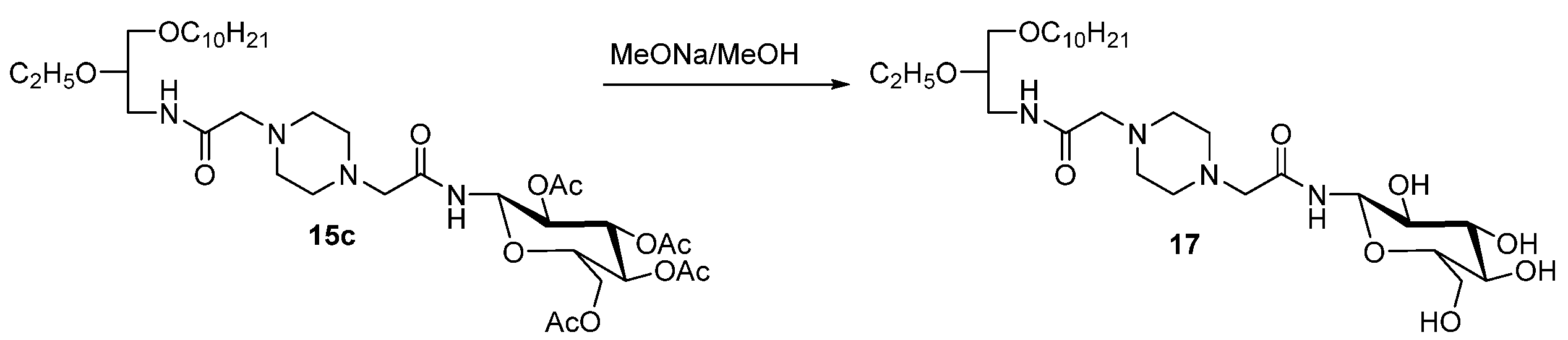
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Synthetic Methods
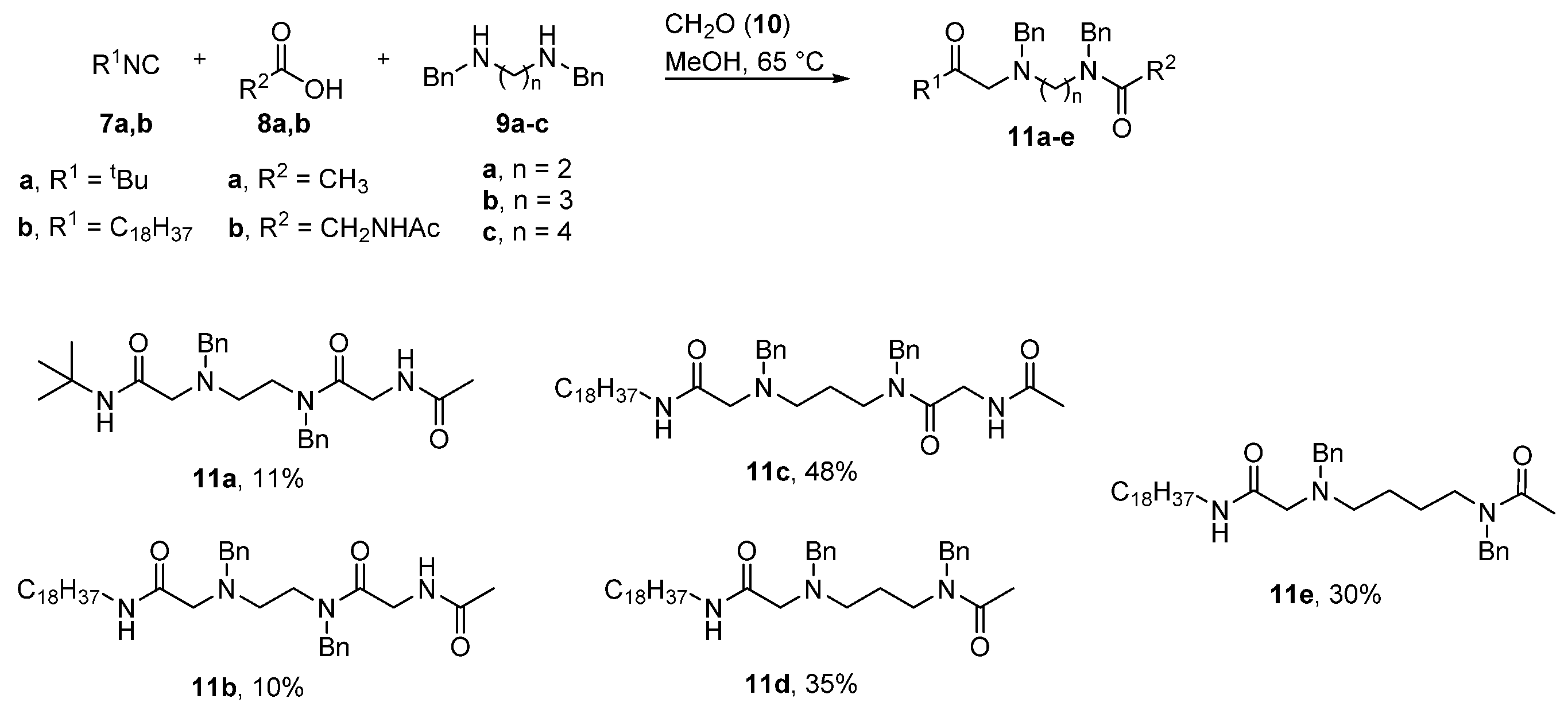
4.2.1. General Procedure for the Synthesis for Compounds 11a–e, 12a–e
1,8-Diamino-N8-Acetyl-N1-Tert-Butyl-1,7-Dioxo-N3,N6-Dibenzyl-3,6-Diazaoctane (11a)
1,8-Diamino-N8-Acetyl-N1-Octadecyl-1,7-Dioxo-N3,N6-Dibenzyl-3,6-Diazaoctane (11b)
1,9-Diamino-N9-Acetyl-N1-Octadecyl-1,8-Dioxo-N3,N7-Dibenzyl-3,7-Diazanonane (11c)
1,6-Diamino-N1-Acetyl-N6-Octadecyl-6-Oxo-N1,N3-Dibenzyl-4-Azahexane (11d)
1,7-Diamino-N1-Acetyl-N7-Octadecyl-7-Oxo-N1,N4-Dibenzyl-5-Azaheptane (11e)
N1-(N-Acetylglycyl)-N4-[(N-Octadecyl)Aminocarbonyl]Methylpiperazin (12a)
N1-Acetyl-N4-[(N-Octadecyl)Aminocarbonyl]Methylpiperazin (12b)
N1-(N-Acetylglycyl)-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (12c)
N1-Acetyl-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (12d)
N1-Acetyl-N4-[N-(Cyclohexyl)Aminocarbonyl]Methylpiperazin (12e)
4.2.2. Synthesis of Compounds 13a,b
N1-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (13a)
N1,N4-bis[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (13b)
4.2.3. General Procedure for the Synthesis of Compounds 15a–c
N1-[N-(Isopropyl)Aminocarbonyl]Methyl-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (15a)
N1-[N-(Pentyl)Aminocarbonyl]Methyl-N4-[N-(rac-(1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (15b)
N1-[N-(2,3,4,6-Tetra-O-Acetyl-β-D-Glucopyranosyl)Aminocarbonyl]Methyl-N4-[N-(rac-1-decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (15c)
4.2.4. General Procedure for Synthesis of Compounds 16a–f
1,9-Diamino-N9-ethyl-N1-Octadecyl-N3,N7-dibenzyl-3,7-Diazanonane (16a)
N1-Ethyl-N4-[(N-Octadecyl)Aminoethyl]Piperazin (16b)
N1-[2-(Ethylamino)Ethyl]-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Amino]Ethylpiperazin (16c)
N1-Ethyl-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Amino]Ethylpiperazin (16d)
N1-[2-[N-(Isopropylamino)ethyl]-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Amino]Ethylpiperazin (16e)
N1-[2-(N-Pentylamino)Ethyl]-N4-[N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Amino]Ethylpiperazin (16f)
N1-[(β-D-Glucopyranosyl)Aminocarbonyl]Methyl-N4-[(N-(rac-1-Decyloxy-2-Ethyloxyprop-3-yl)Aminocarbonyl]Methylpiperazin (17)
4.3. Cell Lines and Culture Conditions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Murray Stewart, T.; Pegg, A.E. Polyamine Metabolism and Cancer: Treatments, Challenges and Opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Moinard, C.; Cynober, L.; de Bandt, J.P. Polyamines: Metabolism and Implications in Human Diseases. Clin. Nutr. 2005, 24, 184–197. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Polyamines: Mysterious Modulators of Cellular Functions. Biochem. Biophys. Res. Commun. 2000, 271, 559–564. [Google Scholar] [CrossRef]
- Bercovich, Z.; Snapir, Z.; Keren-Paz, A.; Kahana, C. Antizyme Affects Cell Proliferation and Viability Solely through Regulating Cellular Polyamines. J. Biol. Chem. 2011, 286, 33778–33783. [Google Scholar] [CrossRef]
- Saab, N.H.; West, E.E.; Bieszk, N.C.; Preuss, C.V.; Mank, A.R.; Casero, R.A.; Woster, P.M. Synthesis and Evaluation of Unsymmetrically Substituted Polyamine Analogues as Modulators of Human Spermidine/Spermine-N1-Acetyltransferase (SSAT) and as Potential Antitumor Agents. J. Med. Chem. 1993, 36, 2998–3004. [Google Scholar] [CrossRef]
- Nichugovskiy, A.; Tron, G.C.; Maslov, M. Recent Advances in the Synthesis of Polyamine Derivatives and Their Applications. Molecules 2021, 26, 6579. [Google Scholar] [CrossRef]
- Perevoshchikova, K.A.; Nichugovskiy, A.I.; Isagulieva, A.K.; Morozova, N.G.; Ivanov, I.V.; Maslov, M.A.; Shtil, A.A. Synthesis of Novel Lipophilic Tetraamines with Cytotoxic Activity. Mendeleev Commun. 2019, 29, 616–618. [Google Scholar] [CrossRef]
- Varlamova, E.A.; Isagulieva, A.K.; Morozova, N.G.; Shmendel, E.V.; Maslov, M.A.; Shtil, A.A. Non-Phosphorus Lipids as New Antitumor Drug Prototypes. Russ. J. Bioorg. Chem. 2021, 47, 965–979. [Google Scholar] [CrossRef]
- Li, J.J. Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications. Choice Rev. Online 2014, 52, 52–1438. [Google Scholar] [CrossRef]
- Khan, A.; Gamble, L.D.; Upton, D.H.; Ung, C.; Yu, D.M.T.; Ehteda, A.; Pandher, R.; Mayoh, C.; Hébert, S.; Jabado, N.; et al. Dual Targeting of Polyamine Synthesis and Uptake in Diffuse Intrinsic Pontine Gliomas. Nat. Commun. 2021, 12, 971. [Google Scholar] [CrossRef] [PubMed]
- Ugi, I. The A-Addition of Immonium Ions and Anions to Isonitriles Accompanied by Secondary Reactions. Angew. Chem. Int. Ed. 1962, 1, 8–21. [Google Scholar] [CrossRef]
- Giovenzana, G.B.; Tron, G.C.; Di Paola, S.; Menegotto, I.G.; Pirali, T. A Mimicry of Primary Amines by Bis-Secondary Diamines as Components in the Ugi Four-Component Reaction. Angew. Chem. 2006, 118, 1117–1120. [Google Scholar] [CrossRef]
- Rabêlo, W.F.; Echemendía, R. Synthesis of Novel 1,4 Naphthoquinone-Based Molecules by an Ugi-Type Four-Component Reaction. Synth. Commun. 2019, 49, 515–521. [Google Scholar] [CrossRef]
- Pirali, T.; Callipari, G.; Ercolano, E.; Genazzani, A.A.; Giovenzana, G.B.; Tron, G.C. A Concise Entry into Nonsymmetrical Alkyl Polyamines. Org. Lett. 2008, 10, 4199–4202. [Google Scholar] [CrossRef]
- Nichugovskiy, A.I.; Khrulev, A.A.; Perevoshchikova, K.A.; Cheshkov, D.A.; Morozova, N.G.; Maslov, M.A. Synthesis of Isonitrile Derivatives of Diglycerides and Carbohydrates as Intermediates for Multicomponent Ugi Reaction. Russ. J. Bioorg. Chem. 2021, 47, 929–938. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds; Springer: Berlin/Heidelberg, Germany, 2020; ISBN 978-3-662-62438-8. [Google Scholar]
- She, E.X.; Hao, Z. A Novel Piperazine Derivative Potently Induces Caspase-Dependent Apoptosis of Cancer Cells via Inhibition of Multiple Cancer Signaling Pathways. Am. J. Transl. Res. 2013, 5, 622–633. [Google Scholar]
- Tuncbilek, M.; Bilget Guven, E.; Onder, T.; Cetin Atalay, R. Synthesis of Novel 6-(4-Substituted Piperazine-1-Yl)-9-(β-d-Ribofuranosyl)Purine Derivatives, Which Lead to Senescence-Induced Cell Death in Liver Cancer Cells. J. Med. Chem. 2012, 55, 3058–3065. [Google Scholar] [CrossRef]
- Sarı, C.; Nalçaoğlu, A.; Değirmencioğlu, İ.; Celep Eyüpoğlu, F. Tumor-Selective New Piperazine-Fragmented Silicon Phthalocyanines Initiate Cell Death in Breast Cancer Cell Lines. J. Photochem. Photobiol. B Biol. 2021, 216, 112143. [Google Scholar] [CrossRef] [PubMed]
- İbiş, K.; Nalbat, E.; Çalışkan, B.; Kahraman, D.C.; Cetin-Atalay, R.; Banoglu, E. Synthesis and Biological Evaluation of Novel Isoxazole-Piperazine Hybrids as Potential Anti-Cancer Agents with Inhibitory Effect on Liver Cancer Stem Cells. Eur. J. Med. Chem. 2021, 221, 113489. [Google Scholar] [CrossRef]
- Saab, A.M.; Dobmeier, M.; Koenig, B.; Fabri, E.; Finotti, A.; Borgatti, M.; Lampronti, I.; Bernardi, F.; Efferth, T.; Gambari, R. Antiproliferative and Erythroid Differentiation of Piperazine and Triphenyl Derivatives against K-562 Human Chronic Myelogenous Leukemia. Anticancer Res. 2013, 33, 3027–3032. [Google Scholar] [PubMed]
- Olsen, C.A.; Witt, M.; Jaroszewski, J.W.; Franzyk, H. Solid-Phase Polyamine Synthesis Using Piperazine and Piperidine Building Blocks. Org. Lett. 2003, 5, 4183–4185. [Google Scholar] [CrossRef] [PubMed]
- Bałczewski, P.; Zurawiński, R.; Mikina, M.; Dudziński, B. Synthesis of Polyamino Nitriles, En Route to Acylpolyamine Neurotoxins, via the Regioselective Michael Cyanoethylation of Unprotected Polyamines. Unusual Behaviour of 1-(2-Aminoethyl)Piperazine. Tetrahedron 2009, 65, 8727–8732. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. Multicomponent Reactions with Isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Mumm, O. Umsetzung von Säureimidchloriden Mit Salzen Organischer Säuren Und Mit Cyankalium. Ber. Dtsch. Chem. Ges. 1910, 43, 886–893. [Google Scholar] [CrossRef]
- La Spisa, F.; Feo, A.; Mossetti, R.; Tron, G.C. An Efficient Synthesis of Symmetric and Unsymmetric Bis-(β-Aminoamides) via Ugi Multicomponent Reaction. Org. Lett. 2012, 14, 6044–6047. [Google Scholar] [CrossRef]
- Volkov, A.; Tinnis, F.; Slagbrand, T.; Trillo, P.; Adolfsson, H. Chemoselective Reduction of Carboxamides. Chem. Soc. Rev. 2016, 45, 6685–6697. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Reduction of C=O to CHOH by Metal Hydrides. In Comprehensive Organic Synthesis, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 8, ISBN 9780080977430. [Google Scholar]
- Itsuno, S. Boron Hydride Reduction. ACS Symp. Ser. 2016, 1236, 241–274. [Google Scholar] [CrossRef]
- Simmons, B.J.; Hoffmann, M.; Hwang, J.; Jackl, M.K.; Garg, N.K. Nickel-Catalyzed Reduction of Secondary and Tertiary Amides. Org. Lett. 2017, 19, 1910–1913. [Google Scholar] [CrossRef]
- Hie, L.; Fine Nathel, N.F.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of Amides to Esters by the Nickel-Catalysed Activation of Amide C-N Bonds. Nature 2015, 524, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hager, E.R.; Phillips, D.L.; Dunn, V.R.; Hacker, A.; Frydman, B.; Kink, J.A.; Valasinas, A.L.; Reddy, V.K.; Marton, L.J.; et al. A Novel Polyamine Analog Inhibits Growth and Induces Apoptosis in Human Breast Cancer Cells. Clin. Cancer Res. 2003, 9, 2769–2777. [Google Scholar] [PubMed]
- Nosova, Y.N.; Zenin, I.V.; Maximova, V.P.; Zhidkova, E.M.; Kirsanov, K.I.; Lesovaya, E.A.; Lobas, A.A.; Gorshkov, M.V.; Kovaleva, O.N.; Milaeva, E.R.; et al. Influence of the Number of Axial Bexarotene Ligands on the Cytotoxicity of Pt(IV) Analogs of Oxaliplatin. Bioinorg. Chem. Appl. 2017, 2017, 4736321. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Mönch, D.; Maaß, A.; Gromoll, C.; Hehr, T.; Leibold, T.; Schlitt, H.J.; Dahlke, M.H.; Renner, P. Three Dimensional Cultivation Increases Chemo- and Radioresistance of Colorectal Cancer Cell Lines. PLoS ONE 2021, 16, e0244513. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Perrin, D.D. Purification of Laboratory Chemicals Eighth Edition; Butterworth-Heinemann: Oxford, UK, 2017; ISBN 978-0-12-805457-4. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds/Cell Lines | IC50 (µM) | IC50 (Average) | ||
|---|---|---|---|---|
| A-549 | MCF7 | HCT116 | ||
| 16b | 19.0 ± 0.7 | 12.0 ± 1.5 | 20.0 ± 3.0 | 18.5 |
| 16c | 5.1 ± 1.3 | 3.5 ± 0.6 | 5 ± 1.5 | 4.3 |
| 16d | 3.0 ± 0.4 | 1.0 ± 0.14 | 3.8 ± 0.5 | 2.3 |
| 16e | 5.9 ± 0.6 | 3.7 ± 0.5 | 3 ± 0.8 | 4.1 |
| 16f | 5.9 ± 0.5 | 4.6 ± 0.9 | 4.3 ± 1.2 | 4.8 |
| 17 | >100 | >100 | >100 | >100 |
| 3 (BENSpm) | 0.5 | 1 | n/d | |
| Cisplatin | 29.0 ± 10 | 14 ± 7 | 7.5 | 16.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nichugovskiy, A.; Maksimova, V.; Trapeznikova, E.; Eshtukova-Shcheglova, E.; Ivanov, I.; Yakubovskaya, M.; Kirsanov, K.; Cheshkov, D.; Tron, G.C.; Maslov, M. Synthesis of Novel Lipophilic Polyamines via Ugi Reaction and Evaluation of Their Anticancer Activity. Molecules 2022, 27, 6218. https://doi.org/10.3390/molecules27196218
Nichugovskiy A, Maksimova V, Trapeznikova E, Eshtukova-Shcheglova E, Ivanov I, Yakubovskaya M, Kirsanov K, Cheshkov D, Tron GC, Maslov M. Synthesis of Novel Lipophilic Polyamines via Ugi Reaction and Evaluation of Their Anticancer Activity. Molecules. 2022; 27(19):6218. https://doi.org/10.3390/molecules27196218
Chicago/Turabian StyleNichugovskiy, Artemiy, Varvara Maksimova, Ekaterina Trapeznikova, Elizaveta Eshtukova-Shcheglova, Igor Ivanov, Marianna Yakubovskaya, Kirill Kirsanov, Dmitry Cheshkov, Gian Cesare Tron, and Mikhail Maslov. 2022. "Synthesis of Novel Lipophilic Polyamines via Ugi Reaction and Evaluation of Their Anticancer Activity" Molecules 27, no. 19: 6218. https://doi.org/10.3390/molecules27196218
APA StyleNichugovskiy, A., Maksimova, V., Trapeznikova, E., Eshtukova-Shcheglova, E., Ivanov, I., Yakubovskaya, M., Kirsanov, K., Cheshkov, D., Tron, G. C., & Maslov, M. (2022). Synthesis of Novel Lipophilic Polyamines via Ugi Reaction and Evaluation of Their Anticancer Activity. Molecules, 27(19), 6218. https://doi.org/10.3390/molecules27196218