
Synthesis and Characterization of Phosphinecarboxamide and Phosphinecarbothioamide, and Their Complexation with Palladium(II) Complex †

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
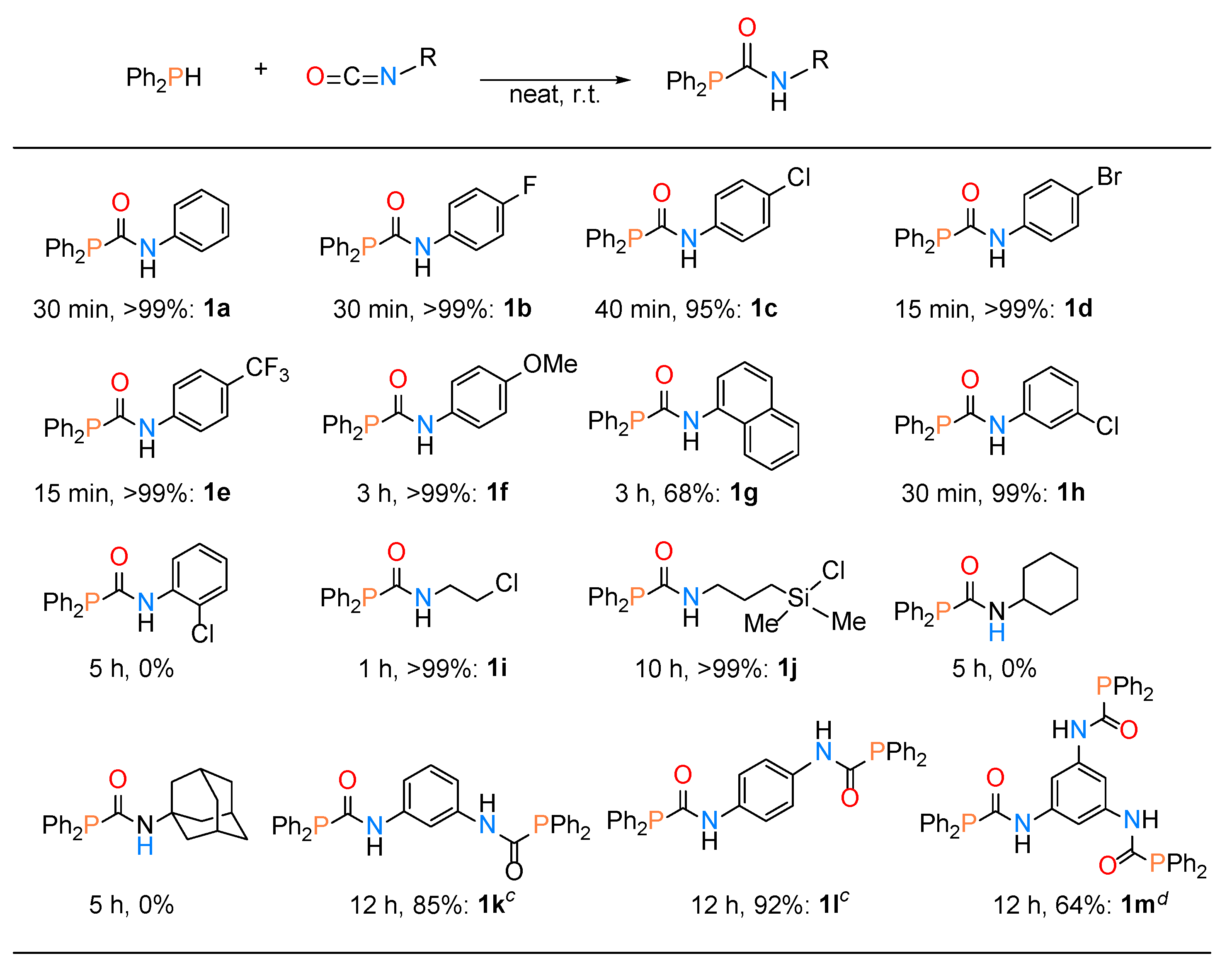
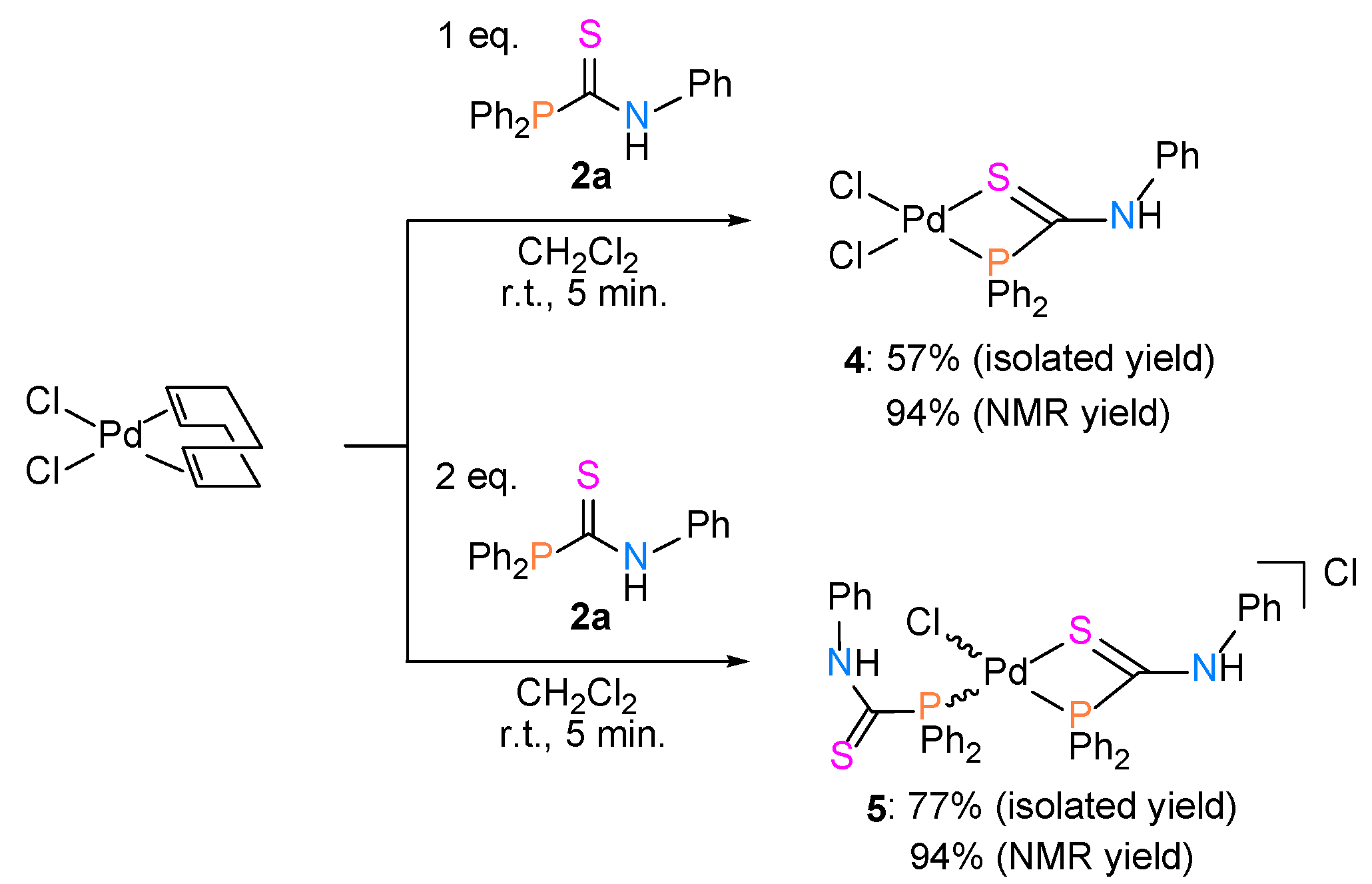
2.1. Synthesis of Phosphinecarboxamide and Phosphinecarbothioamide
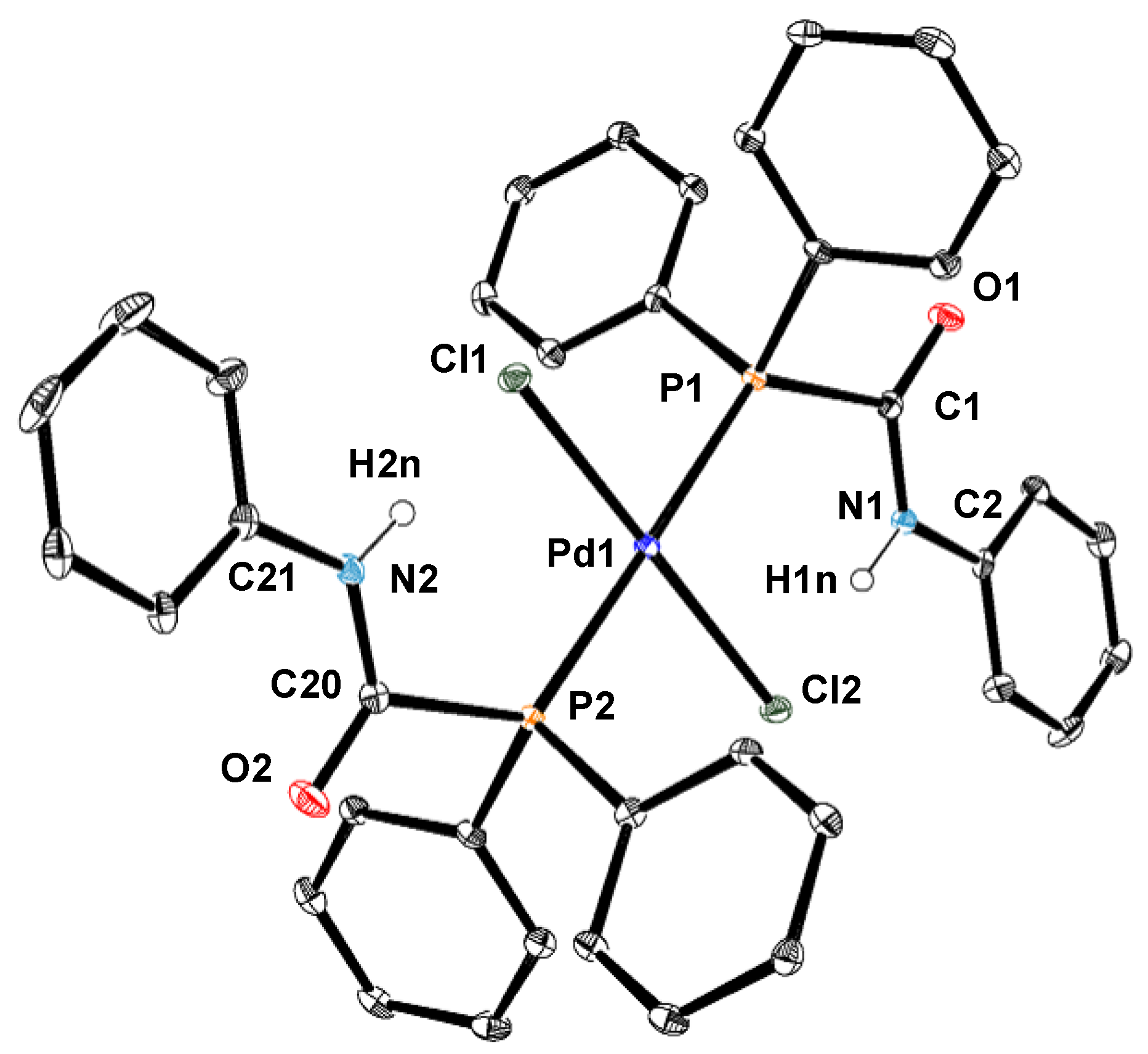
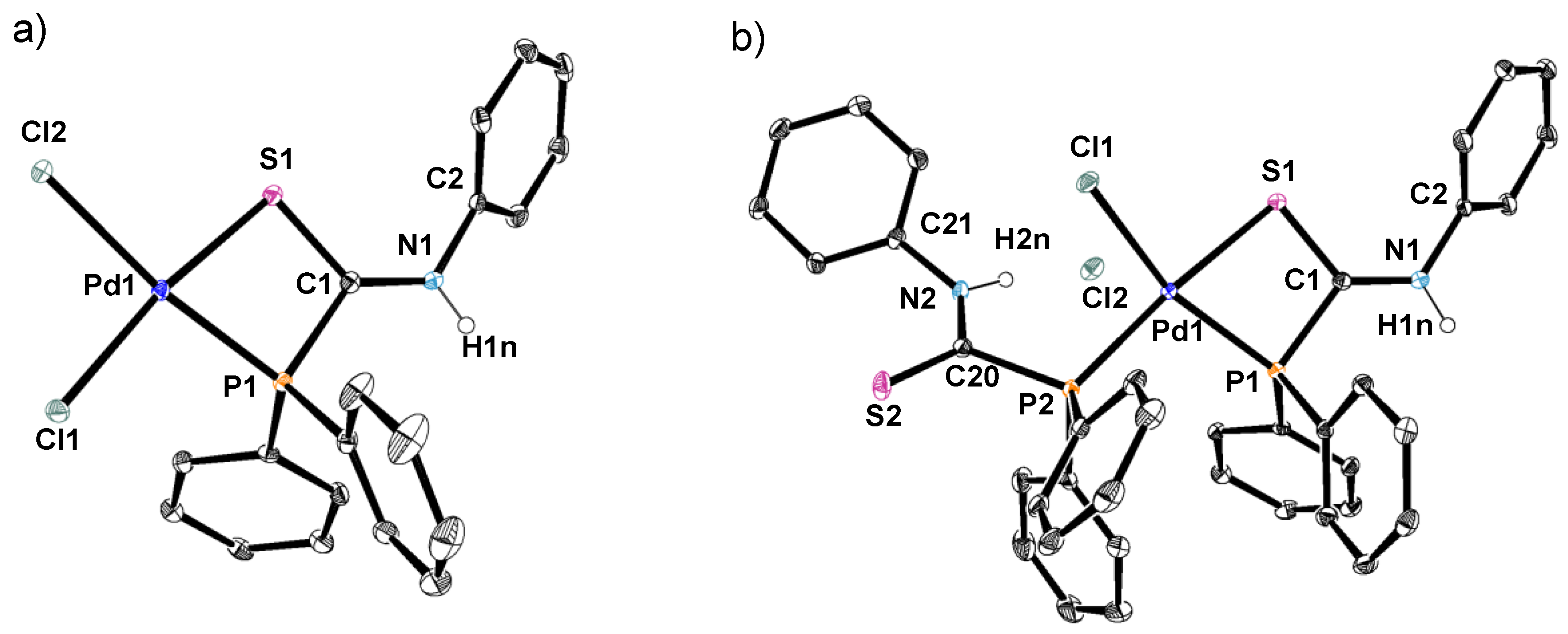
2.2. Synthesis of Palladium(II) Complexes with Phosphinecarboxamide and Phosphinecarbothioamide
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis
3.2.1. General Procedure for Synthesis of Phosphinecarboxamide and Phosphinecarbothioamide
3.2.2. NMR Tube Experiments for Pd(II) Complexes
3.2.3. Synthesis of Pd(II) Complexes
3.3. Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Geeson, M.B.; Jupp, A.R.; McGrady, J.E.; Goicoechea, J.M. On the coordination chemistry of phosphinecarboxamide: Assessing ligand basicity. Chem. Commun. 2014, 50, 12281–12284. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Doyle, R.J.; Lee, H.-Y.; Lu, D.; Salem, G.; Speldewinde, D.J.; Tifan, M.; Willis, A.C. Synthesis of 1,3-azaphosphol-2-ones. Crystal and molecular structures of [SP-4-2]-dichlorobis(3-phenyl-1,3-dihydrobenzo [1,3]azaphosphol-2-one-P)palladium(II) and its chloro(methyl)platinum(II) analogue. Dalton Trans. 2010, 39, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Jupp, A.R.; Trott, G.; de la Garanderie, É.P.; Holl, J.D.G.; Carmichael, D.; Goicoechea, J.M. Exploiting the brønsted acidity of phosphinecarboxamides for the synthesis of new phosphides and phosphines. Chem. Eur. J. 2015, 21, 8015–8018. [Google Scholar] [CrossRef] [PubMed]
- Navrátil, M.; Faria, E.N.; Panahy, G.; Císařová, I.; Goicoechea, J.M.; Štěpnička, P. Novel ferrocenyl functionalised phosphinecarboxamides: Synthesis, characterisation and coordination. Dalton Trans. 2020, 49, 8645–8651. [Google Scholar] [CrossRef]
- Cowan, S.W.; Dakternieks, D.; Gable, R.W.; Hoskins, B.F.; Rolls, C.L.; Tiekink, E.R.T. Studies of some cadmium(II) and mercury(II) complexes with dicyclohexylphosphino-N-phenylthioformamide, LH: Crystal and molecular-structures of [Cdl2(LH)]2, ([HgCl2(LH)]2.CH2Cl2) and HgCl2(LH)2. Aust. J. Chem. 1986, 39, 547–556. [Google Scholar] [CrossRef]
- Malisch, W.; Spörl, A.; Thirase, K.; Fey, O. Übergangsmetall-substituierte phosphane, arsane und stibane, LIX [1], ein addukt des Me3P-substituierten ferrio-diphenylphosphans Cp(OC)(Me3P)Fe-PPh2 mit methylisothiocyanat: Darstellung, protonierung und methylierung. Z. Naturforsch. 1998, 53, 1077–1083. [Google Scholar] [CrossRef]
- Crespo, O.; Fernández, E.J.; Jones, P.G.; Laguna, A.; López-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Pérez, J. Coordination modes of diphenylphosphinothioformamide in its neutral and deprotonated forms at gold(I). Dalton Trans. 2003, 1076–1082. [Google Scholar] [CrossRef]
- Just, B.; Klein, W.; Kope, J.; Steinhäuser, K.G.; Kramolowsky, R. Synthese und koordinationsverhalten ambidenter chelatliganden: III. Kristall-und molekülstrukturen der carbonyl-mangan(I)-komplexe fac-[MnBr(CO)3{Ph2PC(S)NPhH}] und [Mn{S(NPh)CPPh2}(CO)4]. Neutrale phosphinothioformamide und deren deprotonierte, anionische derivate als S,P-chelatliganden. J. Organomet. Chem. 1982, 229, 49–61. [Google Scholar]
- Kunze, U.; Jawad, H.; Hiller, W.; Naumer, R. Phosphinsubstituierte chelatliganden, XIV [1] wasserstoff-brückenbindung in THF-addukten von tetracarbonylchrom- und -molybdänkomplexen mit P,S-koordinierten phosphinothioformamid-liganden. Kristallstruktur von [(CO)4Cr(PPh2C(S)NHMe)]·THF. Z. Naturforsch. 1985, 40, 512–517. [Google Scholar]
- Siasios, G.; Tiekink, E.R.T. Synthesis and crystal structure of [Ni{Ph2PC(S)NPh}2] and [Ni{Ph2P(Y)C(S)NPh}2](Y = S or Se). J. Chem. Soc. Dalton Trans. 1996, 2269–2273. [Google Scholar] [CrossRef]
- Leung, P.-H.; Qin, Y.; He, G.; Mok, K.F.; Vittal, J.J. Coordination chemistry, reactivities, and stereoelectronic properties of chelating phosphine ligands containing thioamide substituents. J. Chem. Soc., Dalton Trans. 2001, 3, 309–314. [Google Scholar] [CrossRef]
- Ambrosius, H.P.M.M.; Willemse, J.; Cras, J.A.; Bosman, W.P.; Noordik, J.H. Different coordination modes of neutral and deprotonated ZC(S)N(H)R ligands (Z = PPh2, R = Ph, Me; Z = NMe2, R = Ph) in molybdenum and tungsten complexes. X-ray structure analysis of [Mo(CO)2[Ph2PC(S)NMe][μ-Ph2PC(S)NMe]]2·CH2Cl2. Inorg. Chem. 1984, 23, 2672–2678. [Google Scholar] [CrossRef]
- Kunze, U.; Jawad, H.; Burghardt, R. Phosphinsubstituierte chelatliganden, XX [1] darstellung und diastereoselektive komplexierung von chiralen phosphinothioformamiden, Ph2P(X)C(S)NHCHMePh (X = 2 e–, O, S). Z. Naturforsch. 1986, 41, 1142–1150. [Google Scholar]
- Ambrosius, H.P.M.M.; Cotton, F.A.; Falvello, L.R.; Hintzenl, H.T.J.M.; Melton, T.J.; Schwotzer, W.; Tomas, M.; van der Linden, J.G.M. Formation of isomeric dimolybdenum(II) compounds using the potentially ambidentate ligands [R2PC(S)NR’]– (R/R’ = Ph/Ph, Ph/Me) and [Me2NC(S)NPh]–. Inorg. Chem. 1984, 23, 1611. [Google Scholar] [CrossRef]
- Buckler, S.A. Reaction of phosphine with Isocyanates. J. Org. Chem. 1959, 24, 1460–1462. [Google Scholar] [CrossRef]
- Behrle, A.C.; Schmidt, J.A.R. Insertion eeactions and catalytic hydrophosphination of heterocumulenes using α-metalated N,N-dimethylbenzylamine rare-earth-metal complexes. Organometallics 2013, 32, 1141–1149. [Google Scholar] [CrossRef]
- Gu, X.; Zhang, L.; Zhu, X.; Wang, S.; Zhou, S.; Wei, Y.; Zhang, G.; Mu, X.; Huang, Z.; Hong, D.; et al. Synthesis of bis(NHC)-based CNC-pincer rare-earth-metal amido complexes and their application for the hydrophosphination of heterocumulenes. Organometallics 2015, 34, 4553–4559. [Google Scholar] [CrossRef]
- Karmel, I.S.; Tamm, M.; Eisen, M.S. Actinide-mediated catalytic addition of E–H bonds (E=N, P, S) to carbodiimides, isocyanates, and isothiocyanates. Angew. Chem. Int. Ed. 2015, 54, 12422–12425. [Google Scholar] [CrossRef]
- Batrice, R.J.; Eisen, M.S. Catalytic insertion of E–H bonds (E = C, N, P, S) into heterocumulenes by amido-actinide complexes. Chem. Sci. 2016, 7, 939–944. [Google Scholar] [CrossRef]
- Sharpe, H.R.; Geer, A.M.; Lewis, W.; Blake, A.J.; Kays, D.L. Iron(II)-catalyzed hydrophosphination of isocyanates. Angew. Chem. Int. Ed. 2017, 56, 4845–4848. [Google Scholar] [CrossRef]
- Zhang, B.; Ma, X.; Yan, B.; Ni, C.; Yu, H.; Yang, Z.; Roesky, H.W. An efficient catalytic method for hydrophosphination of heterocumulenes with diethylzinc as precatalyst without a solvent. Dalton Trans. 2021, 50, 15488–15492. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, L.; Wang, Y.; Yuan, D.; Yao, Y.; Shen, Q. Neutral and cationic zirconium complexes bearing multidentate aminophenolato ligands for hydrophosphination reactions of alkenes and heterocumulenes. Inorg. Chem. 2018, 57, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Downie, T.M.H.; Hall, J.W.; Finn, T.P.C.; Liptrot, D.J.; Lowe, J.P.; Mahon, M.F.; McMullin, C.L.; Whittlesey, M.K. The first ring-expanded NHC-copper(I) phosphides as catalysts in the highly selective hydrophosphination of isocyanates. Chem. Commun. 2020, 56, 13359–13362. [Google Scholar] [CrossRef] [PubMed]
- Schwamm, R.J.; Coles, M.P. Catalytic hydrophosphination of isocyanates by molecular antimony phosphanides. Eur. J. Inorg. Chem. 2022, 2022, e202200064. [Google Scholar] [CrossRef]
- Ito, M.; Iseki, M.; Itazaki, M.; Nakazawa, H. Tetrahedral cage complex with planar vertices: Selective synthesis of Pt4L6 cage complexes involving hydrogen bonds driven by halide binding. Chem. Commun. 2016, 52, 7205–7208. [Google Scholar]
- Matsutani, T.; Itazaki, M.; Akine, S.; Moriuchi, T. Macrocyclic dimer of Fc(NHC(O)PPh2-AuCl)2 induced by aurophilic interactions, and chirality induction into Fc core. J. Organomet. Chem. 2020, 912, 121182. [Google Scholar]
- Itazaki, M.; Matsutani, T.; Nochida, T.; Moriuchi, T.; Nakazawa, H. Convenient synthesis of phosphinecarboxamide and phosphinecarbothioamide by hydrophosphination of isocyanates and isothiocyanates. Chem. Commun. 2020, 56, 443–445. [Google Scholar] [CrossRef]
- Kamitani, M.; Itazaki, M.; Tamiya, C.; Nakazawa, H. Regioselective double hydrophosphination of terminal arylacetylenes catalyzed by an iron complex. J. Am. Chem. Soc. 2012, 134, 11932–11935. [Google Scholar] [CrossRef]
- Itazaki, M.; Katsube, S.; Kamitani, M.; Nakazawa, H. Synthesis of vinylphosphines and unsymmetric diphosphines: Iron-catalyzed selective hydrophosphination reaction of alkynes and vinylphosphines with secondary phosphines. Chem. Commun. 2016, 52, 3163–3166. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, M.; Sun, J.; Guo, D.; Zhang, Y.; Zhou, S.; Wang, S. Hydroamination and hydrophosphination of isocyanates/isothiocyanates under catalyst-free conditions. Eur. J. Org. Chem. 2021, 5213–5218. [Google Scholar] [CrossRef]
- Jupp, A.R.; Goicoechea, J.M. Phosphinecarboxamide: A phosphorus-containing analogue of urea and stable primary phosphine. J. Am. Chem. Soc. 2013, 135, 19131–19134. [Google Scholar] [CrossRef] [PubMed]
- Redfield, D.A.; Nelson, J.H. Equilibrium energetics of cis-trans isomerization for two square-planar palladium(II)-phosphine complexes. Inorg. Chem. 1973, 12, 15–19. [Google Scholar] [CrossRef]
- Redfield, D.A.; Cary, L.W.; Nelson, J.H. Equilibrium thermodynamics and mechanism of cis-trans isomerization for diazidobis(methyldiphenylphosphine)palladium(II) and diazidobis(dimethylphenylphosphine)palladium(II). Inorg. Chem. 1975, 14, 50–59. [Google Scholar] [CrossRef]
- Piper, T.S.; Wilkinson, G. Alkyl and aryl derivatives of π-cyclopentadienyl compounds of chromium, molybdenum, tungsten, and iron. J. Inorg. Nucl. Chem. 1956, 3, 104–124. [Google Scholar] [CrossRef]
- Drew, D.; Doyle, J.R. Cyclic diolefin complexes of platinum and palladium. Inorg. Synth. 1972, 13, 47–55. [Google Scholar]
- Rigaku. REQAB; Version 1.1; Rigaku Corporation: Tokyo, Japan, 1998. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliard, A.; Moliterni, A.G.G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar]
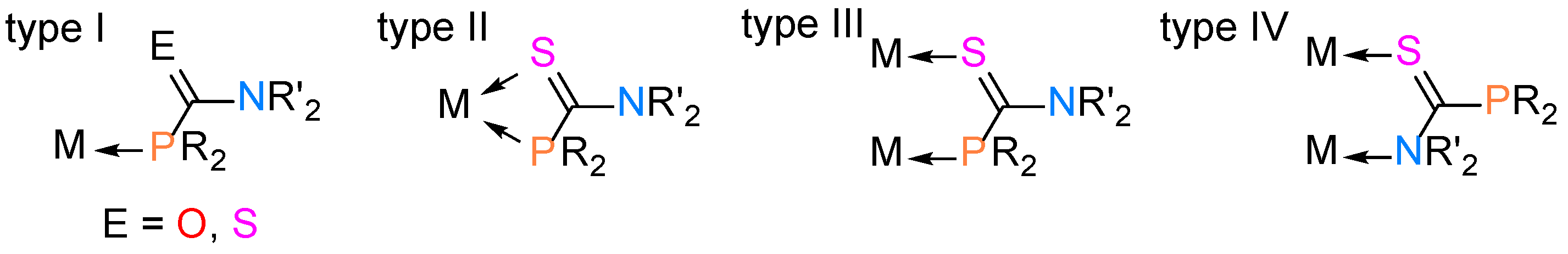
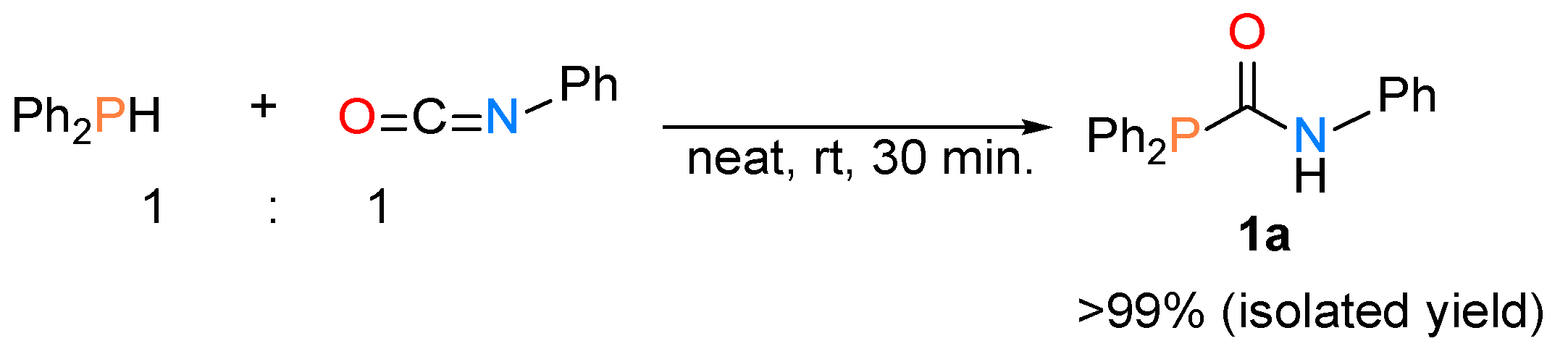



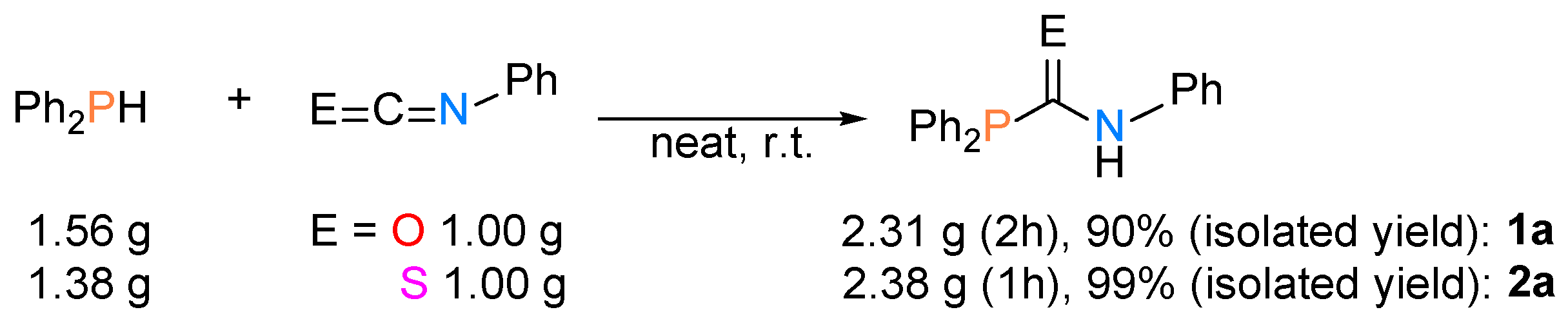




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1i | 2c | 3 | 4·0.5(CH3)2CO | 5·CH2Cl2 | |
|---|---|---|---|---|---|
| empirical formula | C15H15ClNOP | C19H15ClNPS | C38H32Cl2N2O2P2Pd | C20.5H19Cl2NO0.5PSPd | C39H34Cl4N2P2S2Pd |
| formula weight | 291.70 | 355.80 | 787.89 | 527.70 | 904.94 |
| T (K) | 200(2) | 200(2) | 110(2) | 110(2) | 110(2) |
| crystal system | triclinic | monoclinic | orthorhombic | triclinic | monoclinic |
| space group | P21/n | Pca21 | P21/n | ||
| a (Å) | 9.29470(10) | 9.2578(5) | 12.9390(2) | 9.679(3) | 17.288(5) |
| b (Å) | 11.8190(2) | 21.2798(9) | 12.4521(3) | 14.927(4) | 13.508(3) |
| c (Å) | 21.0466(4) | 9.9399(5) | 21.0466(4) | 15.962(4) | 18.189(5) |
| α (°) | 85.569(6) | 106.543(4) | |||
| β (°) | 82.125(7) | 116.3332(15) | 92.7607(7) | 112.506(3) | |
| γ (°) | 67.021(6) | 103.748(3) | |||
| volume (Å3) | 1448.16(9) | 1755.00(15) | 3390.98(12) | 2130.6(10) | 3924.0(17) |
| Z | 4 | 4 | 4 | 4 | 4 |
| ρcalcd (mg m–3) | 1.338 | 1.347 | 1.543 | 1.645 | 1.532 |
| μ (mm–1) | 0.365 | 0.426 | 0.837 | 1.303 | 0.965 |
| F(000) | 608 | 736 | 1600 | 1056 | 1832 |
| crystal size (mm3) | 0.37 × 0.27 × 0.12 | 0.27 × 0.18 × 0.17 | 0.12 × 0.07 × 0.02 | 0.13 × 0.07 × 0.03 | 0.21 × 0.16 × 0.06 |
| reflections collected | 17,154 | 13,335 | 27,555 | 22,263 | 39,512 |
| R(int) | 6564 (0.0361) | 3974 (0.0253) | 5973 (0.0309) | 9665 (0.0288) | 8955 (0.0336) |
| R1 (I > 2σ(I)) | 0.0478 | 0.0503 | 0.0204 | 0.0278 | 0.0346 |
| wR2 (all data) | 0.1187 | 0.1062 | 0.0502 | 0.0600 | 0.0704 |
| goodness of fit | 1.106 | 1.094 | 1.042 | 1.046 | 1.094 |
| CCDC deposition number | 1959302 | 2193371 | 2168755 | 2168782 | 2168783 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itazaki, M.; Okabayashi, K.; Matsutani, T.; Nochida, T.; Moriuchi, T.; Nakazawa, H. Synthesis and Characterization of Phosphinecarboxamide and Phosphinecarbothioamide, and Their Complexation with Palladium(II) Complex. Molecules 2022, 27, 5564. https://doi.org/10.3390/molecules27175564
Itazaki M, Okabayashi K, Matsutani T, Nochida T, Moriuchi T, Nakazawa H. Synthesis and Characterization of Phosphinecarboxamide and Phosphinecarbothioamide, and Their Complexation with Palladium(II) Complex. Molecules. 2022; 27(17):5564. https://doi.org/10.3390/molecules27175564
Chicago/Turabian StyleItazaki, Masumi, Kento Okabayashi, Takanari Matsutani, Tomoya Nochida, Toshiyuki Moriuchi, and Hiroshi Nakazawa. 2022. "Synthesis and Characterization of Phosphinecarboxamide and Phosphinecarbothioamide, and Their Complexation with Palladium(II) Complex" Molecules 27, no. 17: 5564. https://doi.org/10.3390/molecules27175564
APA StyleItazaki, M., Okabayashi, K., Matsutani, T., Nochida, T., Moriuchi, T., & Nakazawa, H. (2022). Synthesis and Characterization of Phosphinecarboxamide and Phosphinecarbothioamide, and Their Complexation with Palladium(II) Complex. Molecules, 27(17), 5564. https://doi.org/10.3390/molecules27175564