
Friedel-Crafts-Type Acylation and Amidation Reactions in Strong Brønsted Acid: Taming Superelectrophiles †


Abstract
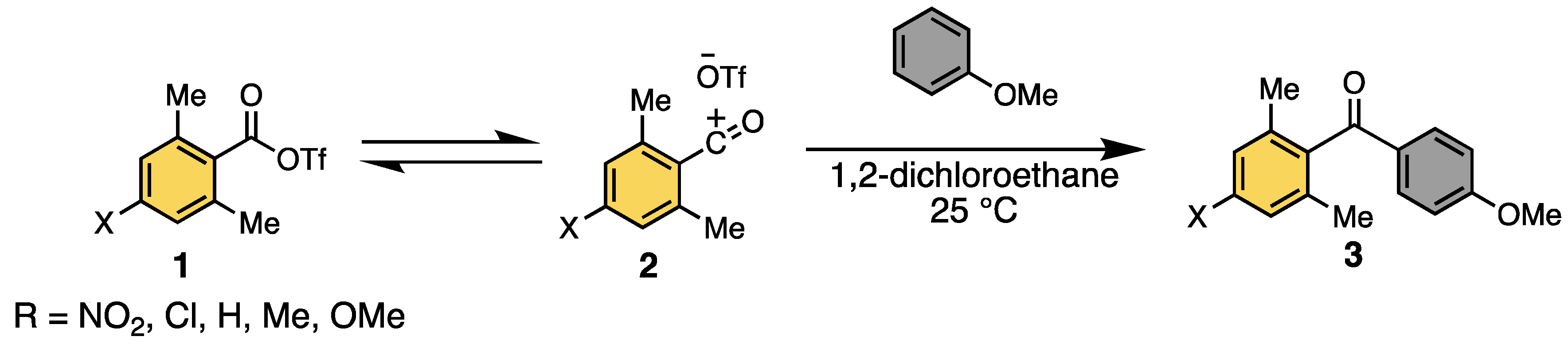
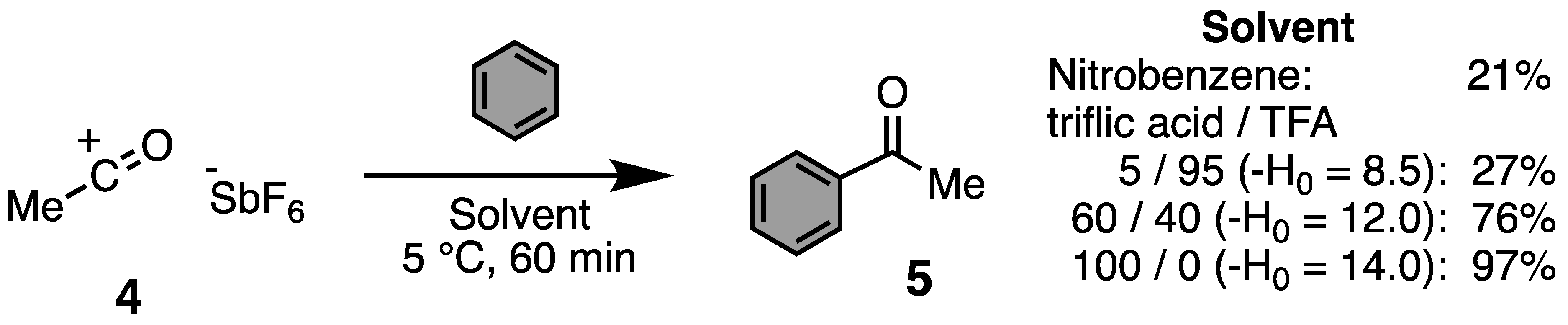
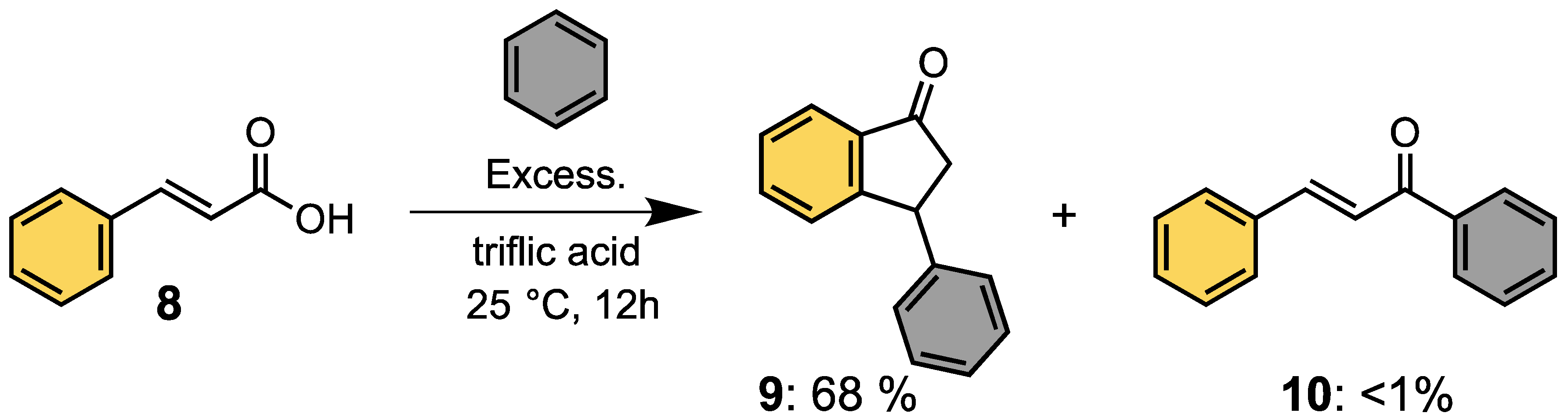
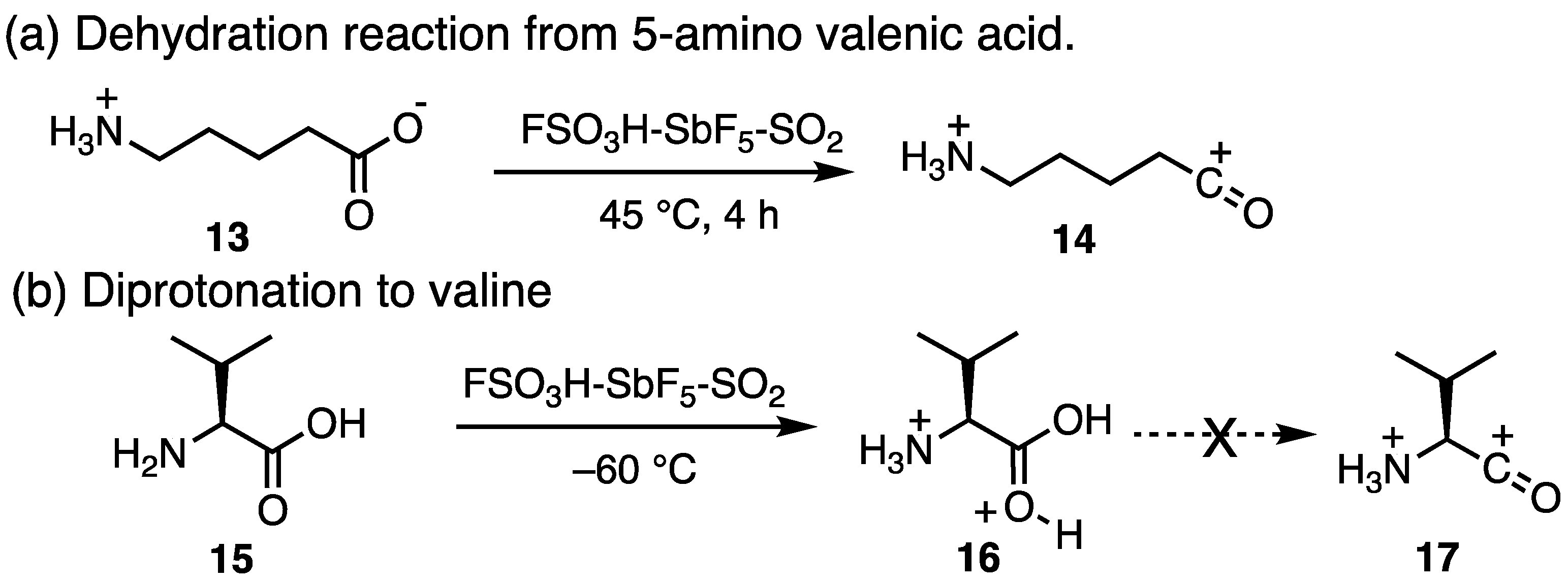
1. Introduction: Acylium Ions
2. Aromatic Amidation
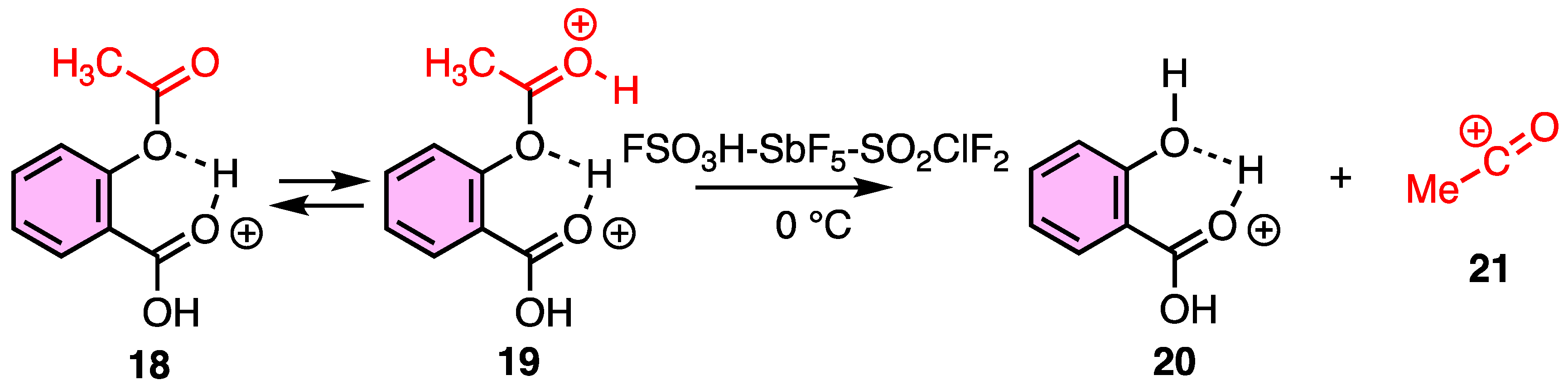
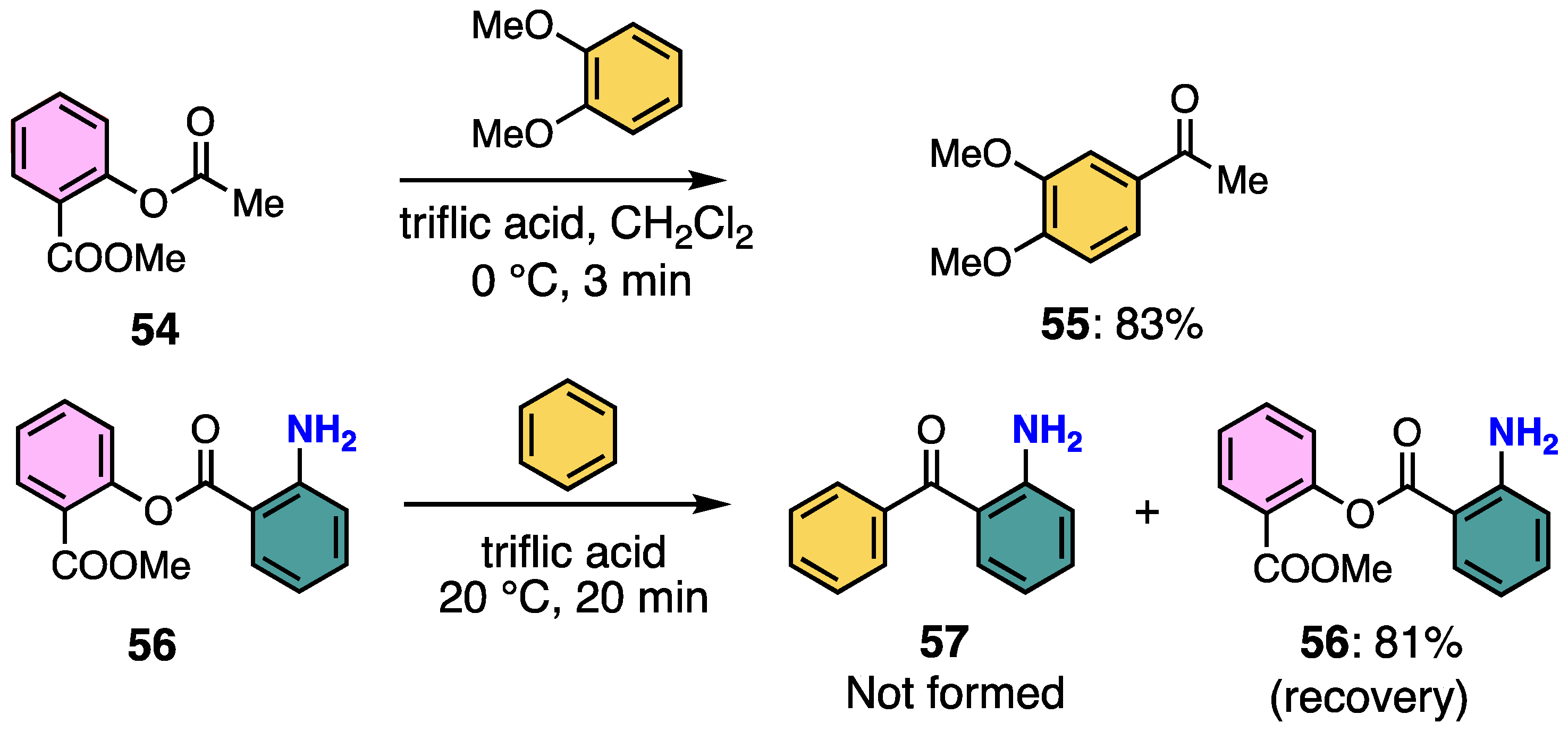
2.1. Utility of Methyl Salicylate as a Leaving Group in Generation of Electrophiles
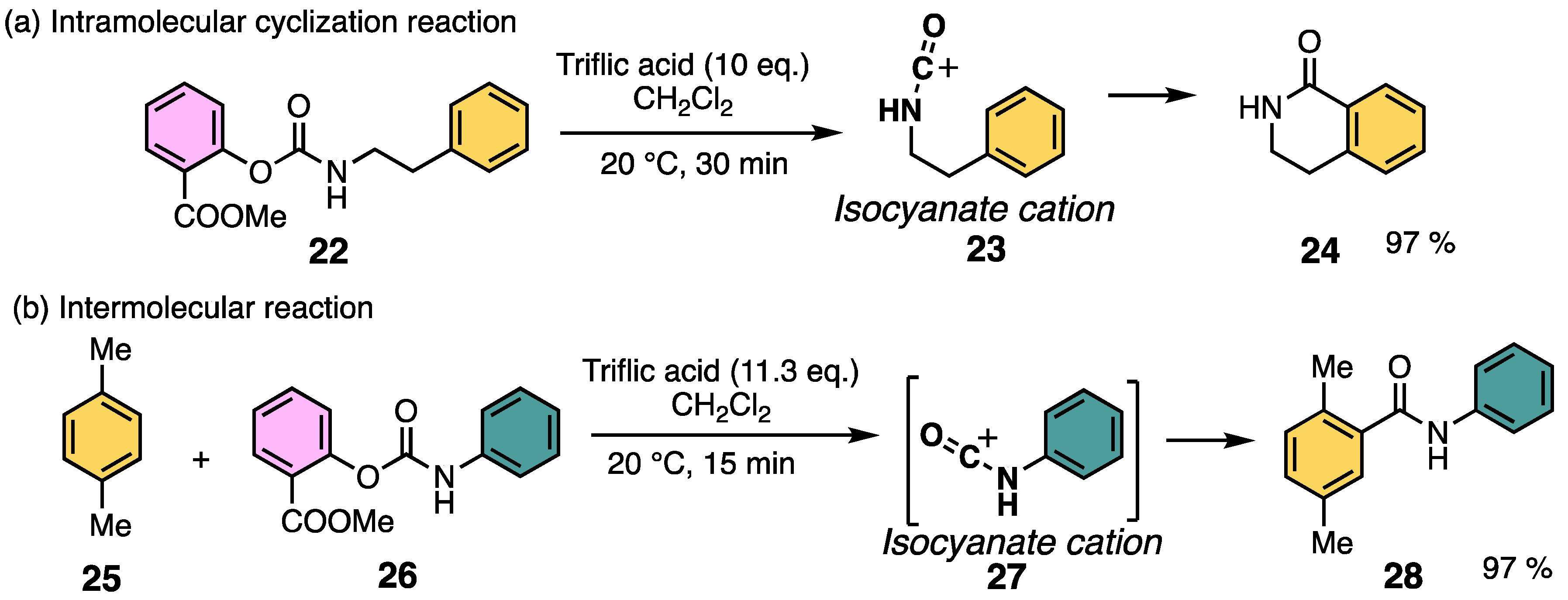
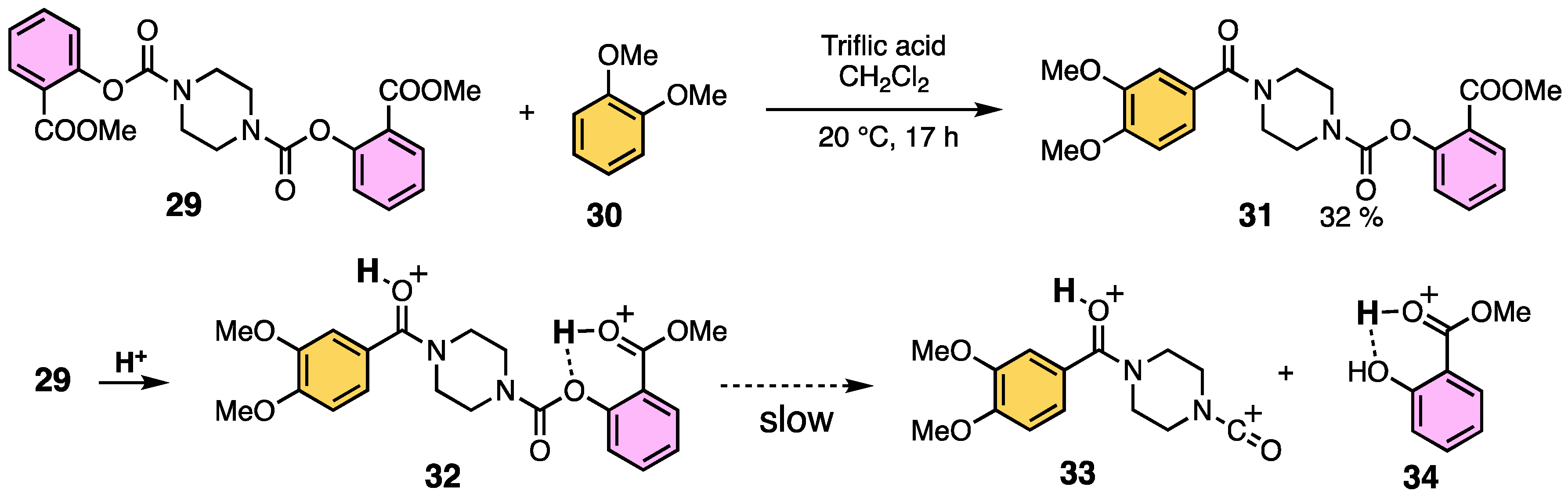
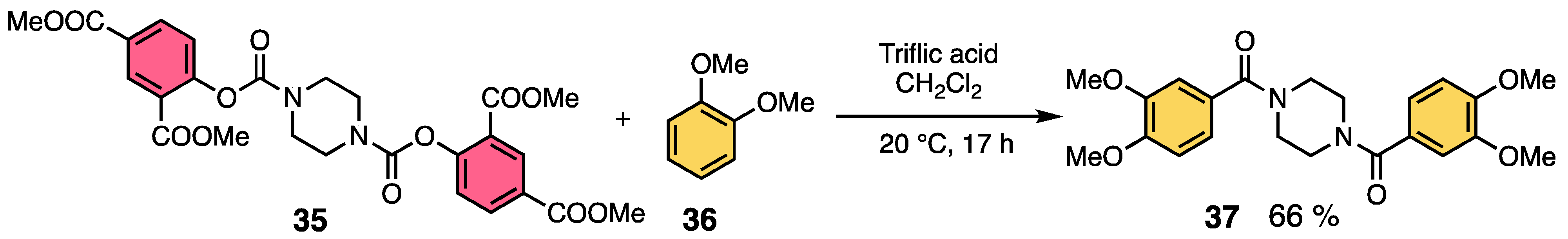
2.2. Aromatic Amidation Reaction Using Methyl Salicylate as a Leaving Group
2.3. Aromatic Acylation Reaction Using Methyl Salicylate as a Leaving Group
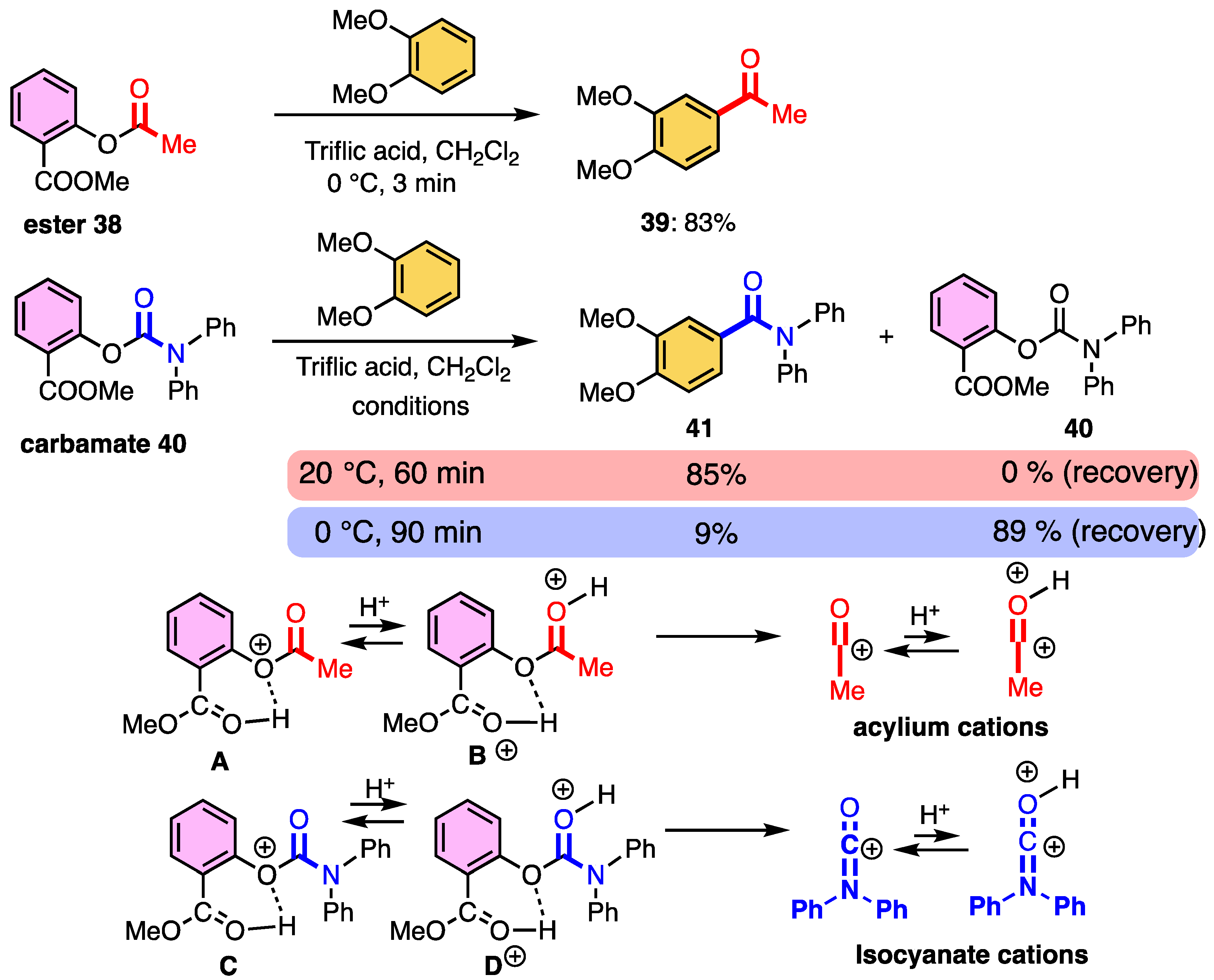
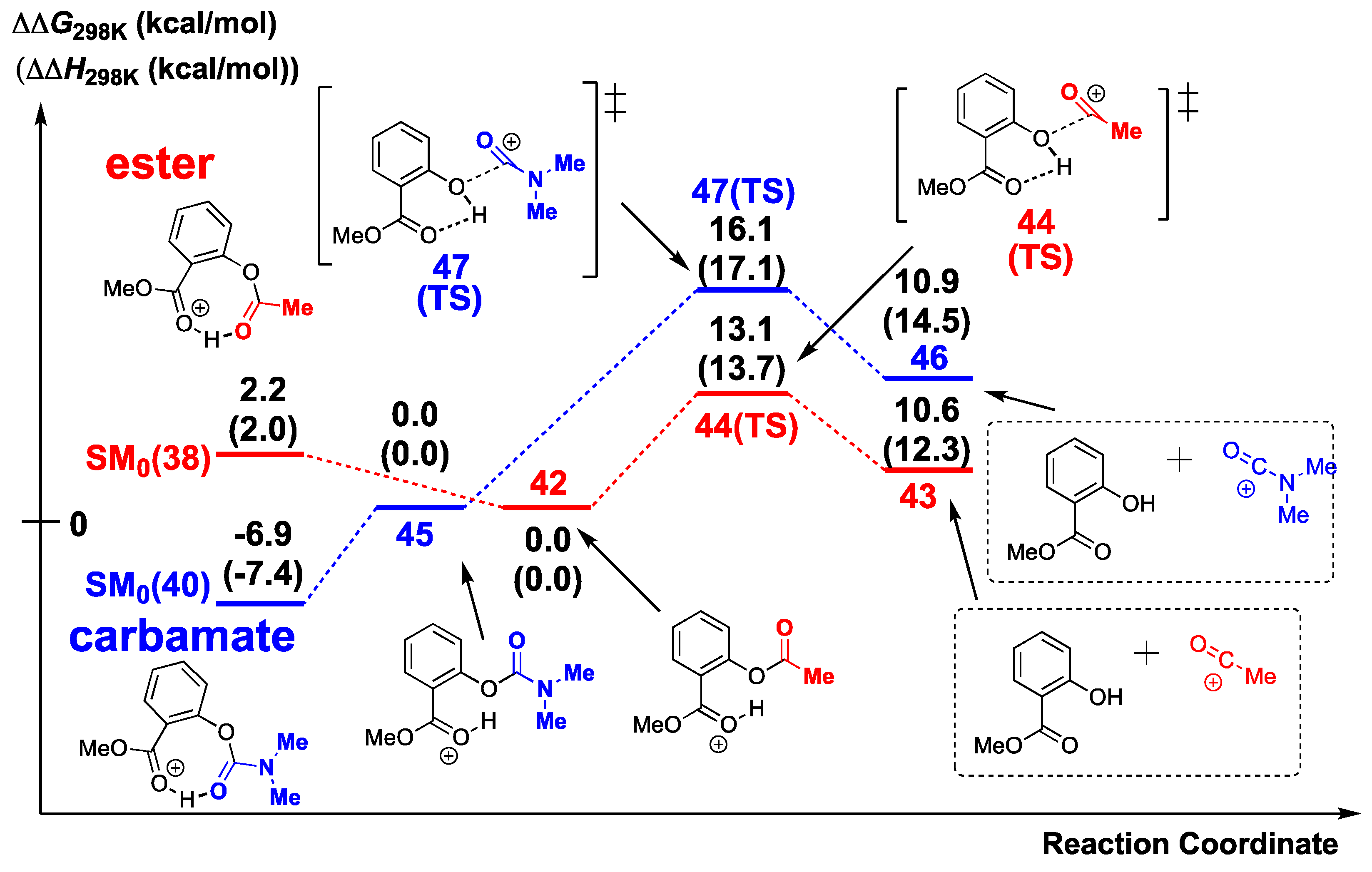
2.4. Difference in Cleavage Ability
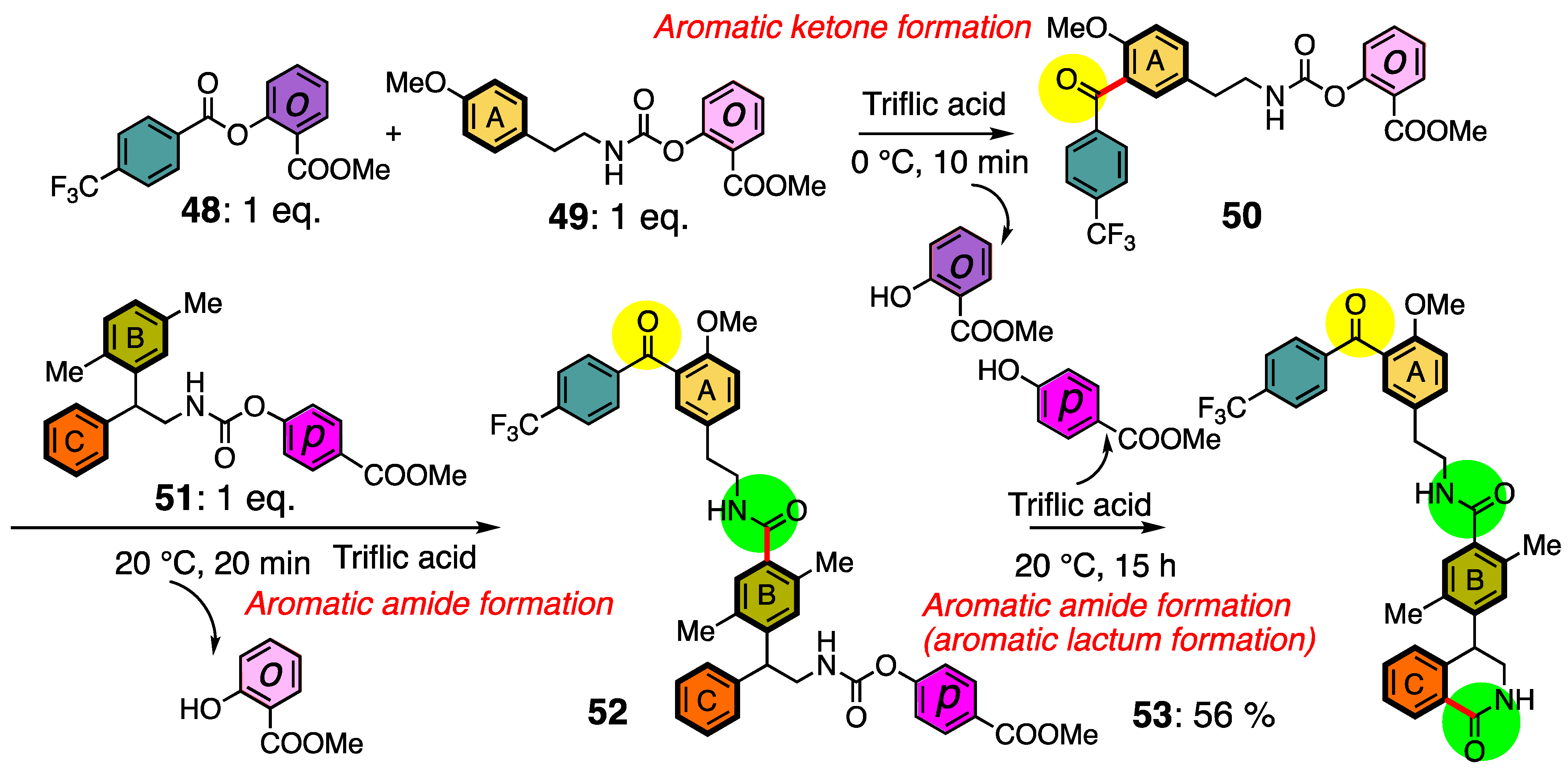
2.5. Tandem Reactions
2.6. Limitations of the Present Reaction System
3. Aromatic Acylation Reaction with Phosphoric Acid Esters and Strong Bronsted Acid
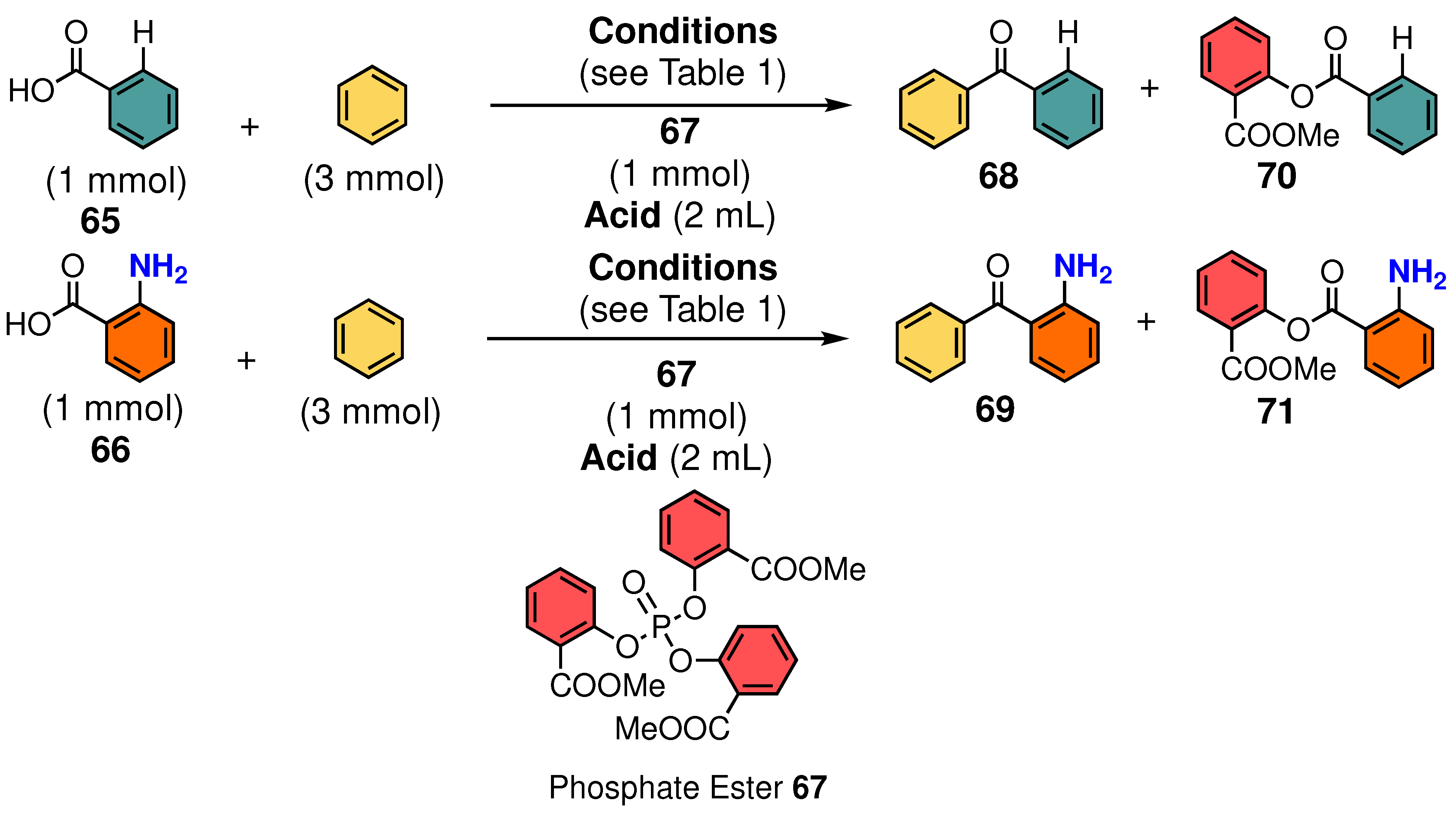
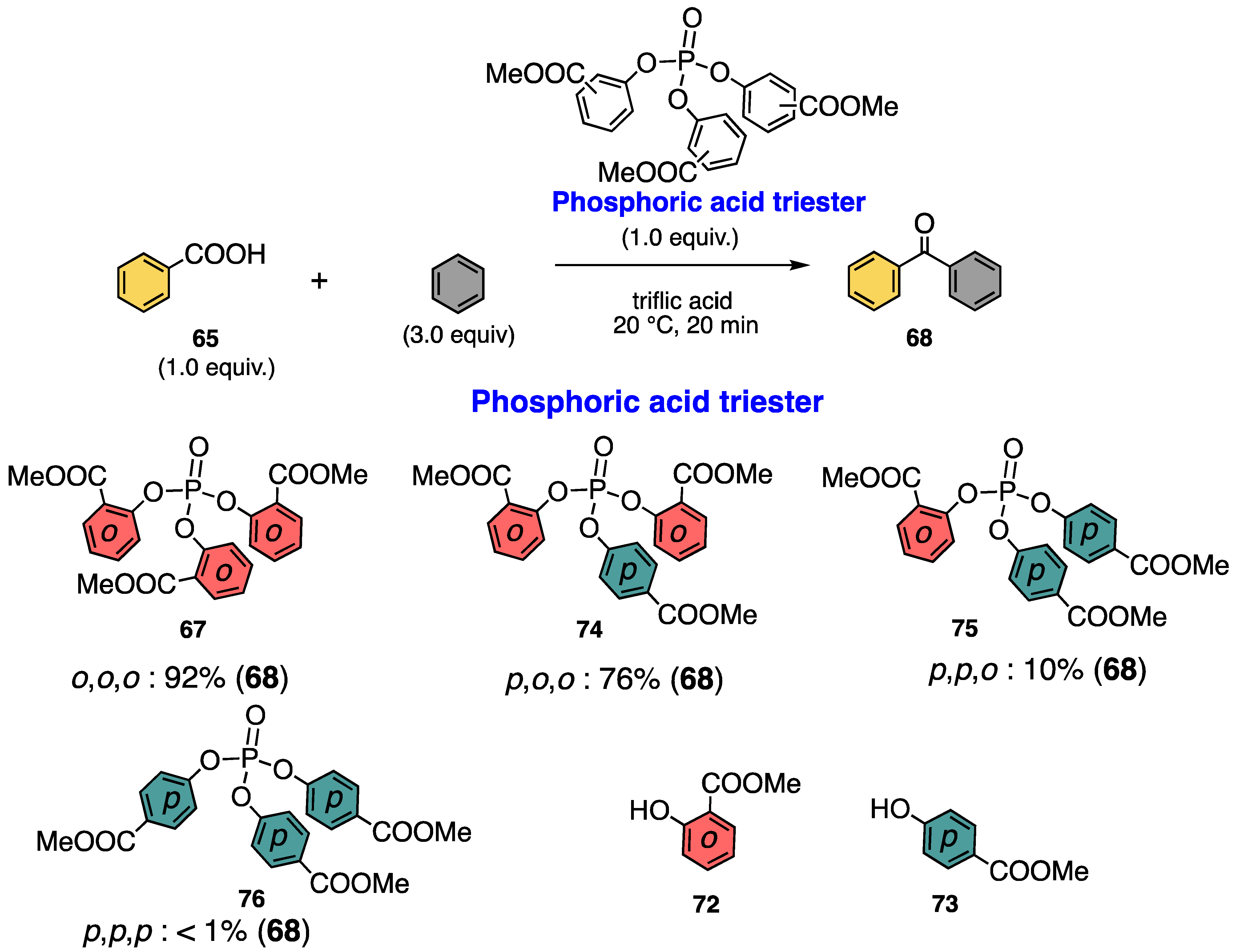
3.1. Phosphoric Acid Esters
3.2. Aromatic Acylation of Aminocarboxylic Acids
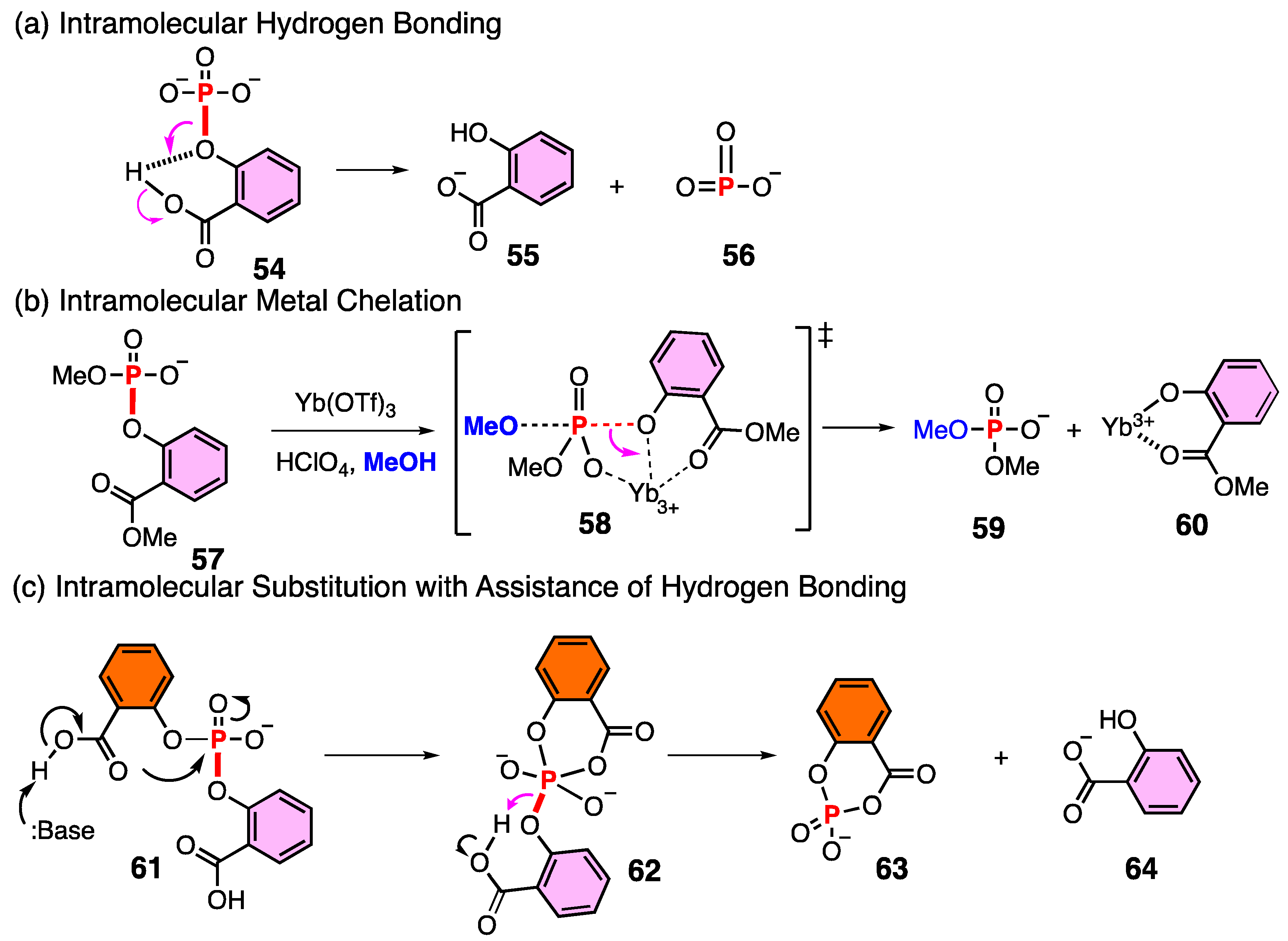
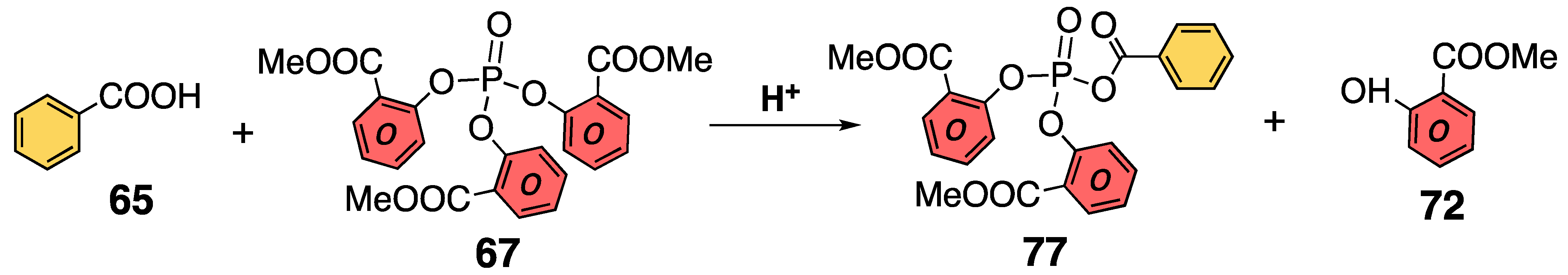
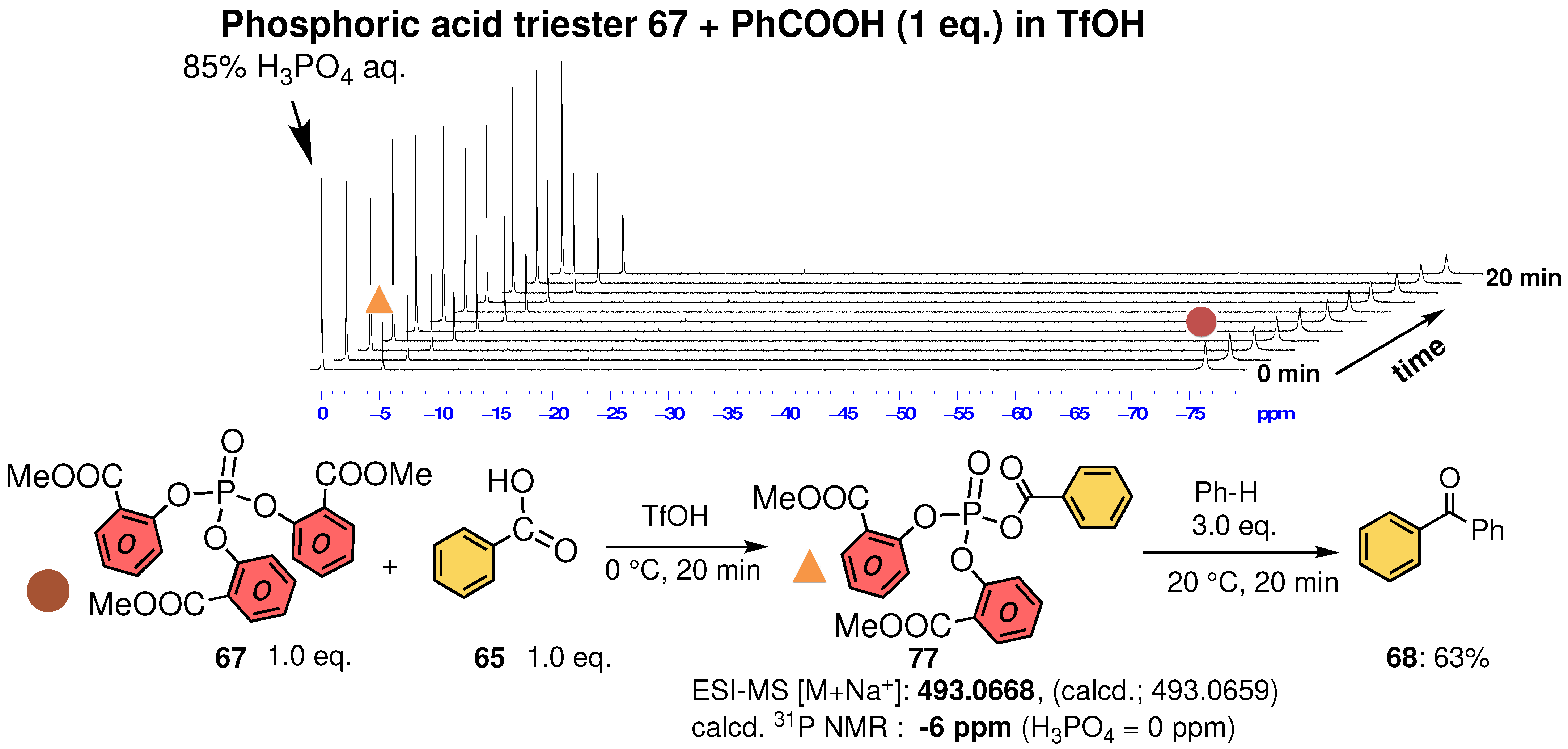
3.3. Characterization of Phosphoric Acid Esters of Methyl Salicylate
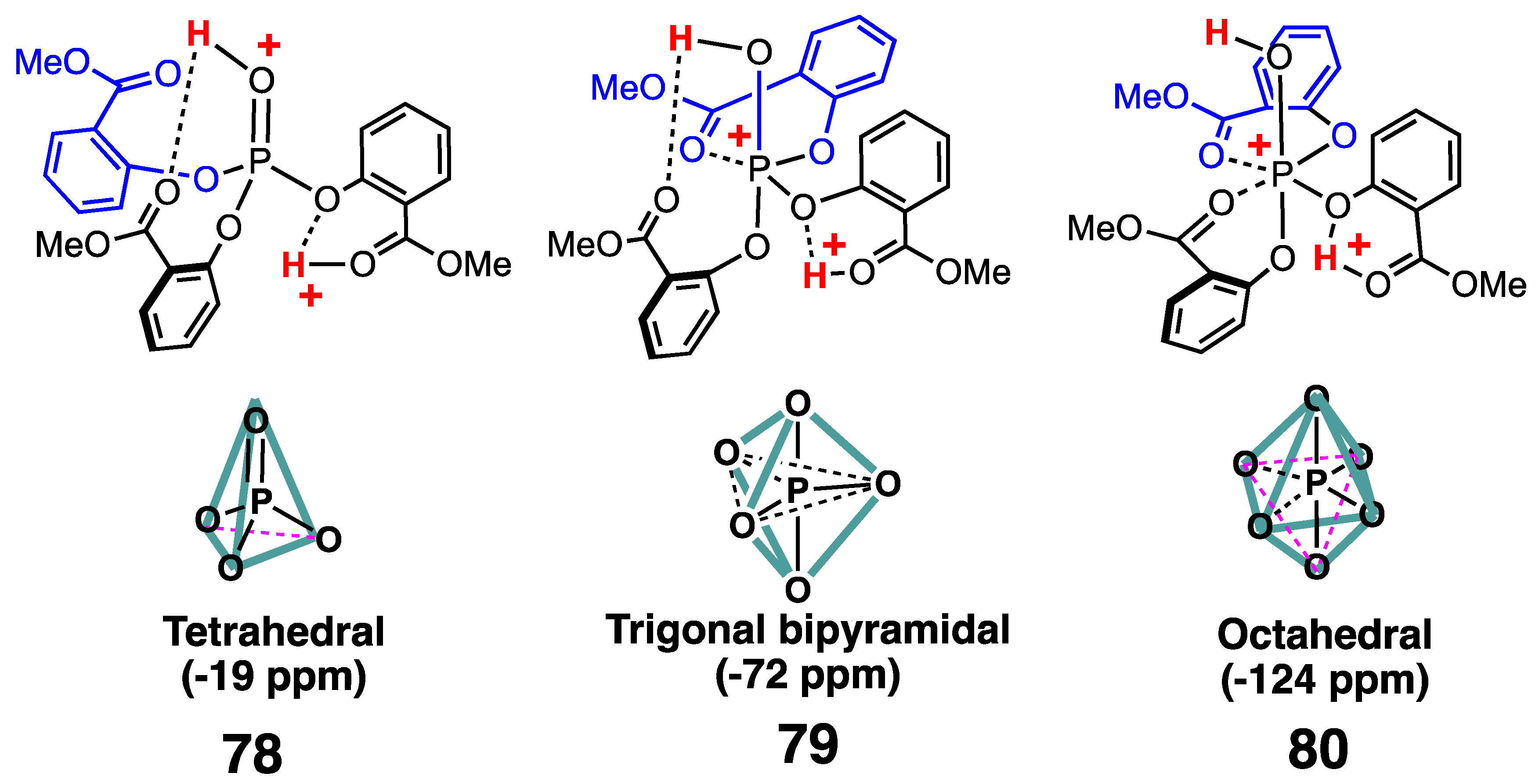
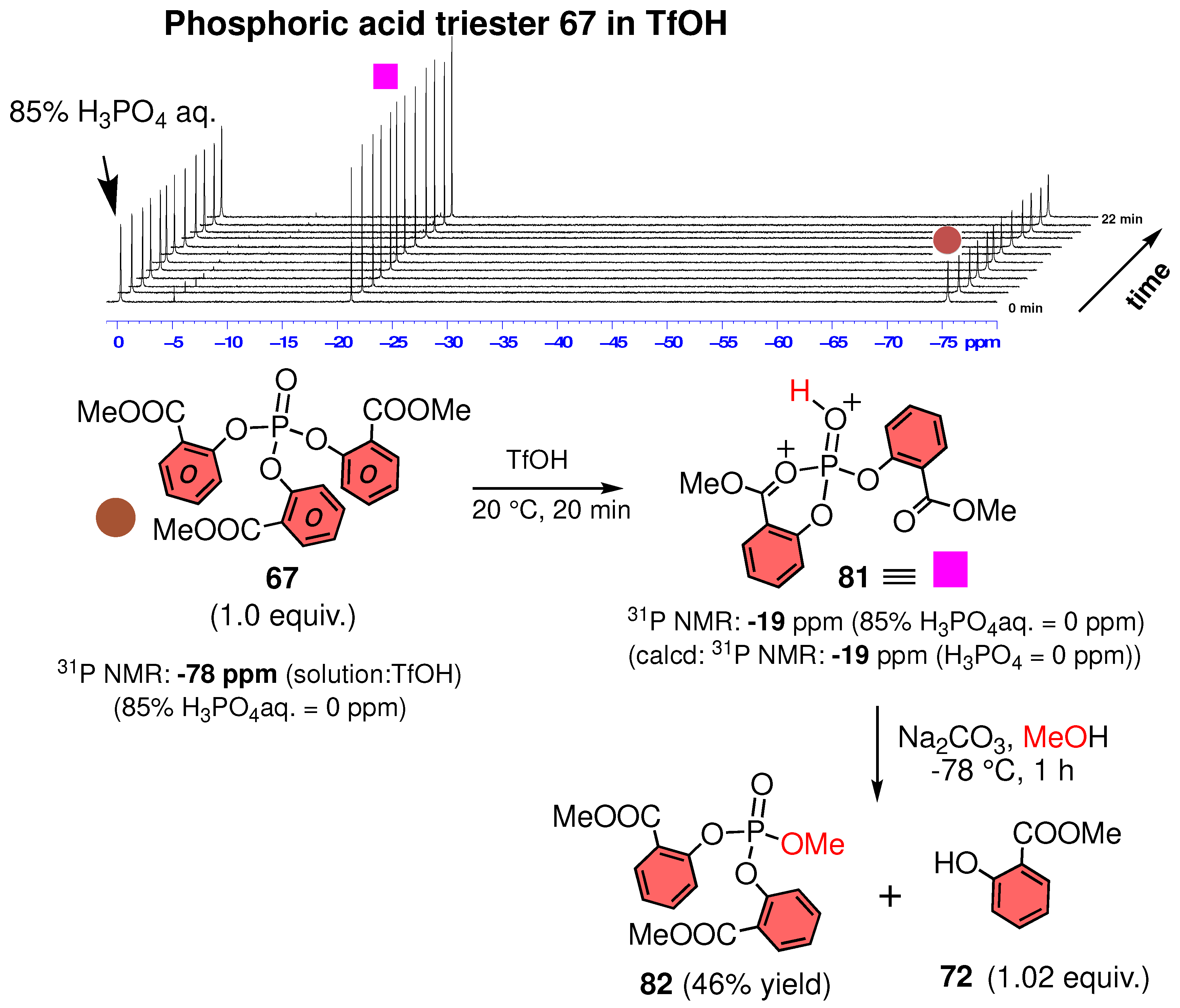
3.4. Structure of Phosphoric Acid Ester in Strong Acid
3.5. Experimental Evidence for the P–O Bond Formation of Carboxylic Acid
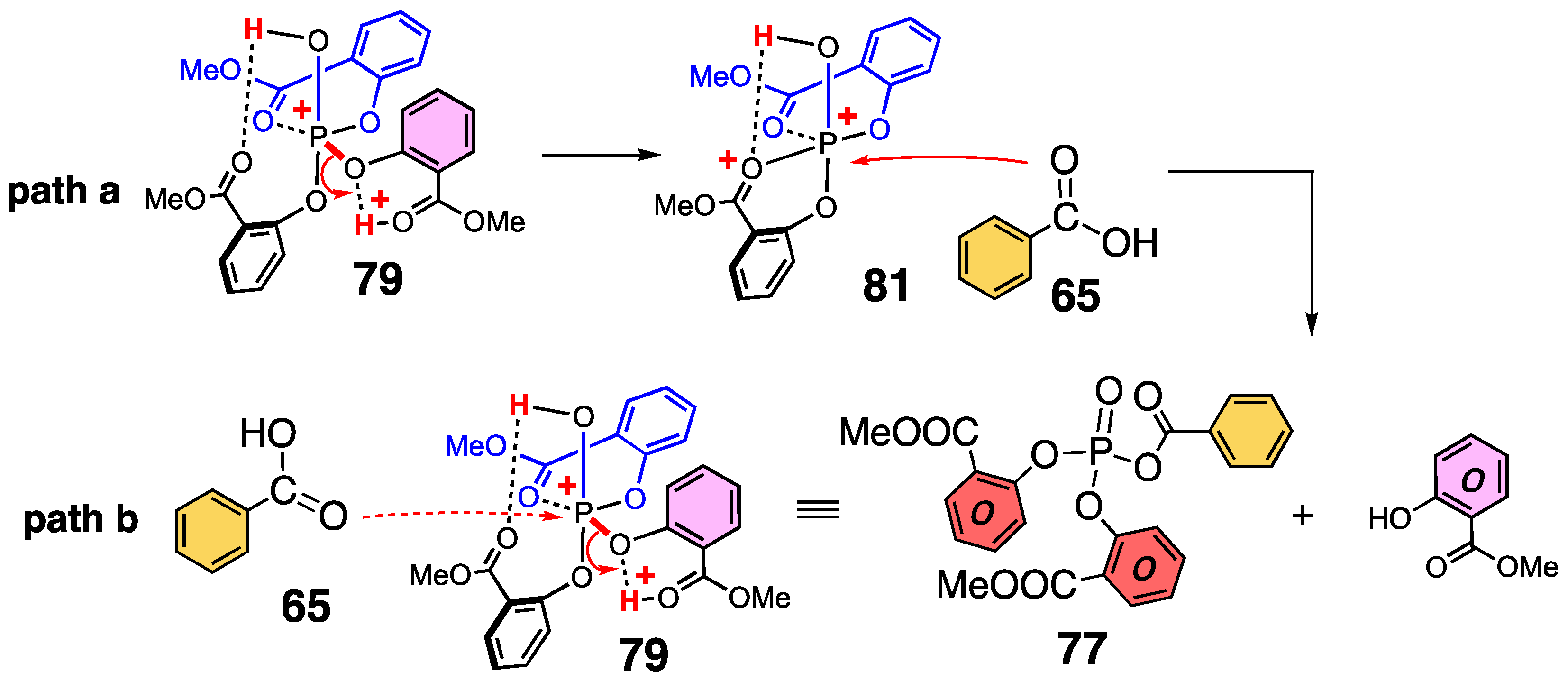
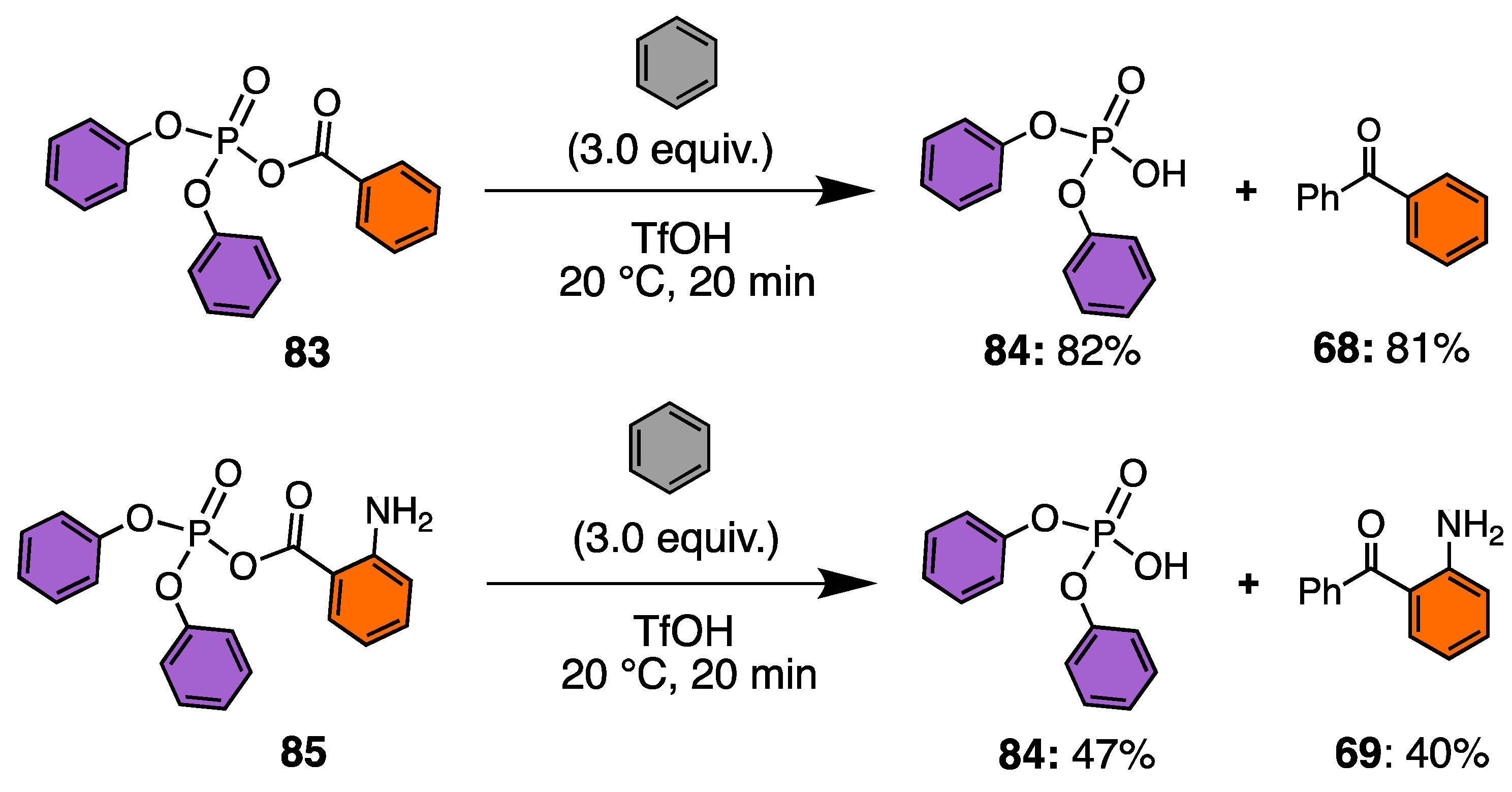
3.6. Reactivity of Acyl Phosphate
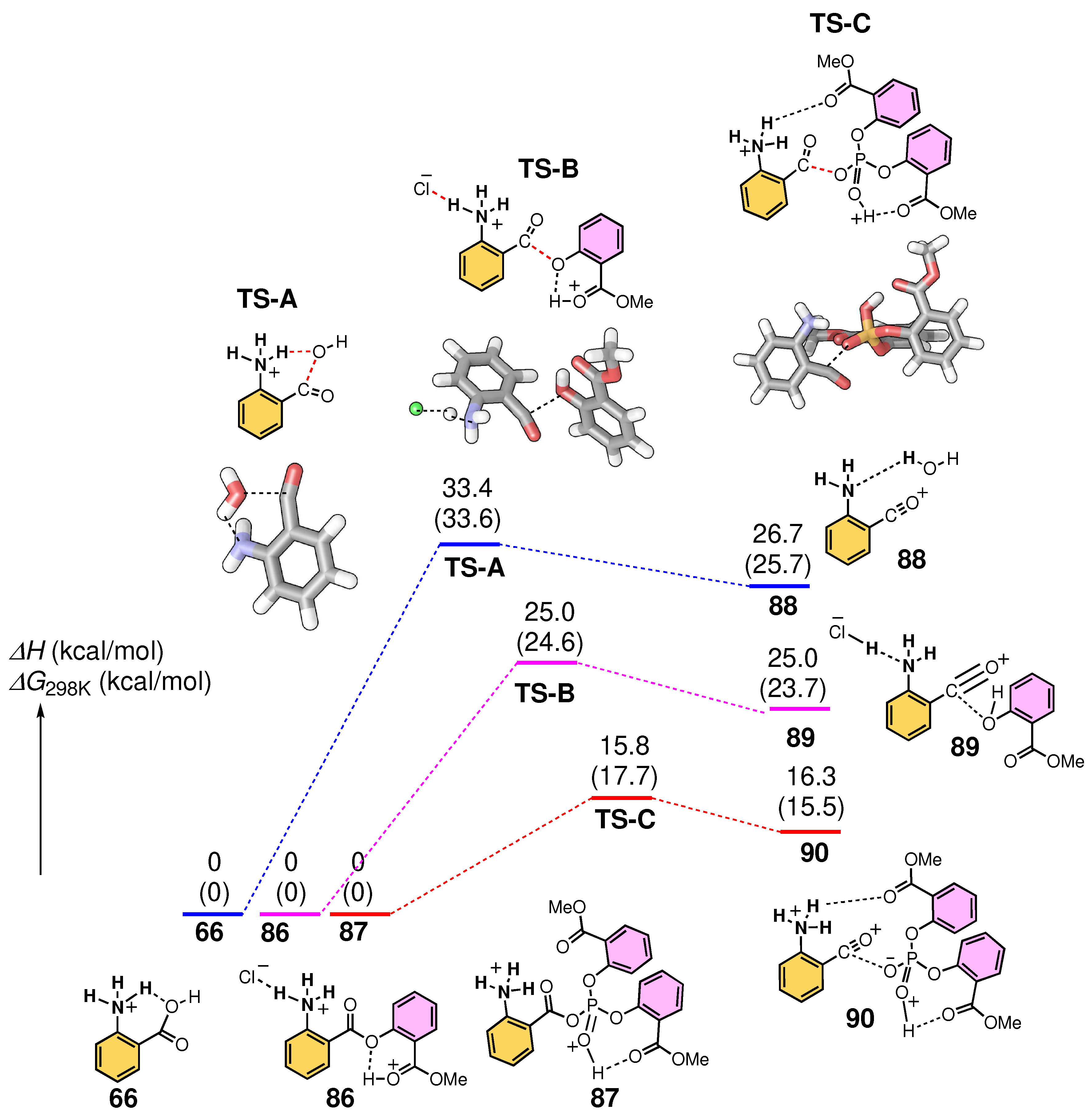
3.7. Computed Reaction Profile of Acyl Phosphate Containing an Anthranilic Acid Moiety
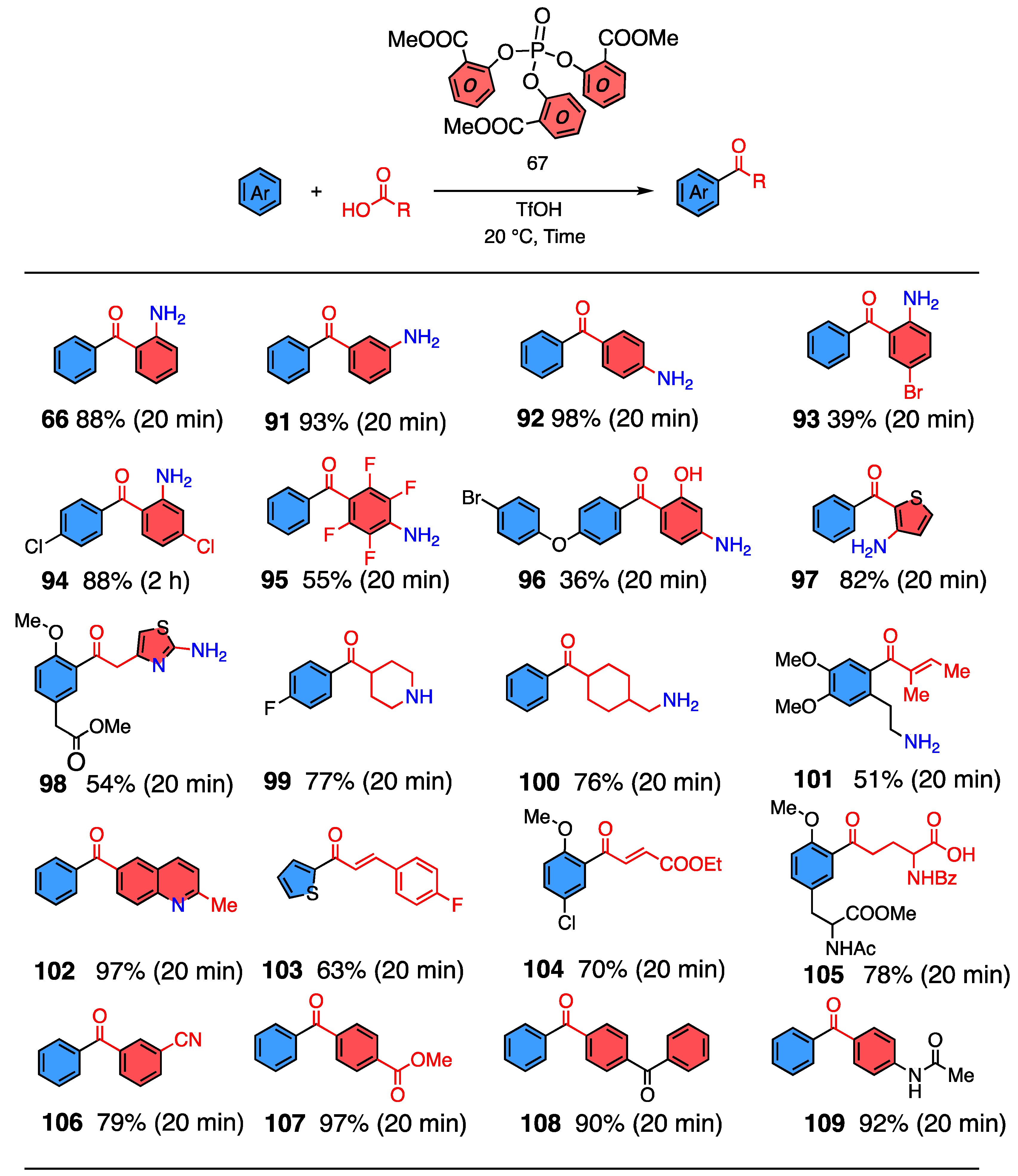
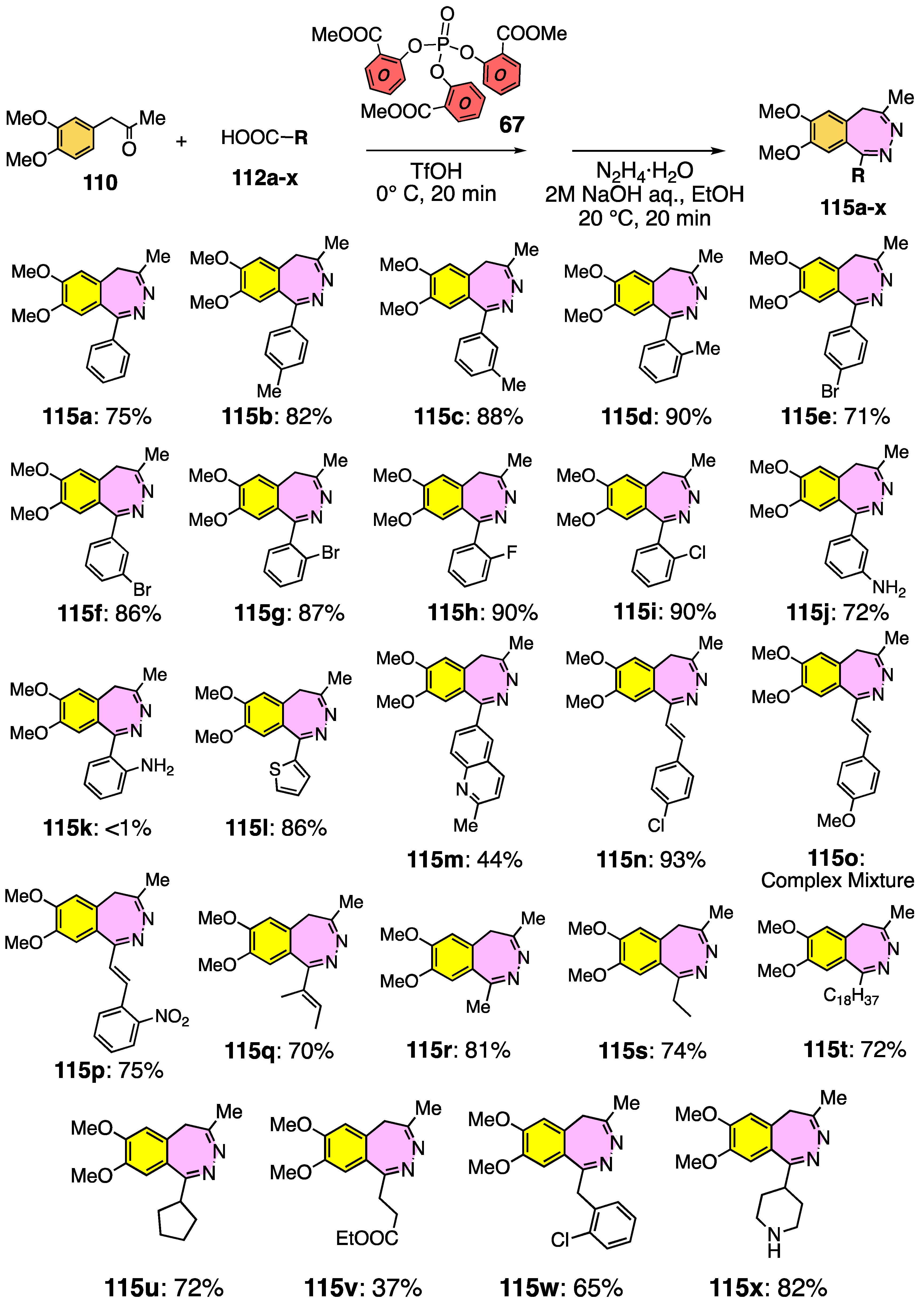
3.8. Substrate Generality
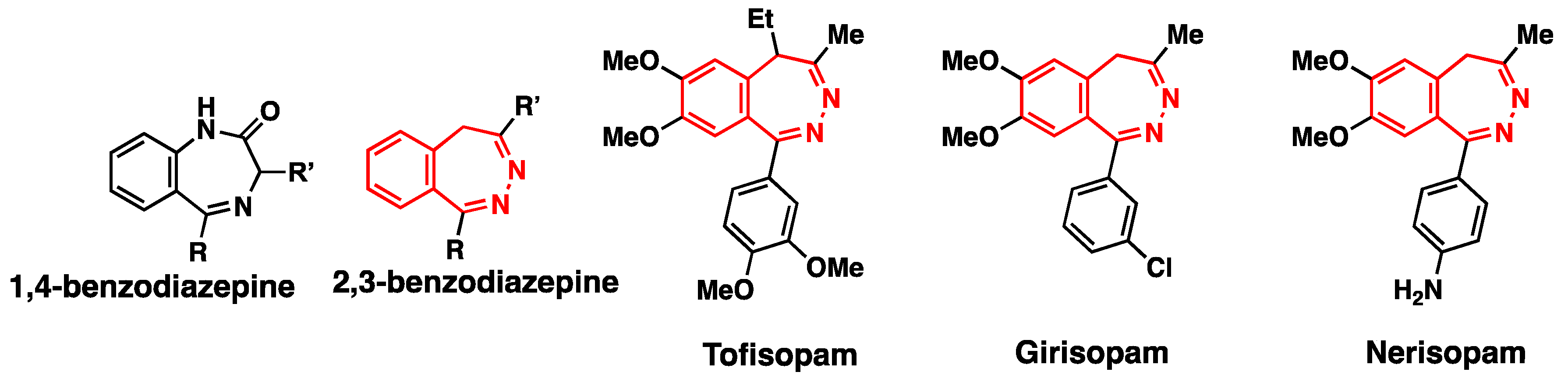
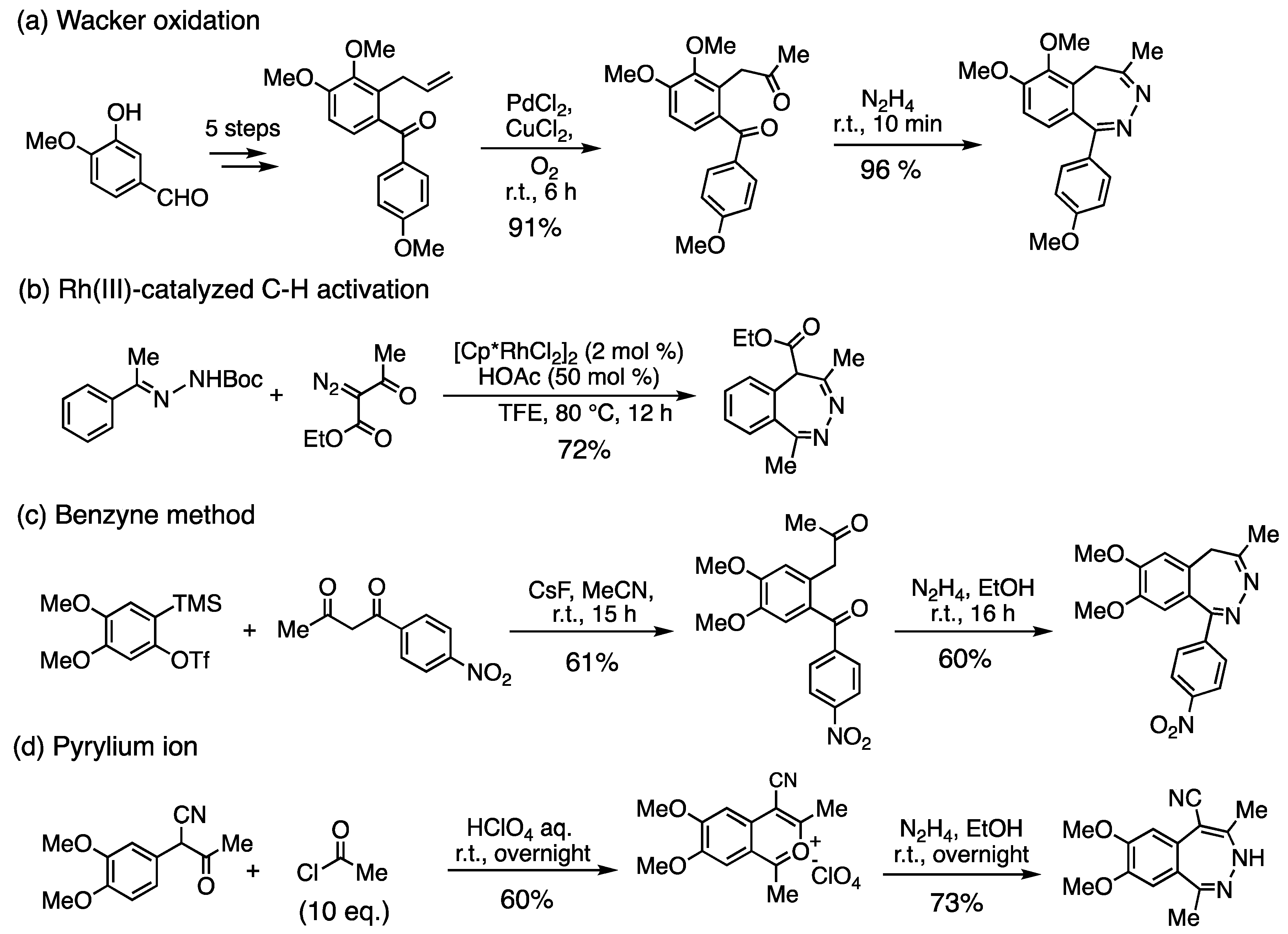
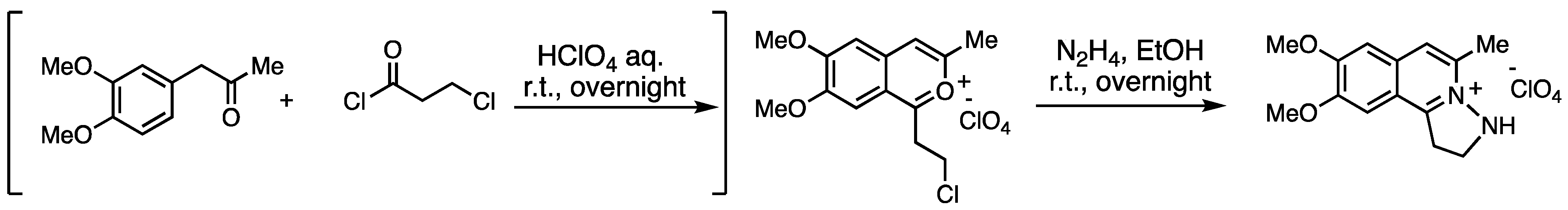
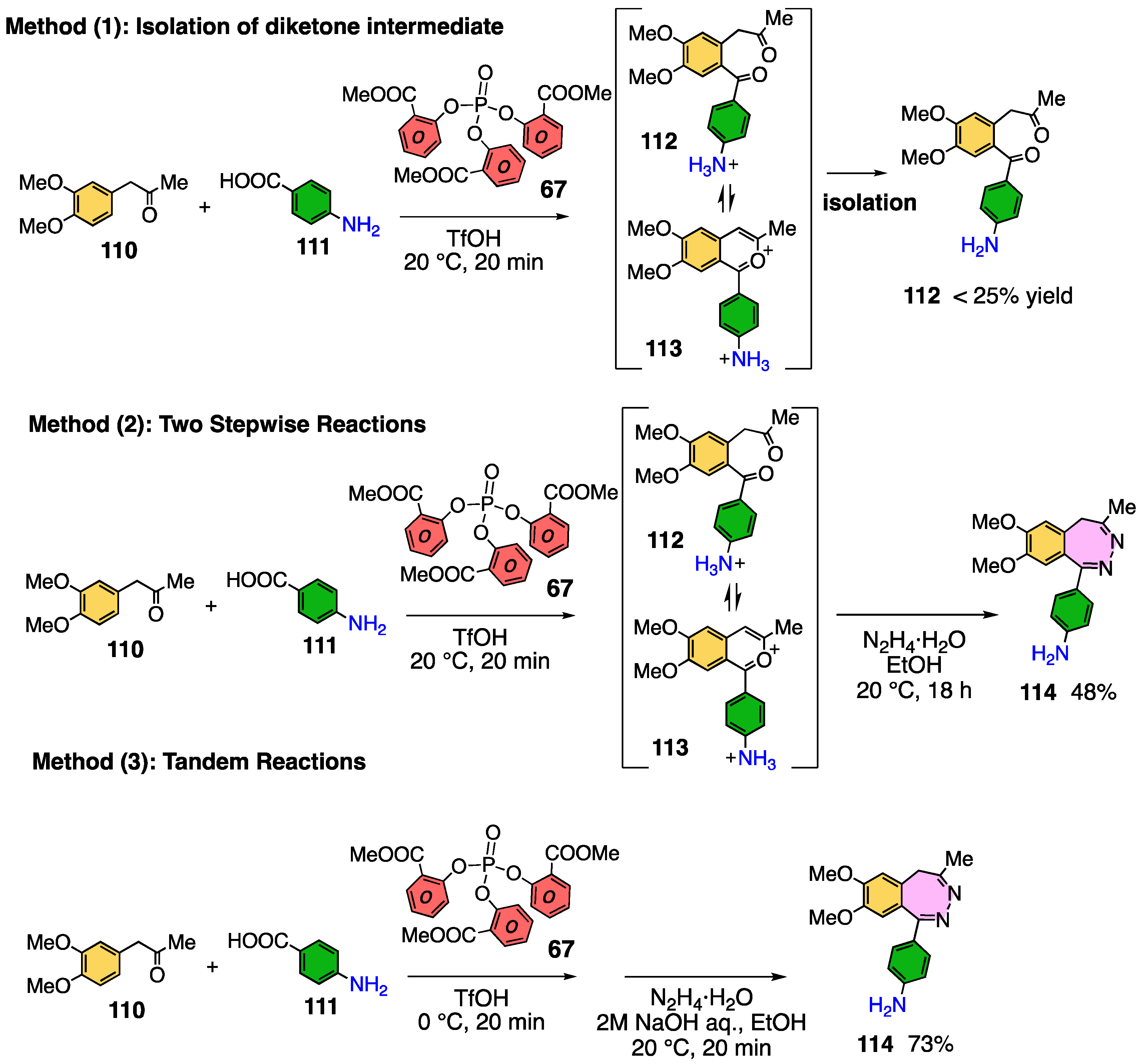
3.9. Application to 2,3-Benzodiazepine Skeleton Construction
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Heravi, M.M.; Zadsirjan, V.; Saedi, P.; Momeni, T. Applications of Friedel–Crafts reactions in total synthesis of natural products. RSC Adv. 2018, 8, 40061–40163. [Google Scholar] [CrossRef] [PubMed]
- Terrasson, V.; de Figueiredo, R.M.; Campagne, J.M. Organocatalyzed Asymmetric Friedel–Crafts Reactions. Eur. J. Org. Chem. 2010, 14, 2635–2655. [Google Scholar] [CrossRef]
- Olah, G.A.; Tolgyesi, W.S.; Kuhn, S.J.; Moffatt, M.E.; Bastien, I.J.; Baker, E.B. Stable Carbonium Ions. IV. Secondary and Tertiary Alkyl and Aralkyl Oxocarbonium Hexafluoroantimonates. Formation and Identification of the Trimethylcarbonium Ion by Decarbonylation of the tert-Butyl Oxocarbonium Ion. J. Am. Chem. Soc. 1963, 85, 1328–1334. [Google Scholar] [CrossRef]
- Olah, G.A.; Iyer, P.S.; Prakash, G.K.S.; Krishnamurthy, V.V. Oxygen-17 NMR spectroscopic study of substituted benzoyl cations. J. Org. Chem. 1984, 49, 4317–4319. [Google Scholar] [CrossRef]
- Boer, F.P. The Crystal Structure of CH3CO+SbF6−. J. Am. Chem. Soc. 1966, 88, 1572–1574. [Google Scholar] [CrossRef]
- Loh, Y.K.; Fuentes, M.A.; Vasko, P.; Aldridge, S. Successive Protonation of an N-Heterocyclic Imine Derived Carbonyl: Superelectrophilic DicationVersus Masked Acylium Ion. Angew. Chem. Int. Ed. 2018, 57, 16559–16563. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, F.; Eberhard, J.K.; Maier, A.H. The First Unequivocal Evidence of the Reacting Electrophile in Aromatic Acylation Reactions. J. Am. Chem. Soc. 1996, 118, 12572–12579. [Google Scholar] [CrossRef]
- Huang, Z.; Jin, L.; Han, H.; Lei, A. The “kinetic capture” of an acylium ion from live aluminum chloride promoted Friedel–Crafts acylation reactions. Org. Biomol. Chem. 2013, 11, 1810–1814. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Sommer, J.; Molnar, A. Superacid Chemistry, 2nd ed.; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Sato, Y.; Yato, M.; Ohwada, T.; Saito, S.; Shudo, K. Involvement of Dicationic Species as the Reactive Intermediates in Gattermann, Houben-Hoesch, and Friedel-Crafts Reactions of Nonactivated Benzenes. J. Am. Chem. Soc. 1995, 117, 3037–3043. [Google Scholar] [CrossRef]
- Olah, G.A.; Klumpp, D.A. Superelectrophiles and Their Chemistry; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Klumpp, D.A.; Anokhin, M.V. Superelectrophiles: Recent Advances. Molecules 2020, 25, 3281. [Google Scholar] [CrossRef]
- Rendy, R.; Zhang, Y.; McElrea, A.; Gomez, A.; Klumpp, D.A. Superacid-Catalyzed Reactions of Cinnamic Acids and the Role of Superelectrophiles. J. Org. Chem. 2004, 69, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Denis, J.-M.; Westerman, P.W. Stable carbocations. CLXV. Carbon-13 NMR spectroscopic study of alkenoyl cations. Importance of delocalized ketone-like carbenium ion resonance forms. J. Org. Chem. 1974, 39, 1206–1210. [Google Scholar] [CrossRef]
- Olah, G.A.; Brydon, D.L.; Porter, R.D. Stable carbonium ions. LXXXIII. Protonation of amino acids, simple peptides, and insulin in superacid solutions. J. Org. Chem. 1970, 35, 317–328. [Google Scholar] [CrossRef]
- Klumpp, D.A. Superelectrophiles: Charge-Charge Repulsive Effects. Chem. Eur. J. 2008, 14, 2004–2015. [Google Scholar] [CrossRef]
- Sumita, A.; Gasonoo, M.; Boblak, H.; Ohwada, T.; Klumpp, D.A. Use of Charge-Charge Repulsion to Enhance π-Electron Delocalization into Anti-Aromatic and Aromatic Systems. Chem. Eur. J. 2017, 23, 2566–2570. [Google Scholar] [CrossRef]
- Cantin, T.; Morgenstern, Y.; Mingot, A.; Kornath, A.; Thibaudeau, S. Evidence and Exploitation of Dicationic Ammonium–Nitrilium Superelectrophiles: Direct Synthesis of Unsaturated Piperidinones. Chem. Commun. 2020, 56, 11110–11113. [Google Scholar] [CrossRef]
- Bonazaba Milandou, L.J.C.; Carreyre, H.; Alazet, S.; Greco, G.; Martin-Mingot, A.; Loumpangou, C.N.; Ouamba, J.-M.; Bouazza, F.; Billard, T.; Thibaudeau, S. Superacid-Catalyzed Trifluoromethylthiolation of Aromatic Amines. Angew. Chem. Int. Ed. 2017, 56, 169–172. [Google Scholar] [CrossRef]
- Ohwada, T.; Shudo, K. Reaction of Diphenylmethyl Cations in a Strong Acid. Participation of Carbodications with Positive Charge Substantially Delocalized over the Aromatic Rings. J. Am. Chem. Soc. 1988, 110, 1862–1870. [Google Scholar] [CrossRef]
- Surana, K.; Chaudhary, B.; Diwaker, M.; Sharma, S. Benzophenone: A ubiquitous scaffold in medicinal chemistry. Med. Chem. Commun. 2018, 9, 1803–1817. [Google Scholar] [CrossRef]
- Hwang, J.P.; Prakash, G.K.S.; Olah, G.A. Trifluoromethanesulfonic Acid Catalyzed Novel Friedel–Crafts Acylation of Aromatics with Methyl Benzoate. Tetrahedron 2000, 56, 7199–7203. [Google Scholar] [CrossRef]
- Craze, G.-A.; Kirby, A.J. The role of the carboxy-group in intramolecular catalysis of acetal hydrolysis. The hydrolysis of substituted 2-methoxymethoxybenzoic acids. J. Chem. Soc. Perkin Trans. 1974, 2, 61–66. [Google Scholar] [CrossRef]
- Olah, G.A.; Westerman, P.W. Stable Carbocations. CLXVII.1 Protonation and Cleavage of Acetylsalicylic Acid and Isomeric Hydroxybenzoic Acids in FSO3H-SbF5 (Magic Acid) Solution. J. Org. Chem. 1974, 39, 1307–1308. [Google Scholar] [CrossRef]
- Kurouchi, H.; Sumita, A.; Otani, Y.; Ohwada, T. Protonation Switching to the Least-Basic Heteroatom of Carbamate through Cationic Hydrogen Bonding Promotes the Formation of Isocyanate Cations. Chem. Eur. J. 2014, 20, 8682–8690. [Google Scholar] [CrossRef] [PubMed]
- Sumita, A.; Kurouchi, H.; Otani, Y.; Ohwada, T. Acid-Promoted Chemoselective Introduction of Amide Functionality onto Aromatic Compounds Mediated by an Isocyanate Cation Generated from Carbamate. Chem. Asian J. 2014, 9, 2995–3004. [Google Scholar] [CrossRef]
- Gund, P. Guanidine, trimethylenemethane, and “Y-delocalization”. Can acyclic compounds have “aromatic” stability? J. Chem. Educ. 1972, 49, 100–103. [Google Scholar] [CrossRef]
- Delebecq, E.; Pascault, J.; Boutevin, B.; Ganachaud, F. On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-isocyanate Polyurethane. Chem. Rev. 2013, 113, 80–118. [Google Scholar] [CrossRef]
- Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.G.; Gagné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered Ureas as Masked Isocyanates: Facile Carbamoylation of Nucleophiles under Neutral Conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar] [CrossRef]
- Adachi, S.; Onozuka, M.; Yoshida, Y.; Ide, M.; Saikawa, Y.; Nakata, M. Smooth Isoindolinone Formation from Isopropyl Carbamates via Bischler–Napieralski-Type Cyclization. Org. Lett. 2014, 16, 358–361. [Google Scholar] [CrossRef]
- Raja, E.K.; Lill, S.O.N.; Klumpp, D.A. Friedel–Crafts-type reactions with ureas and thioureas. Chem. Commun. 2012, 48, 8141–8143. [Google Scholar] [CrossRef]
- Kurouchi, H.; Kawamoto, K.; Sugimoto, H.; Nakamura, S.; Otani, Y.; Ohwada, T. Activation of Electrophilicity of Stable Y-Delocalized Carbamate Cations in Intramolecular Aromatic Substitution Reaction: Evidence for Formation of Diprotonated Carbamates Leading to Generation of Isocyanates. J. Org. Chem. 2012, 77, 9313–9328. [Google Scholar] [CrossRef]
- Sumita, A.; Otani, Y.; Ohwada, T. Tandem buildup of complexity of aromatic molecules through multiple successive electrophile generation in one pot, controlled by varying the reaction temperature. Org. Biomol. Chem. 2016, 14, 1680–1693. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Yamada, T.; Tatekabe, S.; Nagasawa, K. A tandem Friedel–Crafts based method for the construction of a tricyclic pyrroloquinoline skeleton and its application in the synthesis of ammosamide B. Chem. Commun. 2013, 49, 6519–6521. [Google Scholar] [CrossRef] [PubMed]
- Kheira, H.; Li, P.; Xu, J. Synthesis of indenes via aluminum chloride-promoted tandem Friedel–Crafts alkylation of arenes and cinnamaldehydes. J. Mol. Catal. A Chem. 2014, 391, 168–174. [Google Scholar] [CrossRef]
- Zhang, X.; Teo, W.T.; Chan, P.W.H. Ytterbium (III) Triflate Catalyzed Tandem Friedel−Crafts Alkylation/Hydroarylation of Propargylic Alcohols with Phenols as an Expedient Route to Indenols. Org. Lett. 2009, 11, 4990–4993. [Google Scholar] [CrossRef]
- Iakovenko, R.O.; Kazakova, A.N.; Muzalevskiy, V.M.; Lvanov, A.Y.; Boyarskaya, I.A.; Chicca, A.; Petrucci, V.; Gertsch, J.; Krasavin, M.; Starova, G.L.; et al. Reactions of CF3-enones with arenes under superelectrophilic activation: A pathway totrans-1,3-diaryl-1-CF3-indanes, new cannabinoid receptor ligands. Org. Biomol.Chem. 2015, 13, 8827–8842. [Google Scholar] [CrossRef]
- Ramulu, B.V.; Satyanarayana, G. Superacid mediated intramolecular condensation: Facile synthesis of indenones and indanones. RSC Adv. 2015, 5, 70972–70976. [Google Scholar] [CrossRef]
- Sun, F.; Zeng, M.; Gu, Q.; You, S. Enantioselective Synthesis of Fluorene Derivatives by Chiral Phosphoric Acid Catalyzed Tandem Double Friedel–Crafts Reaction. Chem. Eur. J. 2009, 15, 8709–8712. [Google Scholar] [CrossRef]
- Li, Q.; Xu, W.; Hu, J.; Chen, X.; Zhang, F.; Zheng, H. TfOH catalyzed synthesis of 9-arylfluorenes via tandem reaction under warm and efficient conditions. RSC Adv. 2014, 4, 27722–27725. [Google Scholar] [CrossRef]
- Wang, S.-G.; Han, L.; Zeng, M.; Sun, F.-L.; Zhang, W.; You, S.-L. Enantioselective synthesis of fluorene derivatives by chiralN-triflyl phosphoramide catalyzed double Friedel–Crafts alkylation reaction. Org. Biomol. Chem. 2012, 10, 3202–3209. [Google Scholar] [CrossRef]
- Sureshbabu, R.; Saravanan, V.; Dhayalan, V.; Mohanakrishnan, A.K. Lewis Acid Mediated One-Pot Synthesis of Aryl/Heteroaryl-Fused Carbazoles Involving a Cascade Friedel–Crafts Alkylation/Electrocyclization/Aromatization Reaction Sequence. Eur. J. Org. Chem. 2011, 5, 922–935. [Google Scholar] [CrossRef]
- Bianchi, L.; Maccagno, M.; Pani, M.; Petrillo, G.; Scapolla, C.; Tavani, C. A straight access to functionalized carbazoles by tandem reaction between indole and nitrobutadienes. Tetrahedron 2015, 71, 7421–7435. [Google Scholar] [CrossRef]
- Kulkarni, A.; Quang, P.; Török, B. Microwave-Assisted Solid-Acid-Catalyzed Friedel-Crafts Alkylation and Electrophilic Annulation of Indoles Using Alcohols as Alkylating Agents. Synthesis 2009, 23, 4010–4014. [Google Scholar] [CrossRef]
- Huo, C.; Sun, C.; Wang, C.; Jia, X.; Chang, W. Triphenylphosphine-m-sulfonate/Carbon Tetrabromide as an Efficient and Easily Recoverable Catalyst System for Friedel–Crafts Alkylation of Indoles with Carbonyl Compounds or Acetals. J. Org. Chem. Sustain. Chem. Eng. 2013, 1, 549–553. [Google Scholar] [CrossRef]
- Liu, J.; He, T.; Wang, L. FeCl3 as Lewis acid catalyzed one-pot three-component aza-Friedel–Crafts reactions of indoles, aldehydes, and tertiary aromatic amines. Tetrahedron 2011, 67, 3420–3426. [Google Scholar] [CrossRef]
- Ishida, H.; Nukaya, H.; Tsuji, K.; Zenda, H.; Kosuge, T. Studies on Active Principles of Tars. X. The Structures and Some Reactions of Antifungal Constituents in Pix Pini. Chem. Pharm. Bull. 1992, 40, 308–313. [Google Scholar] [CrossRef][Green Version]
- Gangadhararao, G.; Uruvakilli, A.; Swamy, K.-C. Brønsted Acid Mediated Alkenylation and Copper-Catalyzed Aerobic Oxidative Ring Expansion/Intramolecular Electrophilic Substitution of Indoles with Propargyl Alcohols: A Novel One-Pot Approach to Cyclopenta [c]quinolines. Org. Lett. 2014, 16, 6060–6063. [Google Scholar] [CrossRef]
- Xie, X.; Du, X.; Chen, Y.; Liu, Y. One-Pot Synthesis of Indole-Fused Scaffolds via Gold-Catalyzed Tandem Annulation Reactions of 1,2-Bis (alkynyl)-2-en-1-ones with Indoles. J. Org. Chem. 2011, 76, 9175–9181. [Google Scholar] [CrossRef]
- Silvanus, A.C.; Heffernan, S.J.; Liptrot, D.J.; Kociok-Köhn, G.; Andrews, B.I.; Carbery, D.R. Stereoselective Double Friedel−Crafts Alkylation of Indoles with Divinyl Ketones. Org. Lett. 2009, 11, 1175–1178. [Google Scholar] [CrossRef]
- Li, H.; Guillot, R.; Gandon, V.; Jyono; Fujiwara, M.; Kudo, T.; Yokota, M.; Ichikawa, J. Domino Friedel–Crafts-Type Cyclizations of Difluoroalkenes Promoted by the α-Cation-Stabilizing Effect of Fluorine: An Efficient Method for Synthesizing Angular PAHs. Chem. Eur. J. 2011, 17, 12175–12185. [Google Scholar]
- Krajewski, W.W.; Jones, T.A.; Mowbray, S.L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. USA 2005, 102, 10499–10504. [Google Scholar] [CrossRef]
- Gill, H.S.; Pfluegl, G.M.U.; Eisenberg, D. Multicopy crystallographic refinement of a relaxed glutamine synthetase from Mycobacterium tuberculosis highlights flexible loops in the enzymatic mechanism and its regulation. Biochemistry 2002, 41, 9863–9872. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.; Ramos, M.J.; Fernandes, P.A. Reaction Mechanism of Mycobacterium tuberculosis Glutamine Synthetase Using Quantum Mechanics/Molecular Mechanics Calculations. Chem. Eur. J. 2016, 22, 9218–9225. [Google Scholar] [CrossRef] [PubMed]
- Liaw, S.-H.; Eisenberg, D. Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complex. Biochemistry 1994, 33, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Cathopoulis, T.; Chuawong, P.; Hendrickson, T.L. Novel tRNA aminoacylation mechanisms. Mol. BioSyst. 2007, 3, 408–418. [Google Scholar] [CrossRef]
- Ibba, M.; Söll, D. Aminoacyl-trans synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Westheimer, F.H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef]
- McMurry, J.; Begley, T. The Organic Chemistry of Biological Pathways; Roberts and Company Publishers: Englewood, CO, USA, 2005. [Google Scholar]
- Kluger, R. Acyl Phosphate Esters: Charge-Directed Acylation and Artificial Blood. Synlett 2000, 12, 1708–1720. [Google Scholar]
- Smyth, T.P.; Corby, B.W.; Corby, J.O.W. Toward a Clean Alternative to Friedel-Crafts Acylation: In Situ Formation, Observation, and Reaction of an Acyl Bis(trifluoroacetyl)phosphate and Related Structures. J. Org. Chem. 1998, 63, 8946–8951. [Google Scholar] [CrossRef]
- Tzvetkova, S.; Kluger, R. Biomimetic Aminoacylation of Ribonucleotides and RNA with Aminoacyl Phosphate Esters and Lanthanum Salts. J. Am. Chem. Soc. 2007, 129, 15848–15854. [Google Scholar] [CrossRef]
- Wodzinska, J.; Kluger, R. pKa-Dependent Formation of Amides in Water from an Acyl Phosphate Monoester and Amines. J. Org. Chem. 2008, 73, 4753–4754. [Google Scholar] [CrossRef]
- Dhiman, R.S.; Opinska, L.G.; Kluger, R. Biomimetic peptide bond formation in water with aminoacyl phosphate esters. Org. Biomol. Chem. 2011, 9, 5645–5647. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Bearne, S.L. Synthesis of coenzyme A thioesters using methyl acyl phosphates in an aqueous medium. Org. Biomol. Chem. 2014, 12, 9760–9763. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R.; Cameron, L.L. Activation of Acyl Phosphate Monoesters by Lanthanide Ions: Enhanced Reactivity of Benzoyl Methyl Phosphate. J. Am. Chem. Soc. 2002, 124, 3303–3308. [Google Scholar] [CrossRef] [PubMed]
- Popp, F.D.; McEwen, W.E. Polyphosphoric Acids As A Reagent In Organic Chemistry. Chem. Rev. 1958, 58, 321–401. [Google Scholar] [CrossRef]
- Eaton, P.E.; Carlson, G.R.; Lee, J.T. Phosphorus pentoxide-methanesulfonic acid. Convenient alternative to polyphosphoric acid. J. Org. Chem. 1973, 38, 4071–4073. [Google Scholar] [CrossRef]
- Zewge, D.; Chen, C.-Y.; Deer, C.; Domer, P.G.; Hughes, D.L. A Mild and Efficient Synthesis of 4-Quinolones and Quinolone Heterocycles. J. Org. Chem. 2007, 72, 4276–4279. [Google Scholar] [CrossRef]
- Shioiri, T.; Ninomiya, K.; Yamada, S. Diphenylphosphoryl azide. New convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 1972, 94, 6203–6205. [Google Scholar] [CrossRef]
- Castro, B.; Dormoy, J.R.; Evin, G.; Selve, C. Reactifs de couplage peptidique I (1)-l’hexafluorophosphate de benzotriazolyl N-oxytrisdimethylamino phosphonium (B.O.P.). Tetrahedron Lett. 1975, 16, 1219–1222. [Google Scholar] [CrossRef]
- Williams, N.H.; Takasaki, B.; Wall, M.; Chin, J. Structure and Nuclease Activity of Simple Dinuclear Metal Complexes: Quantitative Dissection of the Role of Metal Ions. Acc. Chem. Res. 1999, 32, 485–493. [Google Scholar] [CrossRef]
- Schroeder, G.K.; Lad, C.; Wyman, P.; Wolfenden, R. The time required for water attack at the phosphorus atom of simple phosphodiesters and of DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 4052–4055. [Google Scholar] [CrossRef]
- Johnson, D.W.; Hils, J.E. Phosphate Esters, Thiophosphate Esters and Metal Thiophosphates as Lubricant Additives. Lubricants 2013, 1, 132–148. [Google Scholar] [CrossRef]
- Bender, M.L.; Lawlor, J.M. Isotopic and Kinetic Studies of the Mechanism of Hydrolysis of Salicyl Phosphate. Intramolecular General Acid Catalysis. J. Am. Chem. Soc. 1963, 85, 3010–3017. [Google Scholar] [CrossRef]
- Edwards, D.R.; Liu, C.T.; Garett, G.E.; Neverov, A.A.; Brown, R.S. Leaving Group Assistance in the La3+-Catalyzed Cleavage of Dimethyl (o-Methoxycarbonyl)-aryl Phosphate Triesters in Methanol. J. Am. Chem. Soc. 2009, 131, 13738–13748. [Google Scholar] [CrossRef] [PubMed]
- Abell, K.W.Y.; Kirby, A.J. Intramolecular general acid catalysis of intramolecular nucleophilic catalysis of the hydrolysis of a phosphate diester. J. Chem. Soc. Perkin Trans. 1983, 2, 1171–1174. [Google Scholar] [CrossRef]
- Kirby, A.J.; Noem, F. Fundamentals of Phosphate Transfer. Acc. Chem. Res. 2015, 48, 1806–1814. [Google Scholar] [CrossRef]
- Kirby, A.J.; Mora, J.R.; Noem, F. New light on phosphate transfer from triesters. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 454–463. [Google Scholar] [CrossRef]
- Sumita, A.; Otani, Y.; Ohwada, T. Electrophilic activation of aminocarboxylic acid by phosphate ester promotes Friedel–Crafts acylation by overcoming charge–charge repulsion. Org. Biomol. Chem. 2017, 15, 9398–9407. [Google Scholar] [CrossRef]
- Olah, G.A.; McFarland, C.W. Organophosphorus Compounds. XIII.la Protonation, Cleavage, and Alkylation of Thiophosphates and Thiophosphites. J. Org. Chem. 1975, 40, 2582–2587. [Google Scholar] [CrossRef]
- Holmes, R.R. Comparison of Phosphorus and Silicon: Hypervalency, Stereochemistry, and Reactivity. Chem. Rev. 1996, 96, 927–950. [Google Scholar] [CrossRef]
- Kirby, A.J.; Hollfelder, F. From Enzyme Models to Model Enzymes; Royal Chemical Society (RCS) Publishing: Cambridge, UK, 2009. [Google Scholar]
- Wadsworth, W.S. The phosphoryl cation as an intermediate in the reaction of benzoyl phosphates. J. Chem. Soc. Perkin Trans. 1972, 2, 1686–1689. [Google Scholar] [CrossRef]
- Blonski, C.; Belghith, H.; Klaébé, A.; Perié, J.-J. The N-phosphobiotin route: A possible new pathway for biotin coenzyme. J. Chem. Soc. Perkin Trans. 1987, 2, 1369–1374. [Google Scholar] [CrossRef]
- Kanto, J.; Kangas, L.; Leppänen, T.; Mansikka, M.; Sibakov, M.L. Tofizopam: A benzodiazepine derivative without sedative effect. Int. Clin. Pharm. Ther. Toxic. 1982, 20, 309–312. [Google Scholar]
- Rudolph, U.; Knoflach, F. Beyond classical benzodiazepines: Novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 2011, 10, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Espahbodinia, M.; Ettari, R.; Wen, W.; Wu, A.; Shen, Y.-C.; Niu, L.; Grasso, S.; Zappala, M. Development of novel N-3-bromoisoxazolin-5-yl substituted 2,3-benzodiazepines as noncompetitive AMPAR antagonists. Bioorg. Med. Chem. 2017, 25, 3631–3637. [Google Scholar] [CrossRef]
- Chan, C.-K.; Tsai, Y.-L.; Chan, Y.-L.; Chang, M.-Y. Synthesis of Substituted 2,3-Benzodiazepines. J. Org. Chem. 2016, 81, 9836–9847. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Guo, S.; Zha, S.; Zhu, J. Synthesis of 2,3-Benzodiazepines via Rh (III)-Catalyzed C–H Functionalization of N-Boc Hydrazones with Diazoketoesters. Org. Lett. 2017, 19, 3640–3643. [Google Scholar] [CrossRef]
- Okuma, K.; Tanabe, T.; Nagahora, N.; Shioji, K. Synthesis of 2,3-Benzodiazepines and 2,3-Benzodiazepin-4-ones from Arynes and β-Diketones. Bull. Chem. Soc. Jpn. 2015, 88, 1064–1073. [Google Scholar] [CrossRef]
- Nikolyukin, Y.A.; Bogza, S.L.; Dulenko, V.I. Synthesis of functionally substituted benzo[c]pyrylium salts. Chem. Heterocycl. Compd. 1990, 26, 397–402. [Google Scholar] [CrossRef]
- Bogza, S.L.; Nikolyyukin, Y.A.; Zubritskii, M.Y.; Dulenko, V.I.; Afonin, A.A.; Dulenko, V.I. Functionally substituted benzo[c]pyrilium salts. Reactions of 4-carbethoxybenzo[c]pyrilium salts with benzylamine. Chem. Heterocycl. Comp. 1995, 31, 272–275. [Google Scholar] [CrossRef]
- Horváth, E.J.; Horváth, K.; Hámori, T.; Fekete, M.I.K.; Sólyom, S.; Palkovits, M. Anxiolytic 2,3-benzodiazepines, their specific binding to the basal ganglia. Prog. Neurobiol. 2000, 60, 309–342. [Google Scholar] [CrossRef]
- Kurita, J.; Enkaku, M.; Tsuchiya, T. Studies on Diazepines. XIX. Photochemical Synthesis of 2, 3-Benzodiazepines from Isoquinoline N-Imides. Chem. Pharm. Bull. 1982, 30, 3764–3769. [Google Scholar] [CrossRef][Green Version]
- Sumita, A.; Lee, J.; Otani, Y.; Ohwada, T. Facile Synthesis of 2,3-Benzodiazepines Using One-pot Two-step Phosphate-Assisted Acylation-Hydrazine Cyclization Reactions. Org. Bio Chem. 2018, 16, 4013–4020. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G. A short biomimetic synthesis of the isoquinoline and the naphthalene moieties of ancistrocladus alkaloids from common β-polycarbonyl precursors. Tetrahedron Lett. 1982, 23, 2009–2012. [Google Scholar] [CrossRef]
- Schiess, P.; Huys-Francotte, M.; Vogel, C. Thermolytic Ring Opening of Acyloxybenzeocyclobutenes: An efficient route to 3-substituted isoquinolines. Tetrahedron Lett. 1985, 26, 3959–3962. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
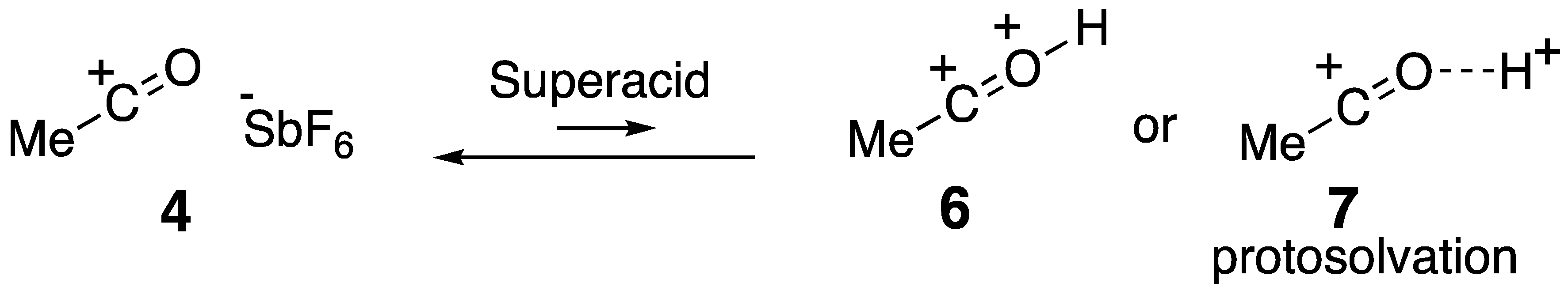{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
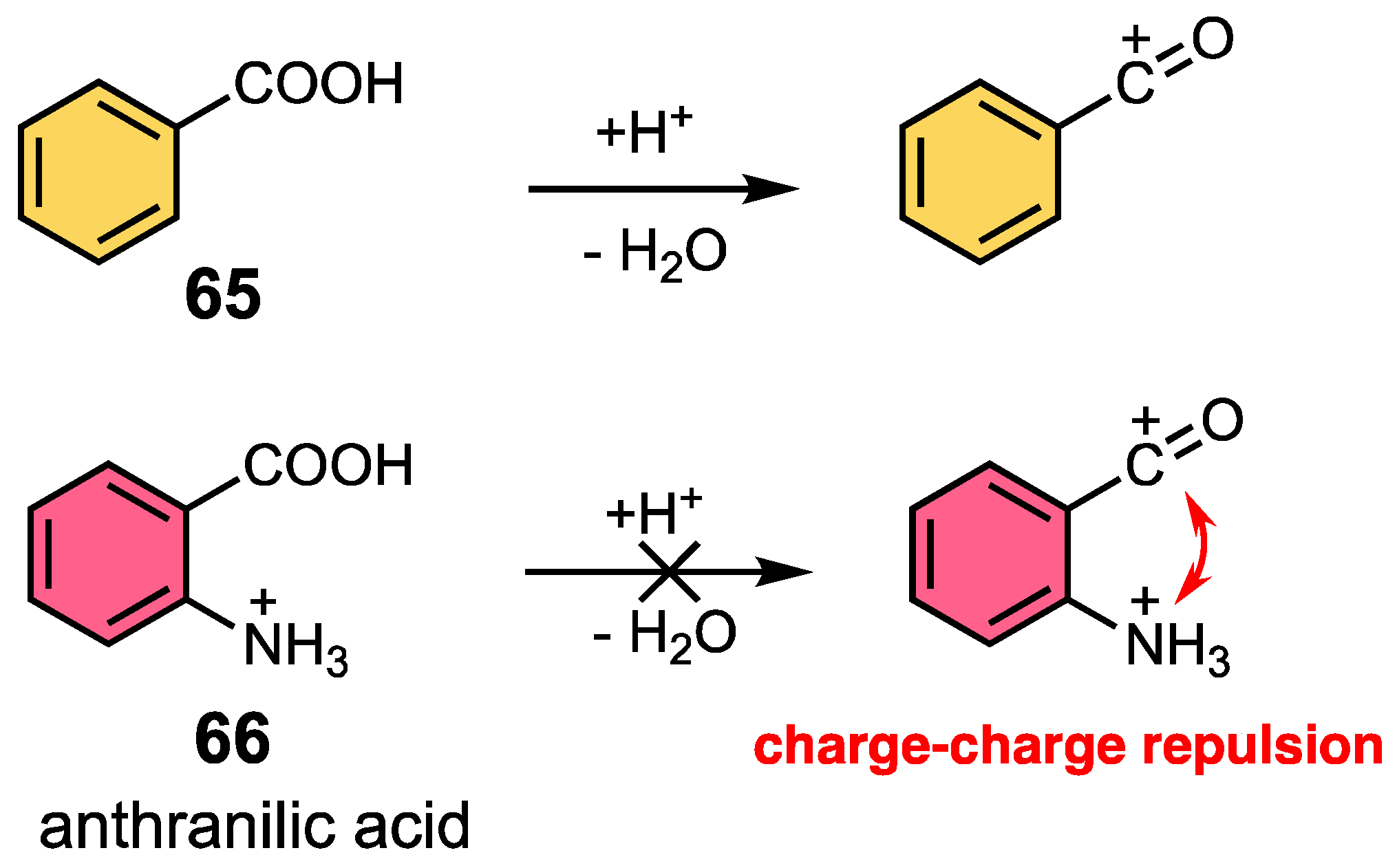{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Carboxylic Acid | Phosphate Ester | Acid | Temp. | Time | Yield of 68 or 69 | Yield of 70 or 71 |
|---|---|---|---|---|---|---|---|
| 1 | H (65) | 67 | TfOH | 20 °C | 20 min | 92% (68) | <1% (70) |
| 2 | H (65) | – | TfOH | 20 °C | 20 min | <1% (68) | <1% (70) |
| 3 | H (65) | – | TfOH | 20 °C | 24 h | 48% (68) | <1% (70) |
| 4 | H (65) | 67 | TFA | 20 °C | 48 h | <1% (68) | 81% (70) |
| 5 | NH2 (66) | 67 | TfOH | 20 °C | 20 min | 88% (69) | <1% (71) |
| 6 | NH2 (66) | – | TfOH | 20 °C | 24 h | <7% (69) | <1% (71) |
| 7 | NH2 (66) | – | TfOH | 20 °C | 20 min | <1% (69) | <1% (71) |
| 8 | NH2 (66) | 67 | TfOH + TFA a | 20 °C | 20 min | 10% (69) | 38% (71) |
| 9 | NH2 (66) | 67 | TFA | 20 °C | 20 min | <1% (69) | <1% (71) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumita, A.; Ohwada, T. Friedel-Crafts-Type Acylation and Amidation Reactions in Strong Brønsted Acid: Taming Superelectrophiles. Molecules 2022, 27, 5984. https://doi.org/10.3390/molecules27185984
Sumita A, Ohwada T. Friedel-Crafts-Type Acylation and Amidation Reactions in Strong Brønsted Acid: Taming Superelectrophiles. Molecules. 2022; 27(18):5984. https://doi.org/10.3390/molecules27185984
Chicago/Turabian StyleSumita, Akinari, and Tomohiko Ohwada. 2022. "Friedel-Crafts-Type Acylation and Amidation Reactions in Strong Brønsted Acid: Taming Superelectrophiles" Molecules 27, no. 18: 5984. https://doi.org/10.3390/molecules27185984
APA StyleSumita, A., & Ohwada, T. (2022). Friedel-Crafts-Type Acylation and Amidation Reactions in Strong Brønsted Acid: Taming Superelectrophiles. Molecules, 27(18), 5984. https://doi.org/10.3390/molecules27185984