
Novel N-normetazocine Derivatives with Opioid Agonist/Sigma-1 Receptor Antagonist Profile as Potential Analgesics in Inflammatory Pain

 , , ,
, , ,  , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
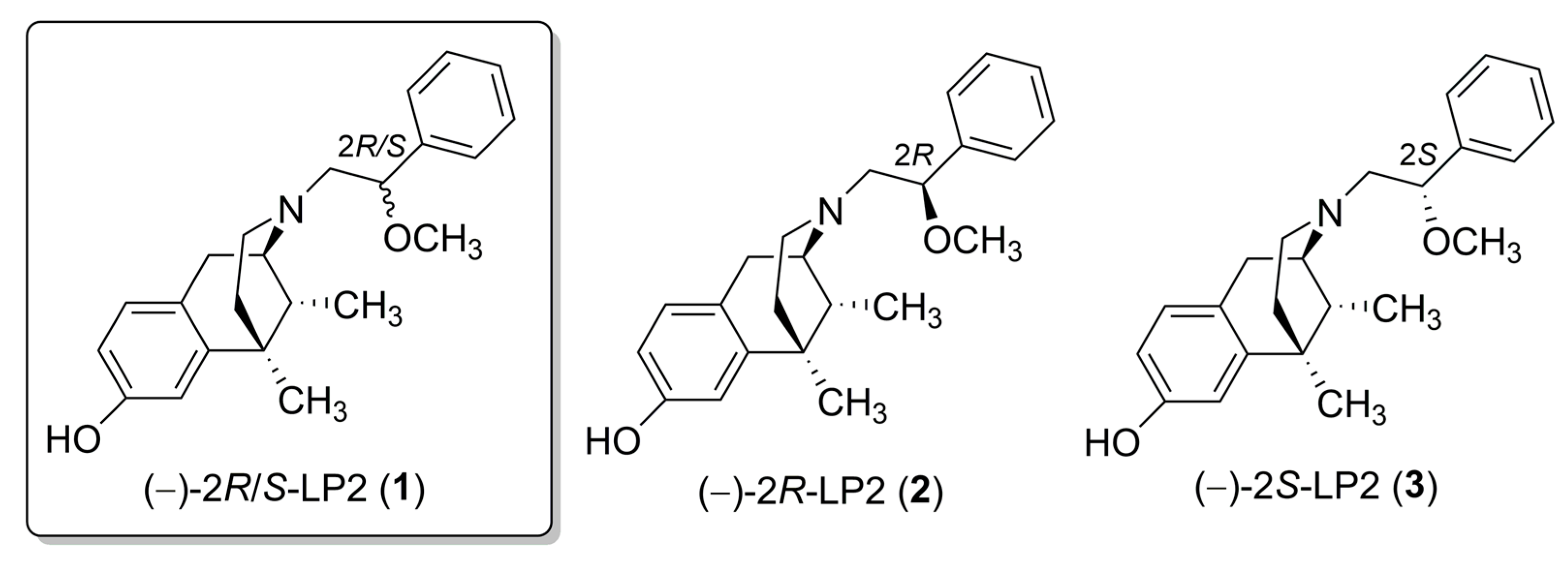
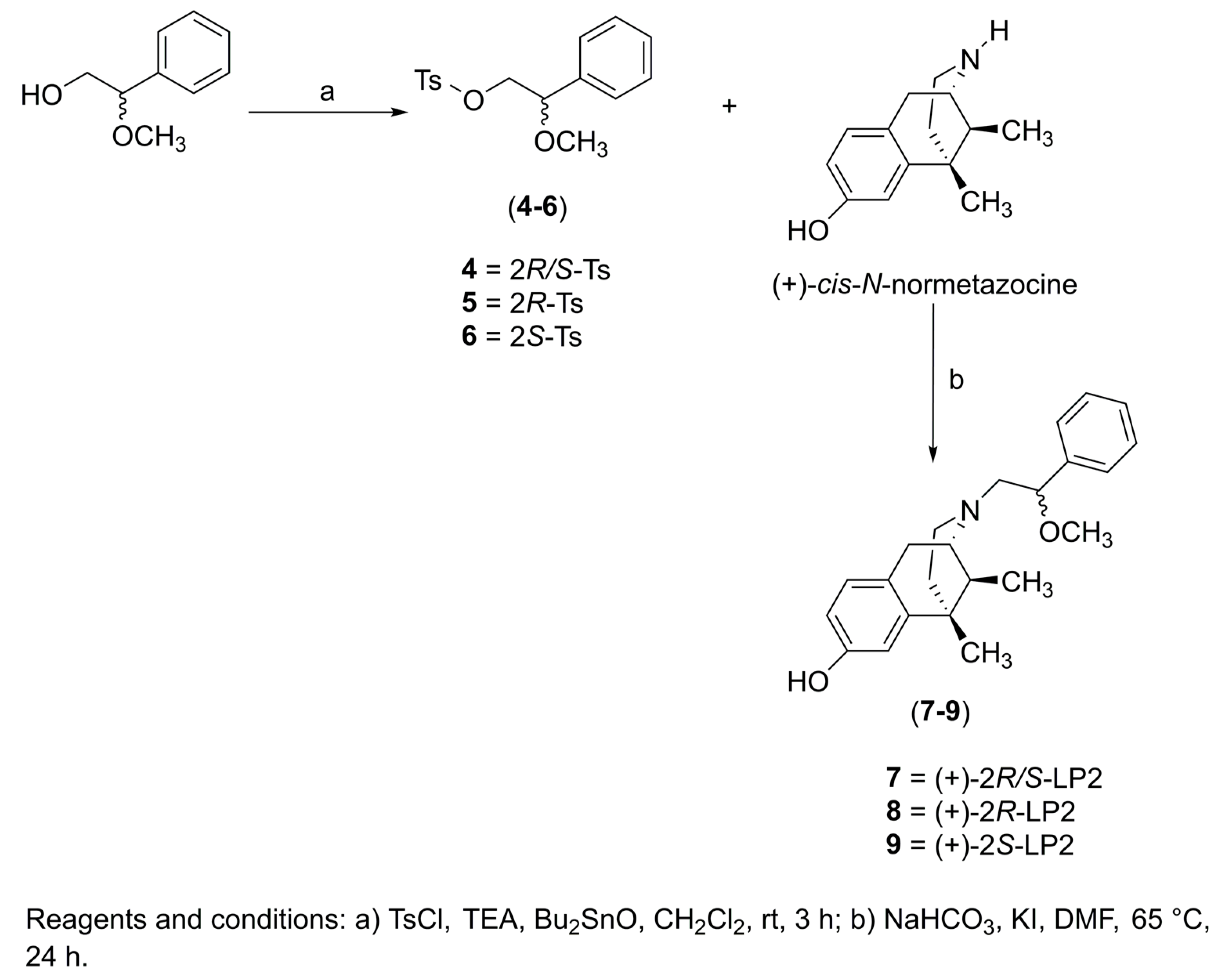
2.1. Chemistry
2.2. Radioligand Binding Assays
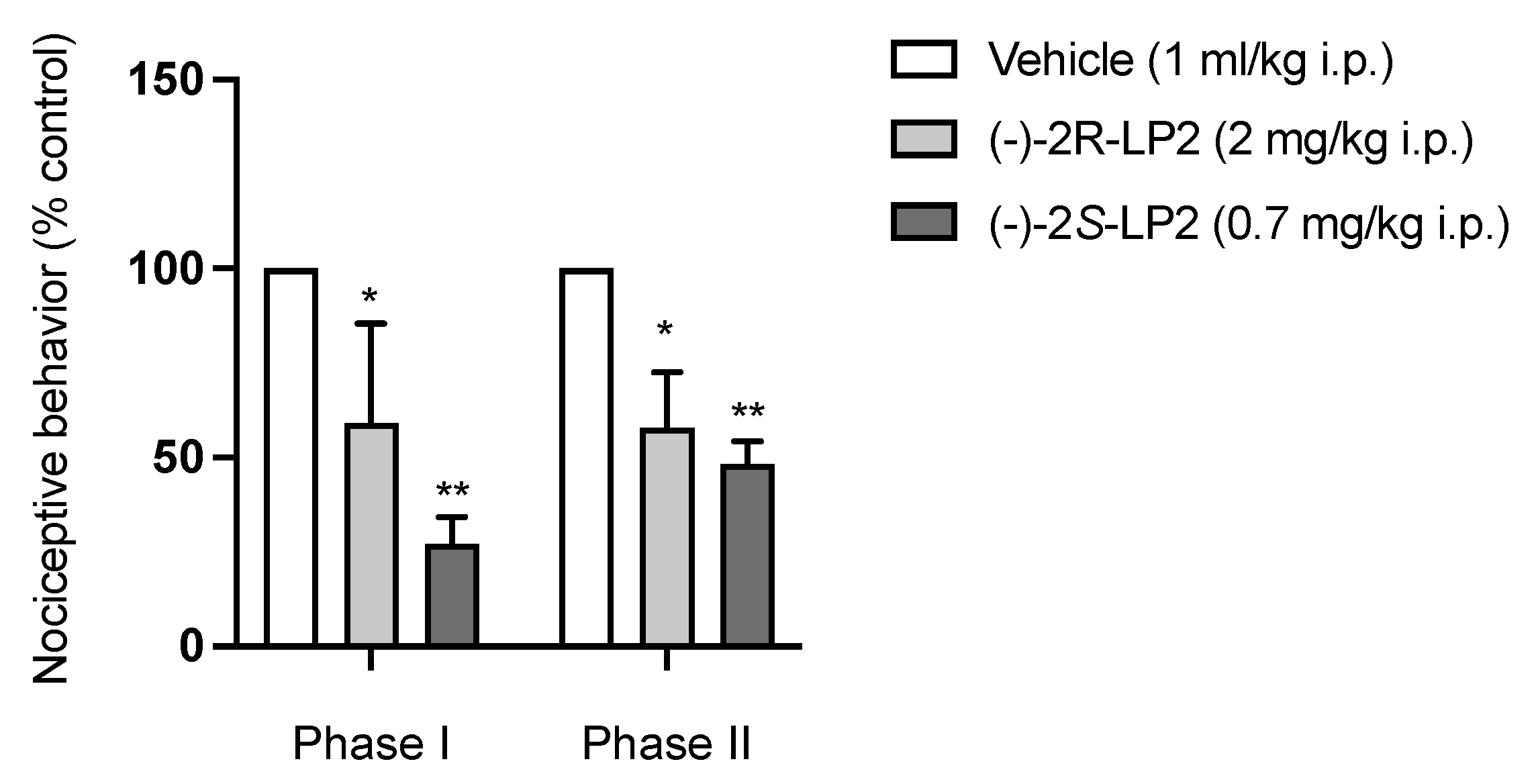
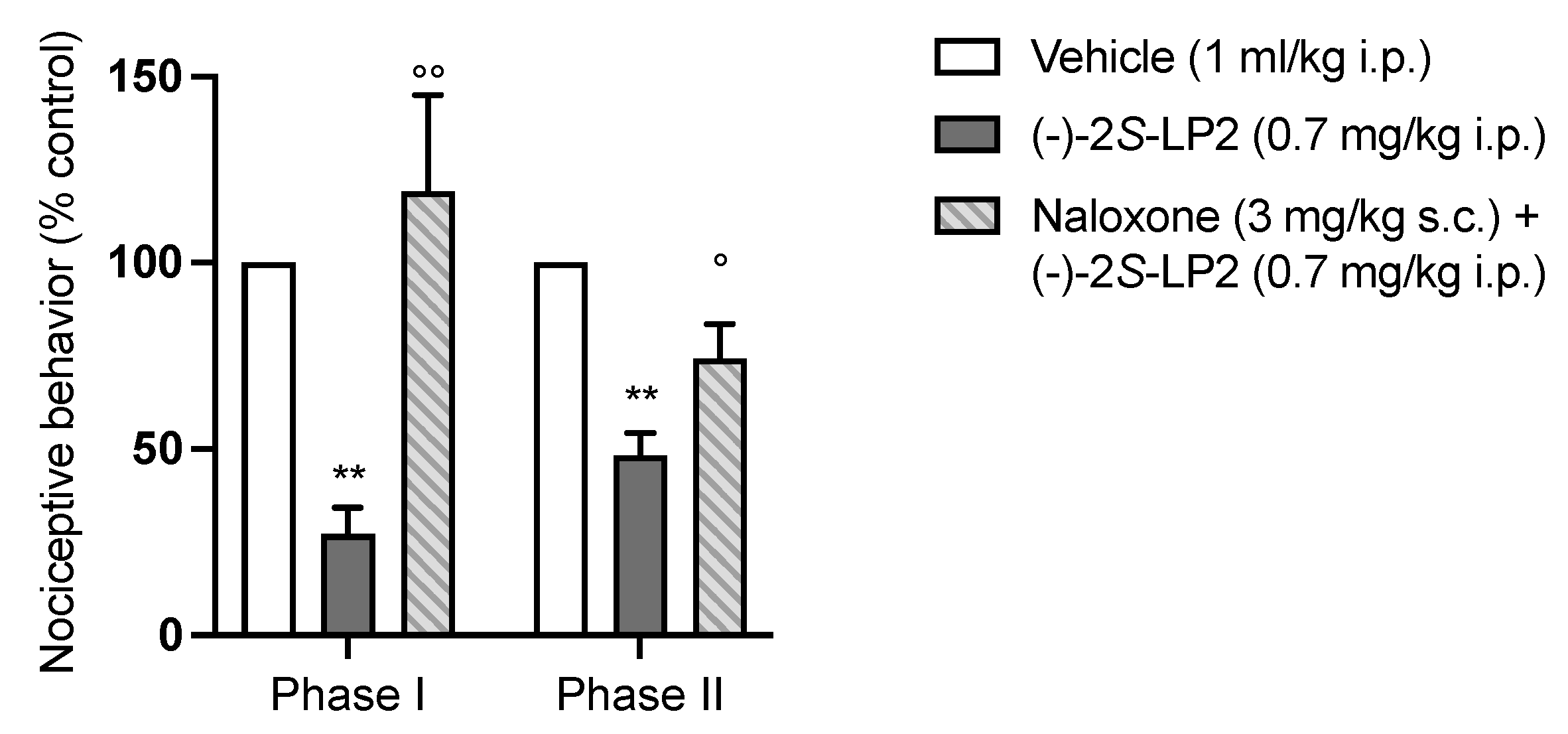
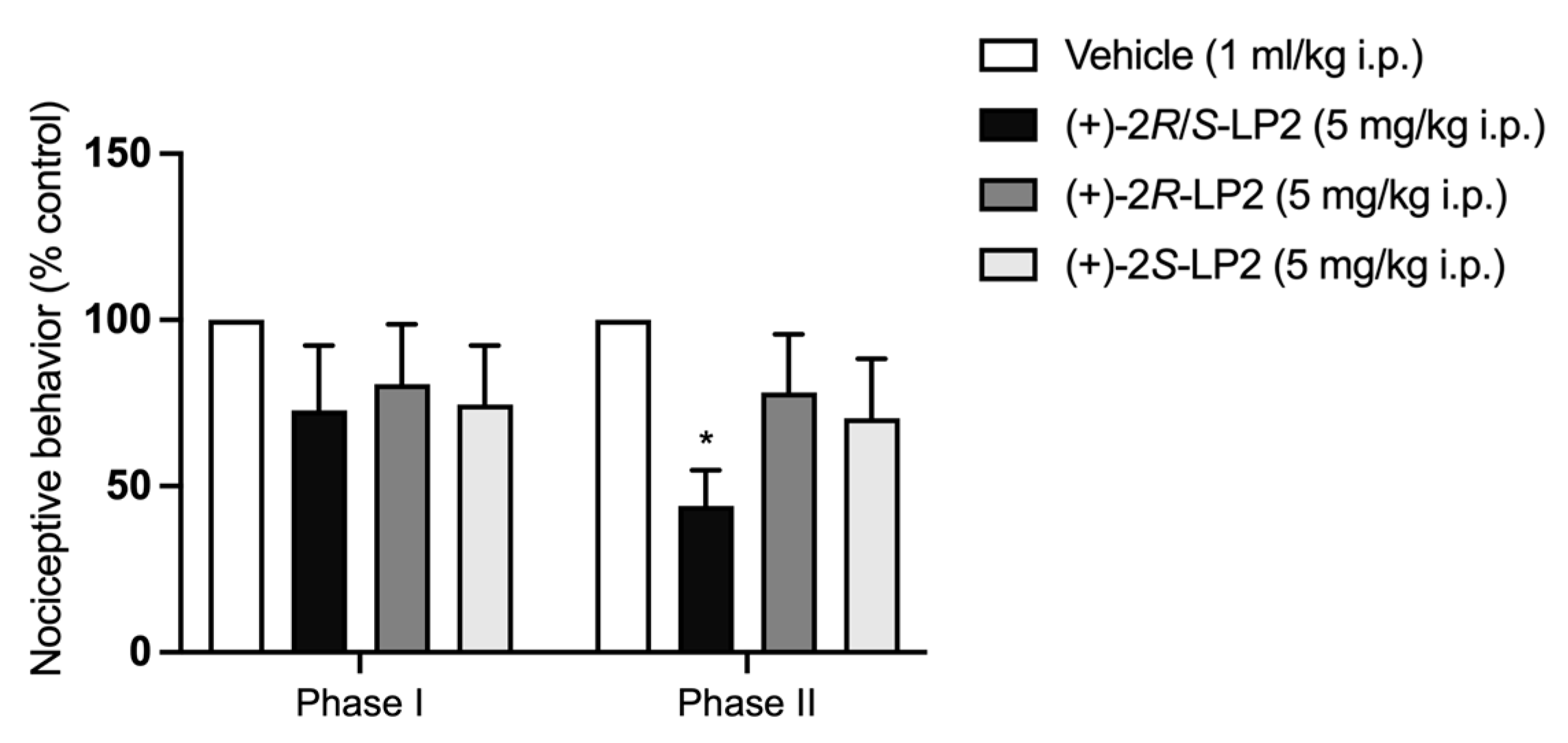
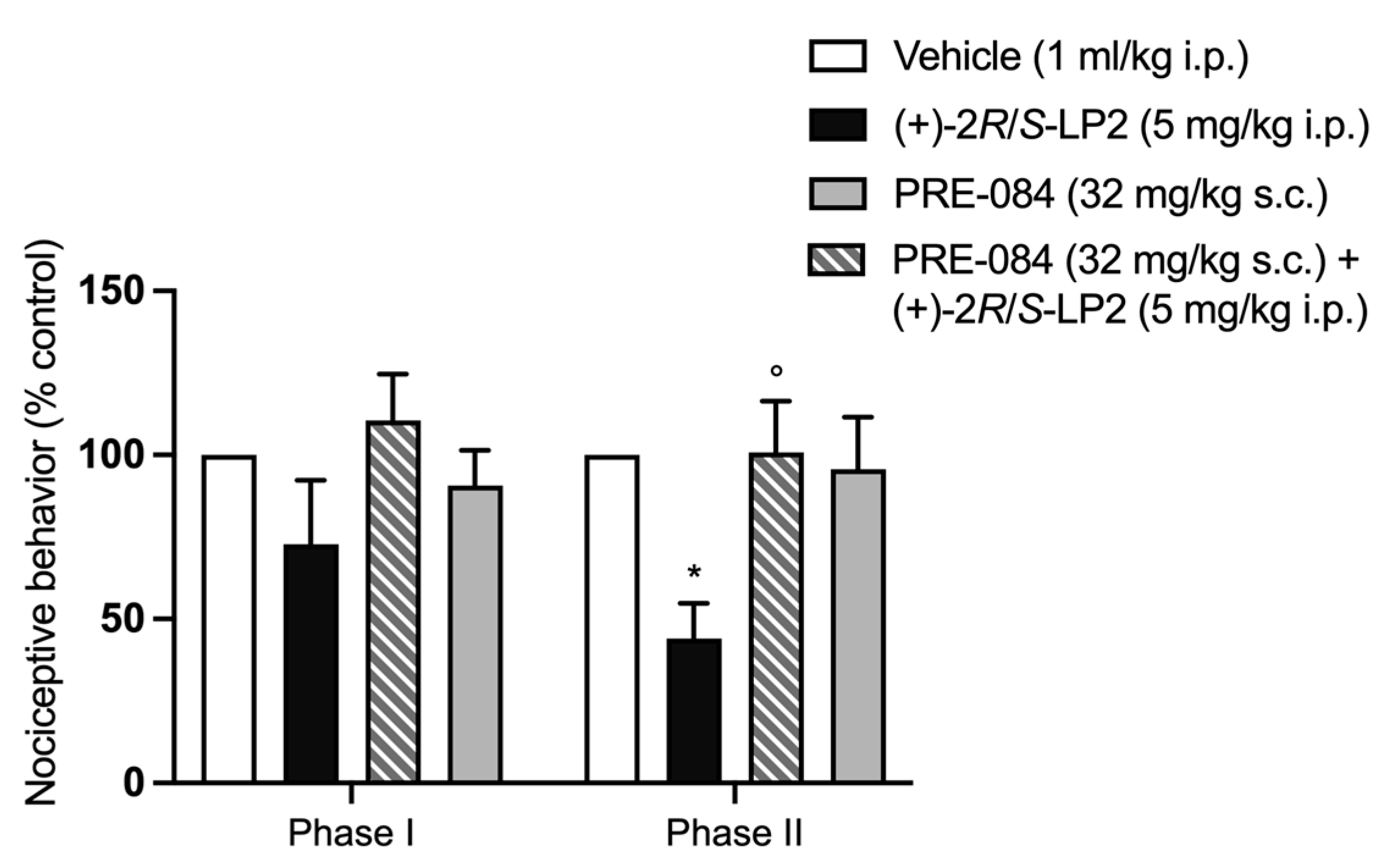
2.3. Formalin Test
3. Materials and Methods
3.1. General Remarks
3.2. General Procedure for the Synthesis of Compounds 7−9
- (2S,6S,11S)-3-(2-methoxy-2-phenylethyl)-6,11-dimethyl-1,2,3,4,5,6-hexahydro-2,6-methanobenzo[d]azocin-8-ol (7)
- (2S,6S,11S)-3-((R)-2-methoxy-2-phenylethyl)-6,11-dimethyl-1,2,3,4,5,6-hexahydro-2,6-methanobenzo[d]azocin-8-ol (8)
- (2S,6S,11S)-3-((S)-2-methoxy-2-phenylethyl)-6,11-dimethyl-1,2,3,4,5,6-hexahydro-2,6-methanobenzo[d]azocin-8-ol (9)
3.3. Radioligand Binding Assays
Animals
3.4. Radioligand Binding Assays for σ1R and σ2R
3.4.1. Materials
3.4.2. Preparation of Membrane Homogenates from Pig Brain
3.4.3. Preparation of Membrane Homogenates from Rat Liver
3.4.4. Protein Determination
3.4.5. σ1. R Ligand Binding Assays
3.4.6. σ2. R Ligand Binding Assays
3.4.7. Data Analysis
3.5. Radioligand Binding Assays for Opioid Receptors
3.5.1. Materials
3.5.2. Preparation of Membrane Homogenates from Sprague-Dawley Rat Brains for MOR and DOR Binding Assays or Guinea Pig Brains for KOR Binding Assays
3.5.3. Protein Determination
3.5.4. Opioid Receptor Ligand Binding Assays
3.5.5. Data Analysis
3.6. In Vivo Pharmacology
3.6.1. Animals
3.6.2. Behavioral Experiment
3.6.3. Drugs
3.6.4. Mouse Formalin Test
3.6.5. Data Analysis
4. Conclusions
5. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas. A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Scadding, G.K. Topical nasal lysine aspirin in aspirin-sensitive and aspirin-tolerant chronic rhinosinusitis with nasal polyposis. Expert Rev. Clin. Immunol. 2014, 10, 657–665. [Google Scholar] [CrossRef]
- Vicario, N.; Parenti, R.; Arico, G.; Turnaturi, R.; Scoto, G.M.; Chiechio, S.; Parenti, C. Repeated activation of delta opioid receptors counteracts nerve injury-induced TNF-alpha up-regulation in the sciatic nerve of rats with neuropathic pain: A possible correlation with delta opioid receptors-mediated antiallodynic effect. Mol. Pain 2016, 12, 1744806916667949. [Google Scholar] [CrossRef]
- Ji, R.R.; Zhang, Q.; Law, P.Y.; Low, H.H.; Elde, R.; Hokfelt, T. Expression of mu-, delta-, and kappa-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J. Neurosci. 1995, 15, 8156–8166. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Vekariya, R.H.; Ananthan, S.; Streicher, J.M. A Novel Mu-Delta Opioid Agonist Demonstrates Enhanced Efficacy with Reduced Tolerance and Dependence in Mouse Neuropathic Pain Models. J. Pain 2020, 21, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Nastase, A.F.; Griggs, N.W.; Anand, J.P.; Fernandez, T.J.; Harland, A.A.; Trask, T.J.; Jutkiewicz, E.M.; Traynor, J.R.; Mosberg, H.I. Synthesis and Pharmacological Evaluation of Novel C- 8 Substituted Tetrahydroquinolines as Balanced-Affinity Mu/Delta Opioid Ligands for the Treatment of Pain. ACS Chem. Neurosci. 2018, 9, 1840–1848. [Google Scholar] [CrossRef]
- Podolsky, A.T.; Sandweiss, A.; Hu, J.; Bilsky, E.J.; Cain, J.P.; Kumirov, V.K.; Lee, Y.S.; Hruby, V.J.; Vardanyan, R.S.; Vanderah, T.W. Novel fentanyl-based dual μ/δ-opioid agonists for the treatment of acute and chronic pain. Life Sci. 2013, 93, 1010–1016. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Prezzavento, O.; Arena, E.; Aricò, G.; Georgoussi, Z.; Parenti, R.; Cantarella, G.; Parenti, C. Development of novel LP1-based analogues with enhanced delta opioid receptor profile. Bioorg. Med. Chem. 2017, 25, 4745–4752. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Montenegro, L.; Caraci, F.; Chiechio, S.; Parenti, C. Simultaneous targeting of MOR/DOR: A useful strategy for inflammatory pain modulation. Eur. J. Pharmacol. 2019, 847, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous Activation of Mu and Delta Opioid Receptors Reduces Allodynia and Astrocytic Connexin 43 in an Animal Model of Neuropathic Pain. Mol. Neurobiol. 2019, 56, 7338–7354. [Google Scholar] [CrossRef]
- Vicario, N.; Denaro, S.; Turnaturi, R.; Longhitano, L.; Spitale, F.M.; Spoto, S.; Marrazzo, A.; Zappalà, A.; Tibullo, D.; Li Volti, G.; et al. Mu and Delta Opioid Receptor Targeting Reduces Connexin 43-Based Heterocellular Coupling during Neuropathic Pain. Int. J. Mol. Sci. 2022, 23, 5864. [Google Scholar] [CrossRef] [PubMed]
- Merlos, M.; Burgueño, J.; Portillo-Salido, E.; Plata-Salamán, C.L.; Vela, J.M. Pharmacological Modulation of the Sigma 1 Receptor and the Treatment of Pain. Adv. Exp. Med. Biol. 2017, 964, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptors. Pharmacol. Ther. 2009, 124, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Campisi, A.; Parenti, C.; Ronsisvalle, S.; Aricò, G.; Arena, E.; Pistolozzi, M.; Scoto, G.M.; Bertucci, C.; Vanella, A.; et al. Synthesis and resolution of cis-(+/-)-methyl (1R,2S/1S,2R)-2-[(4-hydroxy-4-phenylpiperidin-1-yl)methyl]-1-(4-methylphenyl)cyclopropanecarboxylate [(+/-)-PPCC)]: New sigma receptor ligands with neuroprotective effect. J. Med. Chem. 2010, 53, 5881–5885. [Google Scholar] [CrossRef]
- Alonso, G.; Phan, V.; Guillemain, I.; Saunier, M.; Legrand, A.; Anoal, M.; Maurice, T. Immunocytochemical localization of the sigma-1 receptor in the adult rat central nervous system. Neuroscience 2000, 97, 155–170. [Google Scholar] [CrossRef]
- Morales-Lázaro, S.L.; González-Ramírez, R.; Rosenbaum, T. Molecular Interplay Between the Sigma-1 Receptor, Steroids, and Ion Channels. Front. Pharmacol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Ortíz-Rentería, M.; Juárez-Contreras, R.; González-Ramírez, R.; Islas, L.D.; Sierra-Ramírez, F.; Llorente, I.; Simon, S.A.; Hiriart, M.; Rosenbaum, T.; Morales-Lázaro, S.L. TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain. Proc. Natl. Acad. Sci. USA 2018, 115, E1657–E1666. [Google Scholar] [CrossRef]
- Chien, C.C.; Pasternak, G.W. Selective antagonism of opioid analgesia by a sigma system. J. Pharmacol. Exp. Ther. 1994, 271, 1583–1590. [Google Scholar] [PubMed]
- Zamanillo, D.; Romero, L.; Merlos, M.; Vela, J.M. Sigma 1 receptor: A new therapeutic target for pain. Eur. J. Pharmacol. 2013, 716, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Gris, G.; Merlos, M.; Vela, J.M.; Zamanillo, D.; Portillo-Salido, E. S1RA, a selective sigma-1 receptor antagonist, inhibits inflammatory pain in the carrageenan and complete Freund’s adjuvant models in mice. Behav. Pharmacol. 2014, 25, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Almansa, C.; Vela, J.M. Selective sigma-1 receptor antagonists for the treatment of pain. Future Med. Chem. 2014, 6, 1179–1199. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Maurice, T.; Su, T.P. Ca(2+) signaling via sigma(1)-receptors: Novel regulatory mechanism affecting intracellular Ca(2+) concentration. J. Pharmacol. Exp. Ther. 2000, 293, 788–798. [Google Scholar] [PubMed]
- Roh, D.H.; Choi, S.R.; Yoon, S.Y.; Kang, S.Y.; Moon, J.Y.; Kwon, S.G.; Han, H.J.; Beitz, A.J.; Lee, J.H. Spinal neuronal NOS activation mediates sigma-1 receptor-induced mechanical and thermal hypersensitivity in mice: Involvement of PKC-dependent GluN1 phosphorylation. Br. J. Pharmacol. 2011, 163, 1707–1720. [Google Scholar] [CrossRef]
- Tejada, M.A.; Montilla-Garcia, A.; Sanchez-Fernandez, C.; Entrena, J.M.; Perazzoli, G.; Baeyens, J.M.; Cobos, E.J. Sigma-1 receptor inhibition reverses acute inflammatory hyperalgesia in mice: Role of peripheral sigma-1 receptors. Psychopharmacology 2014, 231, 3855–3869. [Google Scholar] [CrossRef]
- Xu, Q.; Yaksh, T.L. A brief comparison of the pathophysiology of inflammatory versus neuropathic pain. Curr. Opin. Anaesthesiol. 2011, 24, 400–407. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Hayashi, T.; Mori, T.; Su, T.P. Sigma-1 receptor chaperones and diseases. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Cendán, C.M.; Pujalte, J.M.; Portillo-Salido, E.; Montoliu, L.; Baeyens, J.M. Formalin-induced pain is reduced in sigma(1) receptor knockout mice. Eur. J. Pharmacol. 2005, 511, 73–74. [Google Scholar] [CrossRef]
- González-Cano, R.; Merlos, M.; Baeyens, J.M.; Cendán, C.M. σ1 receptors are involved in the visceral pain induced by intracolonic administration of capsaicin in mice. Anesthesiology. 2013, 118, 691–700. [Google Scholar] [CrossRef]
- Turnaturi, R.; Montenegro, L.; Marrazzo, A.; Parenti, R.; Pasquinucci, L.; Parenti, C. Benzomorphan skeleton, a versatile scaffold for different targets: A comprehensive review. Eur. J. Med. Chem. 2018, 155, 492–502. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Turnaturi, R.; Calò, G.; Pappalardo, F.; Ferrari, F.; Russo, G.; Arena, E.; Montenegro, L.; Chiechio, S.; Prezzavento, O.; et al. (2S)-N-2-methoxy-2-phenylethyl-6,7-benzomorphan compound (2S-LP2): Discovery of a biased mu/delta opioid receptor agonist. Eur. J. Med. Chem. 2019, 168, 189–198. [Google Scholar] [CrossRef]
- Pathan, H.; Williams, J. Basic opioid pharmacology: An update. Br. J. Pain 2012, 6, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Carroll-Buatti, M.; Spiegel, K. The binding and analgesic properties of a sigma opiate, SKF 10,047. J. Pharmacol. Exp. Ther. 1981, 219, 192–198. [Google Scholar] [PubMed]
- Carroll, F.I.; Abraham, P.; Parham, K.; Bai, X.; Zhang, X.; Brine, G.A.; Mascarella, S.W.; Martin, B.R.; May, E.L.; Sauss, C.; et al. Enantiomeric N-substituted N-normetazocines: A comparative study of affinities at sigma, PCP, and mu opioid receptors. J. Med. Chem. 1992, 35, 2812–2818. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Arena, E.; Sánchez-Fernández, C.; Turnaturi, R.; Parenti, C.; Marrazzo, A.; Catalano, R.; Amata, E.; Pasquinucci, L.; Cobos, E.J. (+)-and (−)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/σ1 antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 2017, 125, 603–610. [Google Scholar] [CrossRef]
- Turnaturi, R.; Pasquinucci, L.; Chiechio, S.; Grasso, M.; Marrazzo, A.; Amata, E.; Dichiara, M.; Prezzavento, O.; Parenti, C. Exploiting the Power of Stereochemistry in Drug Action: 3-[(2S,6S,11S)-8-Hydroxy-6,11-dimethyl-1,4,5,6-tetrahydro-2,6-methano-3-benzazocin-3(2H)-yl]-N-phenylpropanamide as Potent Sigma-1 Receptor Antagonist. ACS Chem. Neurosci. 2020, 11, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Parenti, C.; Marrazzo, A.; Aricò, G.; Parenti, R.; Pasquinucci, L.; Ronsisvalle, S.; Ronsisvalle, G.; Scoto, G.M. The antagonistic effect of the sigma 1 receptor ligand (+)-MR200 on persistent pain induced by inflammation. Inflamm. Res. 2014, 63, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Kwon, Y.B.; Roh, D.H.; Yoon, S.Y.; Han, H.J.; Kim, K.W.; Beitz, A.J.; Lee, J.H. Intrathecal treatment with sigma1 receptor antagonists reduces formalin-induced phosphorylation of NMDA receptor subunit 1 and the second phase of formalin test in mice. Br. J. Pharmacol. 2006, 148, 490–498. [Google Scholar] [CrossRef]
- Cobos, E.J.; Entrena, J.M.; Nieto, F.R.; Cendán, C.M.; Del Pozo, E. Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr. Neuropharmacol. 2008, 6, 344–366. [Google Scholar] [CrossRef]
- Entrena, J.M.; Cobos, E.J.; Nieto, F.R.; Cendán, C.M.; Gris, G.; Del Pozo, E.; Zamanillo, D.; Baeyens, J.M. Sigma-1 receptors are essential for capsaicin-induced mechanical hypersensitivity: Studies with selective sigma-1 ligands and sigma-1 knockout mice. Pain 2009, 143, 252–261. [Google Scholar] [CrossRef]
- Puente, B.; Nadal, X.; Portillo-Salido, E.; Sánchez-Arroyos, R.; Ovalle, S.; Palacios, G.; Muro, A.; Romero, L.; Entrena, J.M.; Baeyens, J.M.; et al. Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain 2009, 145, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Zammataro, M.; Merlo, S.; Barresi, M.; Parenti, C.; Hu, H.; Sortino, M.A.; Chiechio, S. Chronic Treatment with Fluoxetine Induces Sex-Dependent Analgesic Effects and Modulates HDAC2 and mGlu2 Expression in Female Mice. Front. Pharmacol. 2017, 8, 743. [Google Scholar] [CrossRef] [PubMed]
- Coderre, T.J.; Melzack, R. The role of NMDA receptor-operated calcium channels in persistent nociception after formalin-induced tissue injury. J. Neurosci. 1992, 12, 3671–3675. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Bajorath, J. Modeling Tanimoto Similarity Value Distributions and Predicting Search Results. Mol. Inform. 2017, 36. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki (nM) ± SD a | ||||
|---|---|---|---|---|---|
| σ1R | σ2R | MOR | DOR | KOR | |
| (−)-2R/S-LP2 (1) | 171.00 ± 46.15 | 66.53 ± 10.92 | 1.08 ± 0.10 b | 6.61 ± 0.60 b | 15.22 ± 0.80 b |
| (−)-2R-LP2 (2) | 182.81 ± 20.96 | 66.68 ± 17.89 | 12.30 ± 0.42 c | 151.10 ± 0.60 c | 236.00 ± 0.83 c |
| (−)-2S-LP2 (3) | 112.72 ± 13.49 | 1013.91 ± 191.31 | 0.50 ± 0.03 c | 2.59 ± 0.05 c | 26.50 ± 0.44 c |
| (+)-2R/S-LP2 (7) | 26.61 ± 2.35 | 2393.32 ± 514.13 | 1136 ± 20 | 1035 ± 35 | 1393 ± 40 |
| (+)-2R-LP2 (8) | 47.86 ± 7.52 | 1914.26 ± 325.27 | 789 ± 30 | 1007 ± 25 | 1605 ± 70 |
| (+)-2S-LP2 (9) | 46.99 ± 8.82 | 2217.22 ± 417.17 | 1170 ± 30 | 1040 ± 40 | 1450 ± 29 |
| Haloperidol | 2.6 ± 0.4 | 77 ± 18 | - | - | - |
| (+)-Pentazocine | 4.3 ± 0.5 | 1465 ± 224 | - | - | - |
| DTG | 124 ± 19 | 18 ± 1 | - | - | - |
| BD-1063 | 14 ± 2.7 | 204 ± 31 | - | - | - |
| DAMGO | - | - | 1.16 ± 0.10 | - | - |
| U69,593 | - | - | - | - | 0.34 ± 0.10 |
| Naltrindole | - | - | - | 1.13 ± 0.10 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turnaturi, R.; Chiechio, S.; Pasquinucci, L.; Spoto, S.; Costanzo, G.; Dichiara, M.; Piana, S.; Grasso, M.; Amata, E.; Marrazzo, A.; et al. Novel N-normetazocine Derivatives with Opioid Agonist/Sigma-1 Receptor Antagonist Profile as Potential Analgesics in Inflammatory Pain. Molecules 2022, 27, 5135. https://doi.org/10.3390/molecules27165135
Turnaturi R, Chiechio S, Pasquinucci L, Spoto S, Costanzo G, Dichiara M, Piana S, Grasso M, Amata E, Marrazzo A, et al. Novel N-normetazocine Derivatives with Opioid Agonist/Sigma-1 Receptor Antagonist Profile as Potential Analgesics in Inflammatory Pain. Molecules. 2022; 27(16):5135. https://doi.org/10.3390/molecules27165135
Chicago/Turabian StyleTurnaturi, Rita, Santina Chiechio, Lorella Pasquinucci, Salvatore Spoto, Giuliana Costanzo, Maria Dichiara, Silvia Piana, Margherita Grasso, Emanuele Amata, Agostino Marrazzo, and et al. 2022. "Novel N-normetazocine Derivatives with Opioid Agonist/Sigma-1 Receptor Antagonist Profile as Potential Analgesics in Inflammatory Pain" Molecules 27, no. 16: 5135. https://doi.org/10.3390/molecules27165135
APA StyleTurnaturi, R., Chiechio, S., Pasquinucci, L., Spoto, S., Costanzo, G., Dichiara, M., Piana, S., Grasso, M., Amata, E., Marrazzo, A., & Parenti, C. (2022). Novel N-normetazocine Derivatives with Opioid Agonist/Sigma-1 Receptor Antagonist Profile as Potential Analgesics in Inflammatory Pain. Molecules, 27(16), 5135. https://doi.org/10.3390/molecules27165135