

Ligand-Free Signaling of G-Protein-Coupled Receptors: Relevance to μ Opioid Receptors in Analgesia and Addiction


Abstract
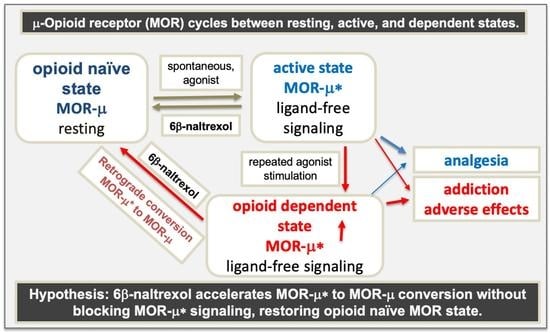
{kind=link}
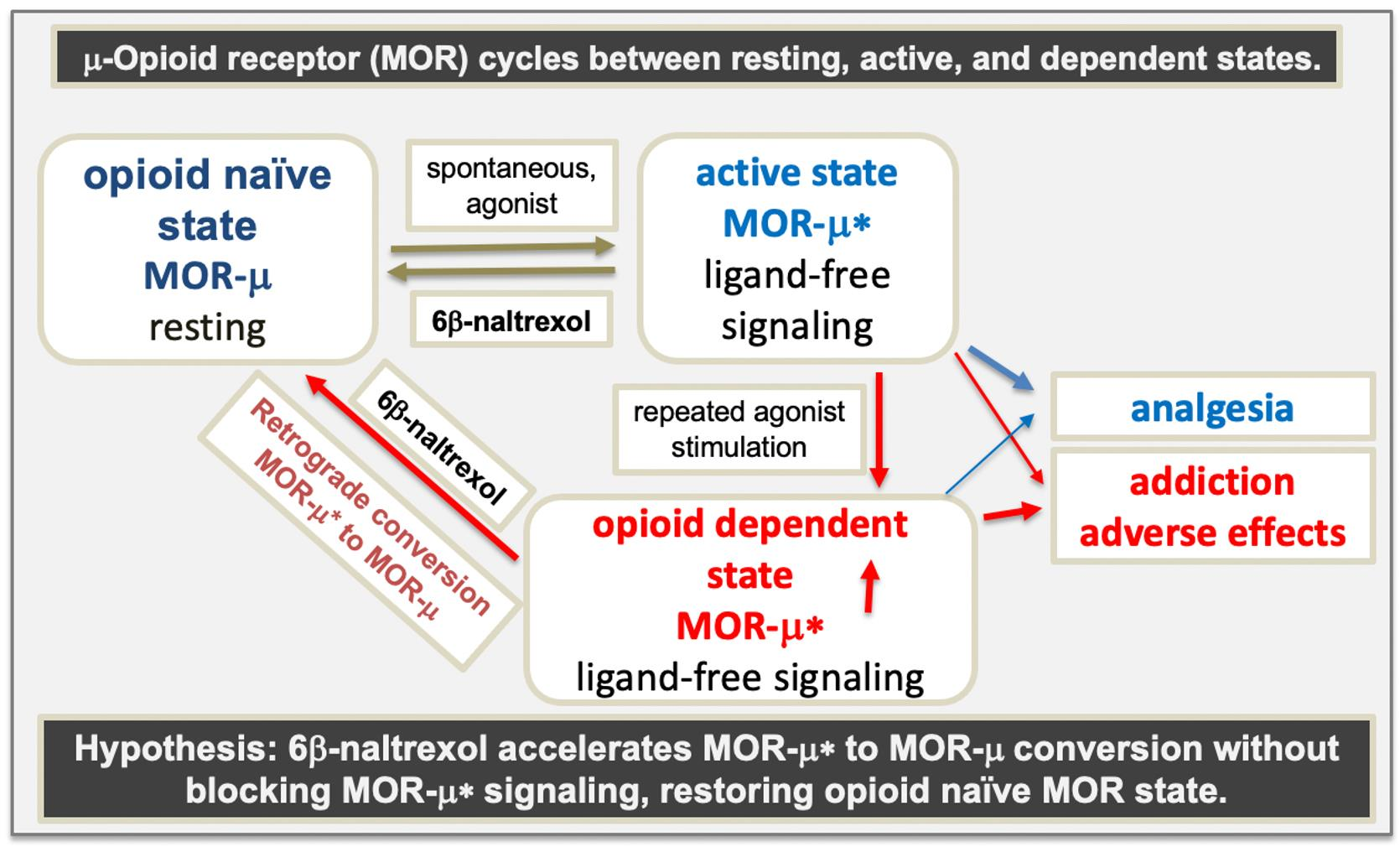
{kind=link}
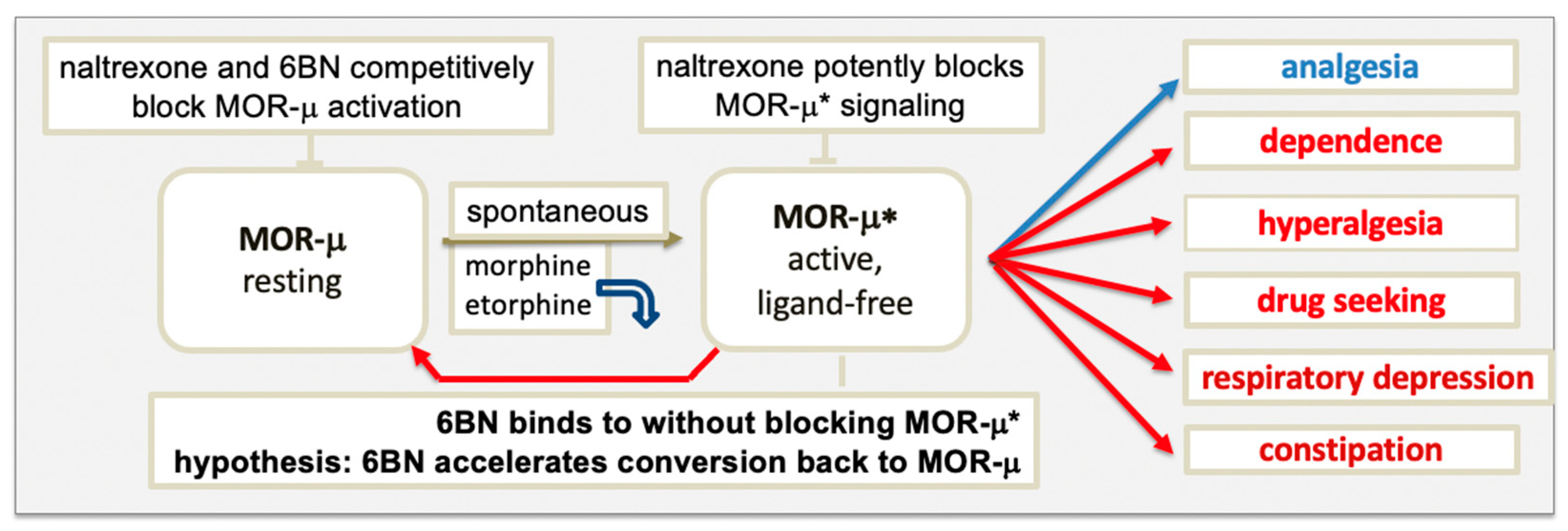
{kind=link}
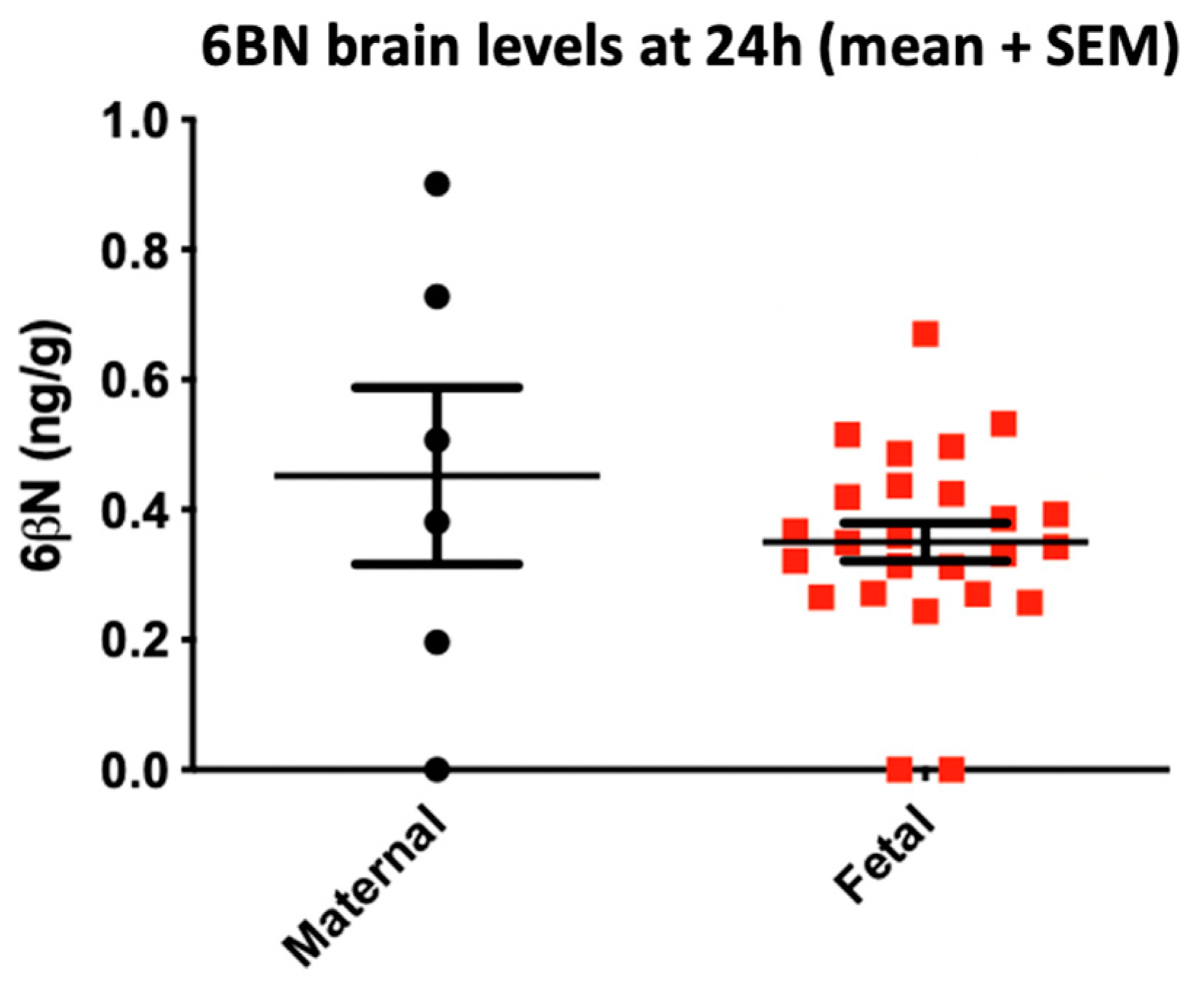{kind=link}
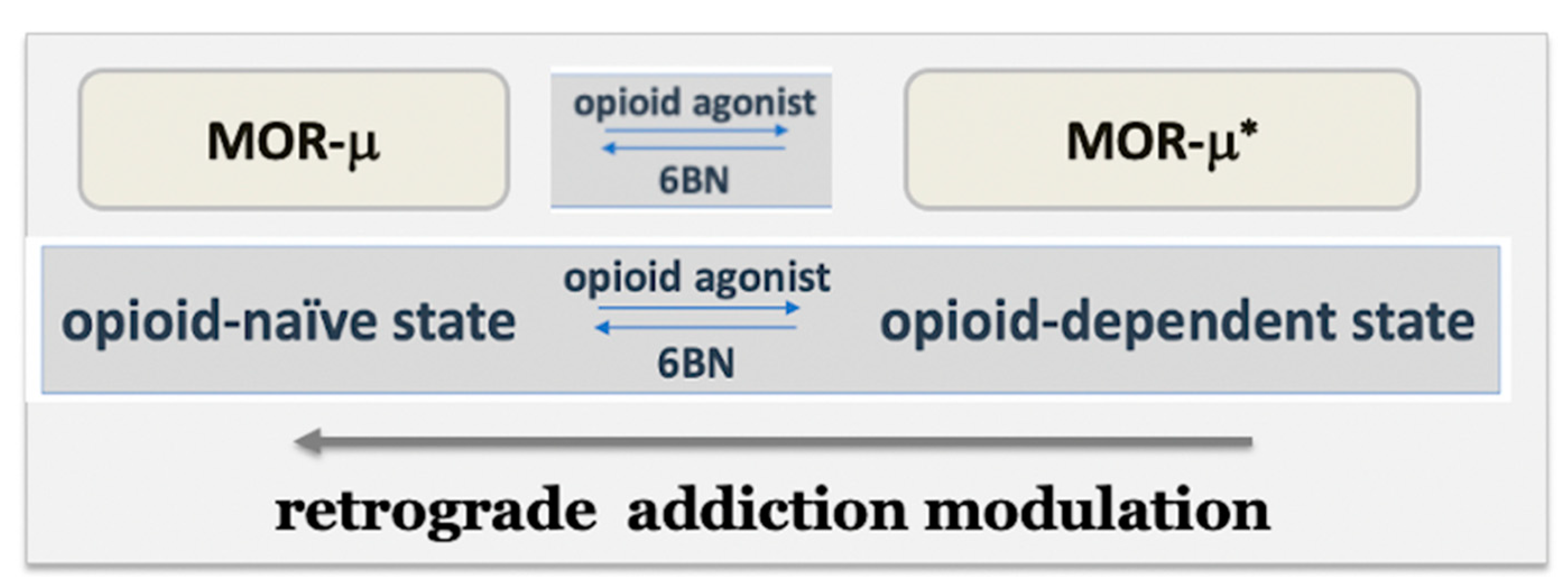{kind=link}
1. Introduction
2. Opioid Pain Therapy, Opioid Use Disorder, and Other Adverse Effects
3. Physiological Role and Regulation of Basal MOR Signaling
3.1. Regulation and Influence in Pain and Dependence
3.2. Neutral Antagonists and Inverse Agonists
3.3. Ligand-Free Signaling of Opioid Receptors in Peripheral Nociceptors
4. Model of μ Opioid Receptor Signaling
4.1. MOR Activation by Agonists
4.2. Blocking MOR Activation and Signaling by Antagonists
4.3. Differences between the New MOR Model and Classical GPCR Multistate Models
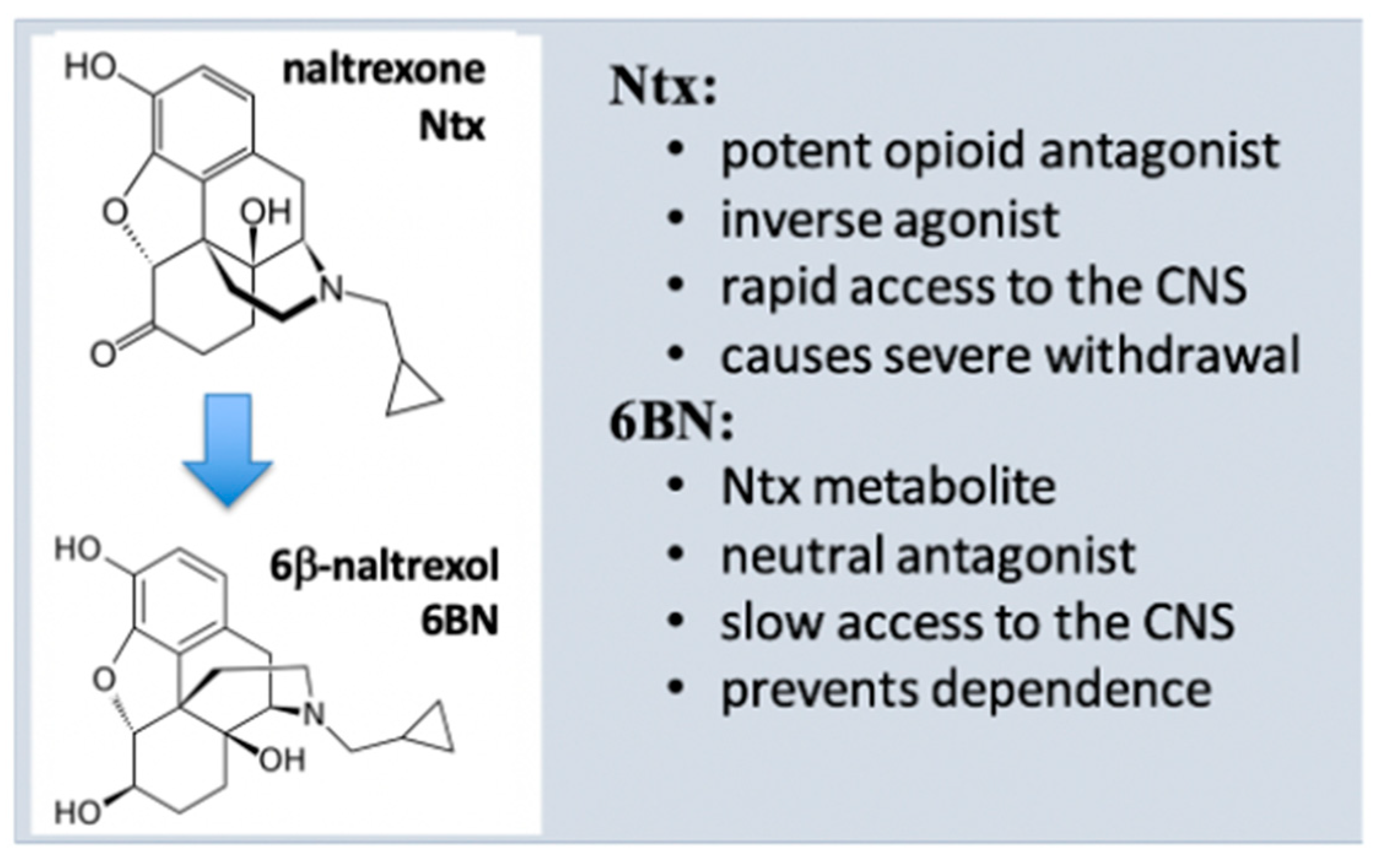
5. Properties of 6β-Naltrexol (6BN)
5.1. 6BN Pharmacology
5.2. Potent Suppression of Opioid Dependence with Low-Dose 6BN (LD-6BN)
5.3. Role of 6β-Naltrexol in the Effects of Very-Low-Dose Naltrexone
5.4. Potential Clinical Uses of 6b-Naltrexol
6. Discussion
7. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parnot, C.; Miserey-Lenkei, S.; Bardin, S.; Corvol, P.; Clauser, E. Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol. Metab. 2002, 13, 336–343. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Aloyo, V.J.; Berg, K.A.; Clarke, W.P.; Spampinato, U.; Harvey, J.A. Inverse agonism at serotonin and cannabinoid receptors. Prog. Mol. Biol. Transl. Sci. 2010, 91, 1–40. [Google Scholar]
- Unal, H.; Karnik, S.S. Domain coupling in GPCRs: The engine for induced conformational changes. Trends Pharm. Sci. 2012, 33, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Pazos, Y.; Casanueva, F.F.; Camiña, J.P. Basic aspects of ghrelin action. Vitam. Horm. 2008, 77, 89–119. [Google Scholar] [PubMed]
- Solomou, S.; Korbonits, M. The role of ghrelin in weight-regulation disorders: Implications in clinical practice. Hormones 2014, 13, 458–475. [Google Scholar] [CrossRef]
- Pimavanserin: Howland, R.H. An Inverse Agonist Antipsychotic Drug. J. Psychosoc. Nurs. Ment. Health Serv. 2016, 54, 21–24. [Google Scholar] [CrossRef]
- Costa, T.; Herz, A. Antagonists with negative intrinsic efficacy at δ opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 7321–7325. [Google Scholar] [CrossRef]
- Wang, Z.; Bilsky, E.J.; Porreca, F.; Sadée, W. Constitutive Receptor Activation as a Regulatory Mechanism Underlying Narcotic Tolerance and Dependence. Life Sci. 1994, 54, 339–350. [Google Scholar] [CrossRef]
- Salsitz, E.A. Chronic Pain, Chronic Opioid Addiction: A Complex Nexus. J. Med. Toxicol. 2016, 12, 54–57. [Google Scholar] [CrossRef]
- Alter, A.; Yeager, C. The Consequences of COVID-19 on the Overdose Epidemic: Overdoses Are Increasing. Washington/Baltimore High Intensity Drug Trafficking Area. Available online: http://www.odmap.org/Content/docs/news/2020/ODMAP-Report-May2020.pdf (accessed on 1 June 2020).
- Ayanga, D.; Shorter, D.; Kosten, T.R. Update on pharmacotherapy for treatment of opioid use disorder. Expert Opin. Pharm. 2016, 17, 2307–2318. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharm 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, R.; Hillhouse, T.M.; Livingston, K.E.; Kochan, K.E.; Meurice, C.; Eans, S.O.; Li, M.-H.; White, A.D.; Roques, B.P.; McLaughlin, J.P.; et al. Positive allosteric modulation of the mu-opioid receptor produces analgesia with reduced side effects. Proc. Natl. Acad. Sci. USA 2021, 118, e2000017118. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef]
- Chakraborty, S.; DiBerto, J.F.; Faouzi, A.; Bernhard, S.M.; Gutridge, A.M.; Ramsey, S.; Zhou, Y.; Provasi, D.; Jilakara, R.; Asher, W.B.; et al. A Novel Mitragynine Analog with Low-Efficacy Mu Opioid Receptor Agonism Displays Antinociception with Attenuated Adverse Effects. J. Med. Chem. 2021, 64, 13873–13892. [Google Scholar] [CrossRef]
- Groer, C.E.; Tidgewell, K.; Moyer, R.A.; Harding, W.W.; Rothman, R.B.; Prisinzano, T.; Bohn, L.M.E. An opioid agonist that does not induce μ-opioid receptor—Arrestin interactions or receptor internalization. Mol. Pharm. 2007, 71, 549–557. [Google Scholar] [CrossRef]
- Bell, J.; Strang, J. Medication Treatment of Opioid Use Disorder. J. Biol. Psychiatry 2020, 87, 82–88. [Google Scholar] [CrossRef]
- Sadee, W.; Oberdick, J.; Wang, Z. Biased opioid antagonists as modulators of opioid dependence: Opportunities to improve pain therapy and opioid use management. Molecules 2020, 25, 4163. [Google Scholar] [CrossRef]
- Walwyn, W.; Evans, C.J.; Hales, T.G. Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J. Neurosci. 2007, 27, 5092–5104. [Google Scholar] [CrossRef]
- Lam, H.; Maga, M.; Pradhan, A.; Evans, C.J.; Maidment, N.T.; Hales, T.G.; Walwyn, W. Analgesic tone conferred by constitutively active mu opioid receptors in mice lacking β-arrestin. Mol. Pain. 2011, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.H.; Hedden, N.S.; Prasoon, P.; Qi, Y.; Taylor, B.K. Post-surgical latent pain sensitization is driven by descending serotonergic facilitation and masked by µ-opioid receptor constitutive activity (MORCA) in the rostral ventromedial medulla. J. Neurosci. 2022, 42, 5811–5870. [Google Scholar] [CrossRef]
- Sullivan, L.C.; Chavera, T.S.; Jamshidi, R.J.; Berg, K.A.; Clarke, W.P. Constitutive Desensitization of Opioid Receptors in Peripheral Sensory Neurons. J. Pharmacol. Exp. Ther. 2016, 359, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Navani, D.M.; Sirohi, S.; Madia, P.A.; Yoburn, B.C. The role of opioid antagonist efficacy and constitutive opioid receptor activity in the opioid withdrawal syndrome in mice. Pharm. Biochem. Behav. 2011, 99, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Shoblock, J.R.; Maidment, N.T. Constitutively active opioid receptors mediate the enhanced conditioned aversive effect of naloxone in morphine-dependent mice. Neuropsychopharm 2006, 31, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Prather, P.L. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol. Pharmacol. 2001, 60, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Shoblock, J.R.; Maidment, N.T. Enkephalin release promotes homeostatic increases in constitutively active mu opioid receptors during morphine withdrawal. Neuroscience 2007, 149, 642–649. [Google Scholar] [CrossRef]
- Raehal, K.M.; Lowery, J.J.; Bhamidipati, C.M.; Paolino, R.M.; Blair, J.R.; Wang, D.; Sadée, W.; Bilsky, E.J. In vivo characterization of 6β-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. J Pharmacol. Exp. Ther. 2005, 313, 1150–1162. [Google Scholar] [CrossRef]
- Sadee, W.; Wang, D.; Bilsky, E.J. Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sci. 2005, 76, 1427–1437. [Google Scholar] [CrossRef]
- Wang, D.; Raehal, K.M.; Lin, E.T.; Lowery, J.J.; Kieffer, B.L.; Bilsky, E.J.; Sadée, W. Basal signaling opioid receptor in mouse brain: Role in narcotic dependence. J. Pharmacol. Exp. Ther. 2004, 308, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Doolen, S.; Donahue, R.R.; Winter, M.K.; Jutras, B.K.L.; He, Y.; Hu, X.; Wieskopf, J.S.; Mogil, J.S.; Storm, D.R.; et al. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science 2013, 341, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Blanco, C.; Wall, M.M.; Olfson, M. Expanding Current Approaches to Solve the Opioid Crisis. JAMA Psychiatry 2022, 79, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Raehal, K.M.; Bilsky, E.J.; Sadée, W. Inverse agonists and neutral antagonists at μ opioid receptor (MOR): Possible role of basal receptor signaling in narcotic dependence. J. Neurochem. 2001, 77, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, S.; Dighe, S.V.; Madia, P.A.; Yoburn, B.C. The relative potency of inverse opioid agonists and a neutral opioid antagonist in precipitated withdrawal and antagonism of analgesia and toxicity. J. Pharmacol. Exp. Ther. 2009, 330, 513–519. [Google Scholar] [CrossRef]
- Bilsky, E.J.; Giuvelis, D.; Osborn, M.D.; Dersch, C.M.; Xu, H.; Rothman, R.B. In vitro and in vivo assessment of m opioid receptor constitutive activity. Methods Enzymol. 2010, 484, 413–443. [Google Scholar]
- Sally, E.J.; Xu, H.; Dersch, C.M.; Hsin, L.W.; Chang, L.T.; Prisinzano, T.E.; Simpson, D.S.; Giuvelis, D.; Rice, K.C.; Jacobson, A.E.; et al. Identification of a novel “almost neutral” micro-opioid receptor antagonist in CHO cells expressing the cloned human mu-opioid receptor. Synapse 2010, 64, 280–288. [Google Scholar] [CrossRef]
- Marczak, E.D.; Jinsmaa, Y.; Li, T.; Bryant, S.D.; Tsuda, Y.; Okada, Y.; Lazarus, L.H. [N-allyl-Dmt1]-endomorphins are μ-opioid receptor antagonists lacking inverse agonist properties. J. Pharmacol. Exp. Ther. 2007, 323, 374–380. [Google Scholar] [CrossRef]
- Tsuruda, P.R.; Vickery, R.G.; Long, D.D.; Armstrong, S.R.; Beattie, D.T. The in vitro pharmacological profile of TD-1211, a neutral opioid receptor antagonist. Naunyn. Schmied. Arch. Pharm. 2013, 386, 479–491. [Google Scholar] [CrossRef]
- Dutta, R.; Lunzer, M.M.; Auger, J.L.; Akgün, E.; Portoghese, P.S.; Binstadt, B.A. A bivalent compound targeting CCR5 and the mu opioid receptor treats inflammatory arthritis pain in mice without inducing pharmacologic tolerance. Arthritis. Res. Ther. 2018, 20, 154. [Google Scholar] [CrossRef]
- Hirayama, S.; Fujii, H. δ Opioid Receptor Inverse Agonists and Their In Vivo Pharmacological Effects. Curr. Top. Med. Chem. 2020, 20, 2889–2902. [Google Scholar] [CrossRef]
- Iwamatsu, C.; Hayakawa, D.; Kono, T.; Honjo, A.; Ishizaki, S.; Hirayama, S.; Gouda, H.; Fujii, H. Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives. Molecules 2020, 25, 3792. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Prather, P.L. Chronic agonist treatment converts antagonists into inverse agonists at delta-opioid receptors. J. Pharmacol. Exp. Ther. 2002, 302, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Muneta-Arrate, I.; Diez-Alarcia, R.; Horrillo, I.; Meana, J.J. Pimavanserin exhibits serotonin 5-HT2A receptor inverse agonism for Gαi1- and neutral antagonism for Gαq/11-proteins in human brain cortex. Eur. Neuropsychopharmacol. 2020, 36, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J.; Millan, M.J. Inability of an opioid antagonist lacking negative intrinsic activity to induce opioid receptor up-regulation in vivo. Br. J. Pharmacol. 1991, 102, 883–886. [Google Scholar] [CrossRef][Green Version]
- Piñeyro, G.; Azzi, M.; deLean, A.; Schiller, P.W.; Bouvier, M. Reciprocal regulation of agonist and inverse agonist signaling efficacy upon short-term treatment of the human delta-opioid receptor with an inverse agonist. Mol. Pharmacol. 2005, 67, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Jeske, N.A. Dynamic Opioid Receptor Regulation in the Periphery. Mol. Pharmacol. 2019, 95, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Tawfik, V.L.; Wang, D.; Sypek, E.I.; Low, S.A.; Dickinson, J.R.; Sotoudeh, C.; Clark, J.D.; Barres, B.A.; Bohlen, C.J.; et al. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 2017, 23, 164–173. [Google Scholar] [CrossRef]
- Walwyn, W.M.; Chen, C.; Kim, H.; Minasyan, A.; Ennes, H.S.; McRoberts, J.A.; Marvizon, J.C.G. Sustained suppression of hyperalgesia during latent sensitization by mu-, delta-, and kappa-opioid receptors and 2A-adrenergic receptors: Role of constitutive activity. Neurobiol. Dis. 2016, 36, 204–221. [Google Scholar]
- Perry, D.C.; Rosenbaum, J.S.; Kurowski, M.; Sadée, W. 3H-Etorphine Receptor Binding In Vivo: Small Fractional Occupancy Elicits Analgesia. Mol. Pharmacol. 1982, 21, 272–279. [Google Scholar]
- Rosenbaum, J.S.; Holford, N.H.G.; Sadée, W. In Vivo Receptor Binding of Opioid Drugs at the µ Site. J. Pharmacol. Exp. Ther. 1985, 233, 735–740. [Google Scholar]
- Monroe Butler, P.M.; Barash, J.A.; Casaletto, K.B.; Cotter, D.L.; La Joie, R.; Geschwind, M.D.; Rosen, H.J.; Kramer, J.H.; Miller, B.L. An Opioid-Related Amnestic Syndrome with Persistent Effects on Hippocampal Structure and Function. J. Neuropsychiatry Clin. Neurosci. 2019, 31, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.D.; Gordon, N.C.; Bornstein, J.C.; Fields, H.L. Analgesic responses to morphine and placebo in individuals with postoperative pain. Proc. Natl. Acad. Sci. USA 1981, 10, 379–389. [Google Scholar] [CrossRef]
- Li, J.-X.; Mc Mahon, L.R.; France, C.P. Comparison of naltrexone, 6α-naltrexol, and 6β-naltrexol in morphine-dependent and in non-dependent rhesus monkeys. Psychopharmacology 2007, 195, 479–486. [Google Scholar] [CrossRef]
- Porter, S.J.; Somogyi, A.A.; White, J.M. In vivo and in vitro potency studies of 6β-naltrexol, the major human metabolite of naltrexone. Addict. Biol. 2002, 7, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.S.; Holford, N.H.G.; Richard, M.L.; Aman, R.A.; Sadee, W. Discrimination of three types of opioid binding sites in rat brain in vivo. Mol. Pharmacol. 1984, 25, 242–248. [Google Scholar] [PubMed]
- Ko, M.C.; Divin, M.F.; Lee, H.; Woods, J.H.; Traynor, J.R. Differential in Vivo Potencies of Naltrexone and 6β-Naltrexol in the Monkey. J. Pharmacol. Exp. Ther. 2006, 316, 772–779. [Google Scholar] [CrossRef]
- Perez, D.M.; Karnik, S.S. Multiple signaling states of G-protein-coupled receptors. Pharmacol. Rev. 2005, 57, 147–161. [Google Scholar] [CrossRef]
- Schafer, C.T.; Fay, J.F.; Janz, J.M.; Farrens, D.L. Decay of an active GPCR: Conformational dynamics govern agonist rebinding and persistence of an active, yet empty, receptor state. Proc. Natl. Acad. Sci. USA 2016, 113, 11961–11966. [Google Scholar] [CrossRef]
- Piñeyro, G.; Azzi, M.; deLéan, A.; Schiller, P.A.; Bouvier, M. Short-term inverse agonist treatment induces reciprocal changes in delta-opioid agonist and inverse agonist binding capacity. Mol. Pharmacol. 2001, 67, 816–827. [Google Scholar]
- Safa, A.; Lau, A.R.; Aten, S.; Schilling, K.; Bales, K.L.; Miller, V.; Fitzgerald, J.; Chen, M.; Hill, K.; Dzwigalski, K.; et al. Pharmacological prevention of neonatal opioid withdrawal in a pregnant guinea pig model. Front. Pharmacol. 2021, 11, 613328. [Google Scholar] [CrossRef]
- Yancey-Wrona, J.; Dallaire, B.; Bilsky, E.J.; Bath, B.; Burkart, J.; Wenster, L.; Magiera, D.; Yang, X.; Phelps, M.A.; Sadee, W. 6β-Naltrexol, a peripherally selective opioid antagonist that inhibits morphine-induced slowing of gastrointestinal transit: An exploratory study. Pain Med. 2011, 12, 1727–1737. [Google Scholar] [CrossRef]
- Wang, D.; Sun, X.; Sadee, W. 2007 Different effects of opioid antagonists on mu, delta, and kappa opioid receptors with and without agonist pretreatment. J. Pharmacol. Exp. Ther. 2011, 321, 544–552. [Google Scholar] [CrossRef]
- Divin, M.F.; Holden Ko, M.C.; Traynor, J.R. Comparison of the opioid receptor antagonist properties of naltrexone and 6beta-naltrexol in morphine-naïve and morphine-dependent mice. Eur. J. Pharmocol. 2008, 583, 48–55. [Google Scholar] [CrossRef]
- Fujimoto, J.M.; Roerig, S.; Wang, R.I.; Chatterjie, N.; Inturrisi, C.E. Narcotic antagonist activity of several metabolites of naloxone and naltrexone tested in morphine dependent mice (38558). Proc. Soc. Exp. Biol. Med. 1975, 148, 443–448. [Google Scholar] [CrossRef]
- Yancey-Wrona, J.E.; Raymond, T.J.; Mercer, H.K.; Sadee, W.; Bilsky, E.J. 6β-Naltrexol preferentially antagonizes opioid effects on gastrointestinal transit compared to antinociception in mice. Life Sci. 2009, 85, 413–420. [Google Scholar] [CrossRef]
- Oberdick, J.; Ling, Y.; Phelps, M.A.; Yudovich, M.S.; Schilling, K.; Sadee, W. Preferential delivery of an opioid antagonist to the fetal brain in pregnant mice. J. Pharmacol. Exp. Ther. 2016, 358, 22–30. [Google Scholar] [CrossRef]
- Perry, D.C.; Mullis, K.B.; Oie, S.; Sadée, W. Opiate Antagonist Receptor Binding In Vivo: Evidence for a New Receptor Binding Model. Brain Res. 1980, 199, 49–61. [Google Scholar] [CrossRef]
- Toljan, K.; Vrooman, B. Low-Dose Naltrexone (LDN)-Review of Therapeutic Utilization. Med. Sci. 2018, 6, 82. [Google Scholar] [CrossRef]
- Leri, F.; Burns, L.H. Ultra-low-dose naltrexone reduces the rewarding potency of oxycodone and relapse vulnerability in rats. Pharm. Biochem. Behav. 2018, 82, 252–262. [Google Scholar] [CrossRef]
- Mannelli, P.; Patkar, A.A.; Peindl, K.; Gorelick, D.A.; Wu, L.-T.; Gottheil, E. Very low dose naltrexone addition in opioid detoxification: A randomized, controlled trial. Addict. Biol. 2009, 14, 204–213. [Google Scholar] [CrossRef]
- Comer, S.D.; Mannelli, P.; Alam, D.; Douaihy, A.; Nangia, N.; Akerman, S.C.; Zavod, A.; Silverman, B.L.; Sullivan, M.A. Transition of Patients with Opioid Use Disorder from Buprenorphine to Extended Release Naltrexone: A Randomized Clinical Trial Assessing Two Transition Regimens. Am. J. Addict. 2020, 29, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.L.; Lavoie, N.R.; Raehal, K.M.; Sadee, W.; Bilsky, E.J. In vivo studies with 6β-naltrexamide, a peripherally selective opioid antagonist that has less inverse agonist activity than naltrexone and naloxone. FASEB J. 2004, 18, A586. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadee, W.; McKew, J.C. Ligand-Free Signaling of G-Protein-Coupled Receptors: Relevance to μ Opioid Receptors in Analgesia and Addiction. Molecules 2022, 27, 5826. https://doi.org/10.3390/molecules27185826
Sadee W, McKew JC. Ligand-Free Signaling of G-Protein-Coupled Receptors: Relevance to μ Opioid Receptors in Analgesia and Addiction. Molecules. 2022; 27(18):5826. https://doi.org/10.3390/molecules27185826
Chicago/Turabian StyleSadee, Wolfgang, and John C. McKew. 2022. "Ligand-Free Signaling of G-Protein-Coupled Receptors: Relevance to μ Opioid Receptors in Analgesia and Addiction" Molecules 27, no. 18: 5826. https://doi.org/10.3390/molecules27185826
APA StyleSadee, W., & McKew, J. C. (2022). Ligand-Free Signaling of G-Protein-Coupled Receptors: Relevance to μ Opioid Receptors in Analgesia and Addiction. Molecules, 27(18), 5826. https://doi.org/10.3390/molecules27185826