
Misuse of Cardiac Lipid upon Exposure to Toxic Trace Elements—A Focused Review


 , , ,
, , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Toxic Trace Elements and Cardiotoxicity
2. Toxic Trace Elements Alter the Lipid Levels in Circulation and the Cardiovascular Risk
3. Lipid Misuse and Cardiotoxicity
4. Epidemiological Evidence of Lipid Alterations after Exposure to Toxic Trace Elements
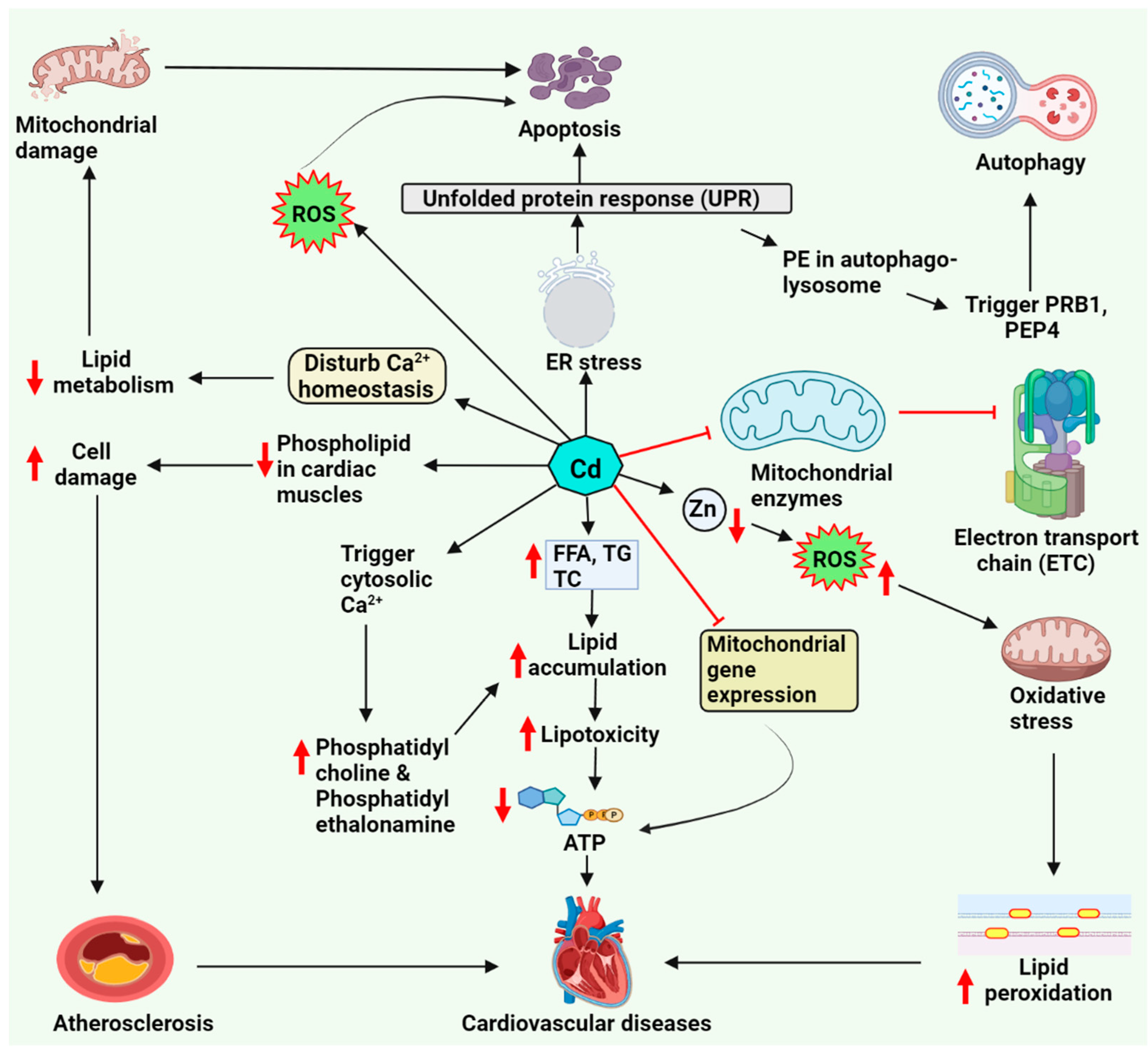
5. Cadmium-Associated Cardiotoxicity via Lipid Metabolism
5.1. Cardiac Lipotoxicity upon Cadmium Exposure
5.2. Role of Zinc and Calcium Homeostasis in Lipid Accumulation
5.3. Lipid-Driven Signaling Pathways upon Cadmium Exposure
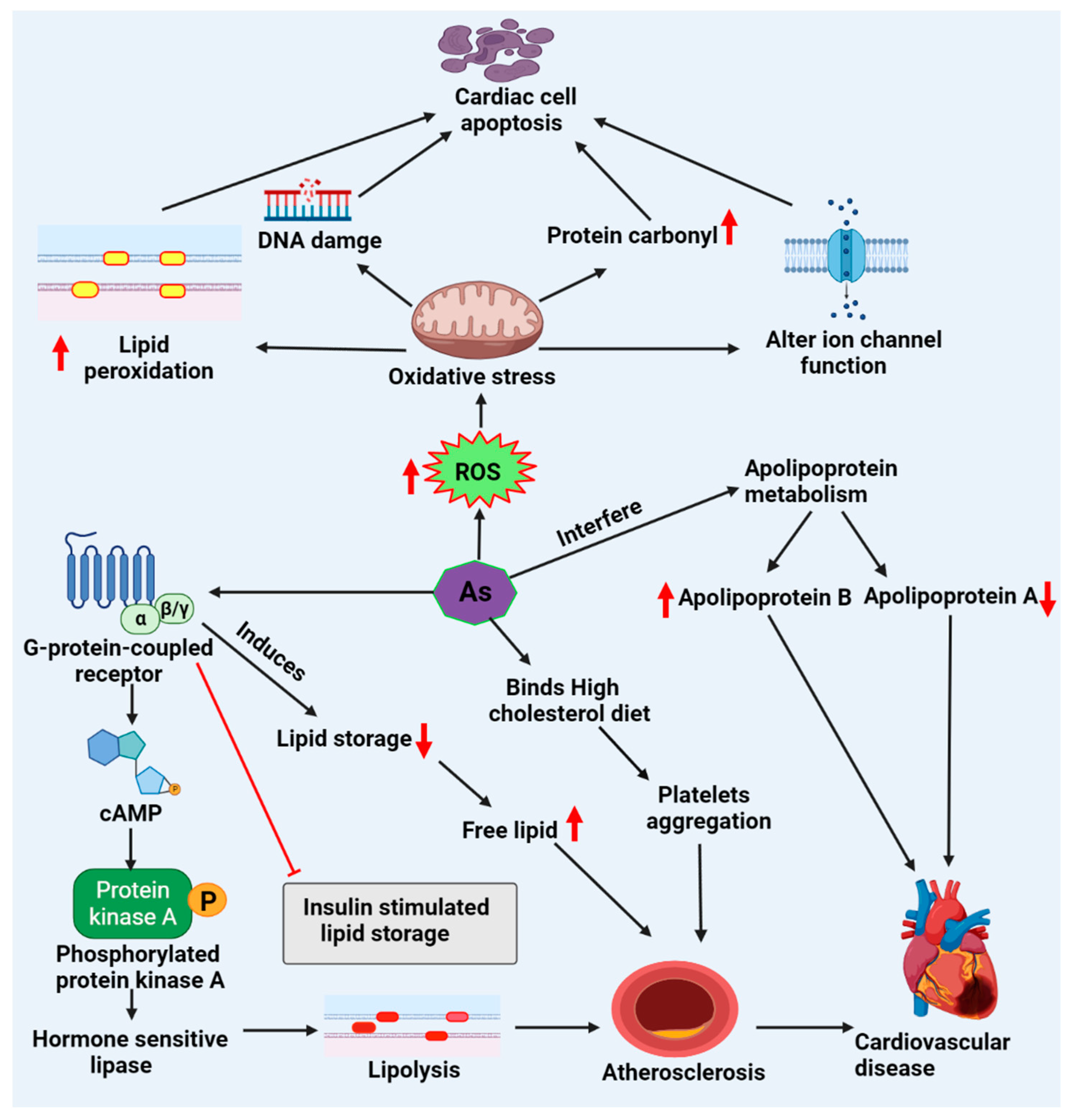
6. Arsenic-Associated Cardiotoxicity via Lipid Metabolism
6.1. Cardiac Lipotoxicity Evidence upon Arsenic Exposure
6.2. Lipid-Driven Signaling Pathways upon Arsenic Exposure
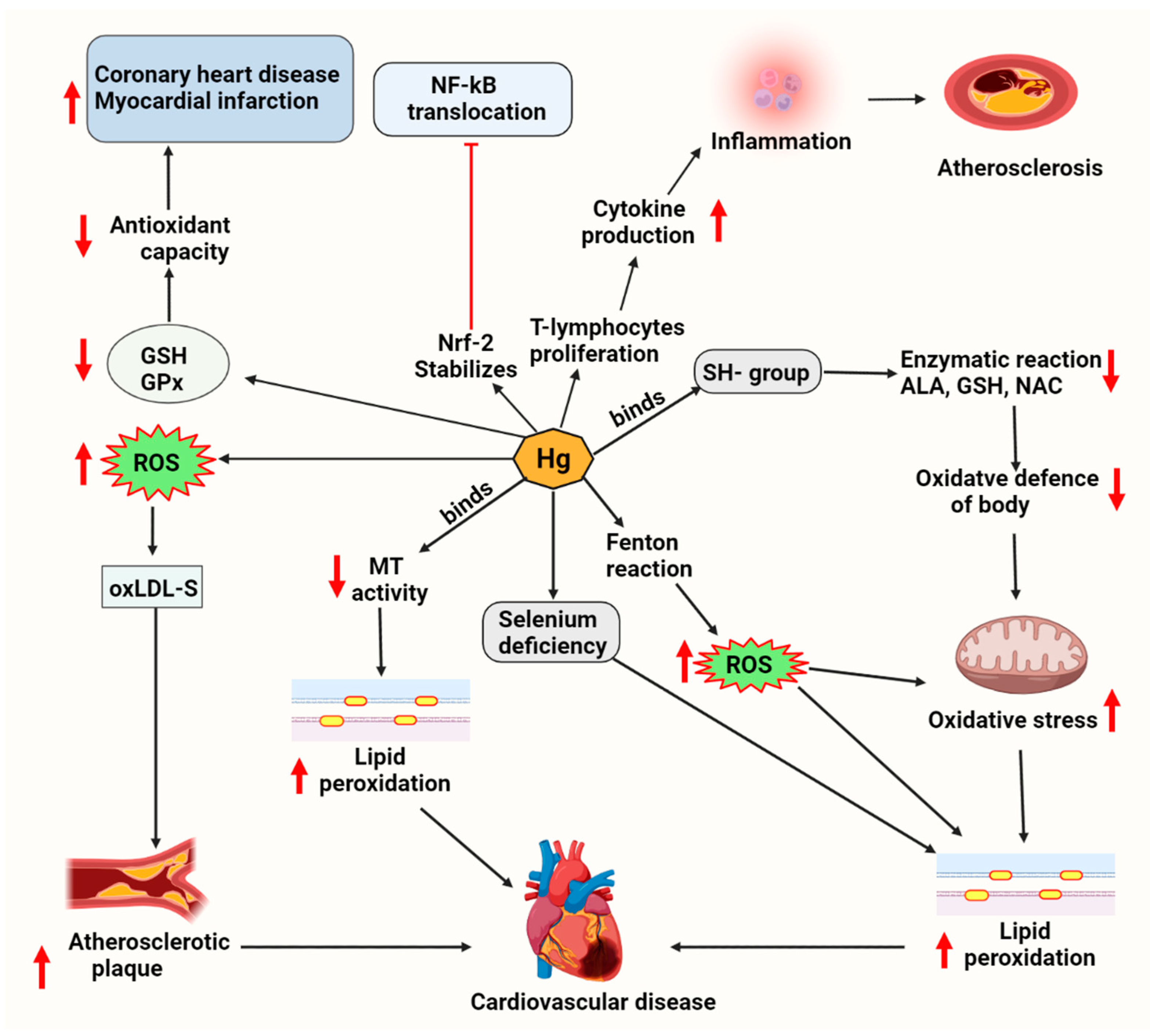
7. Mercury-Associated Cardiotoxicity via Lipid Metabolism
7.1. Cardiac Lipotoxicity Evidence upon Mercury Exposure
7.2. Lipid-Driven Signaling Pathways upon Mercury Exposure
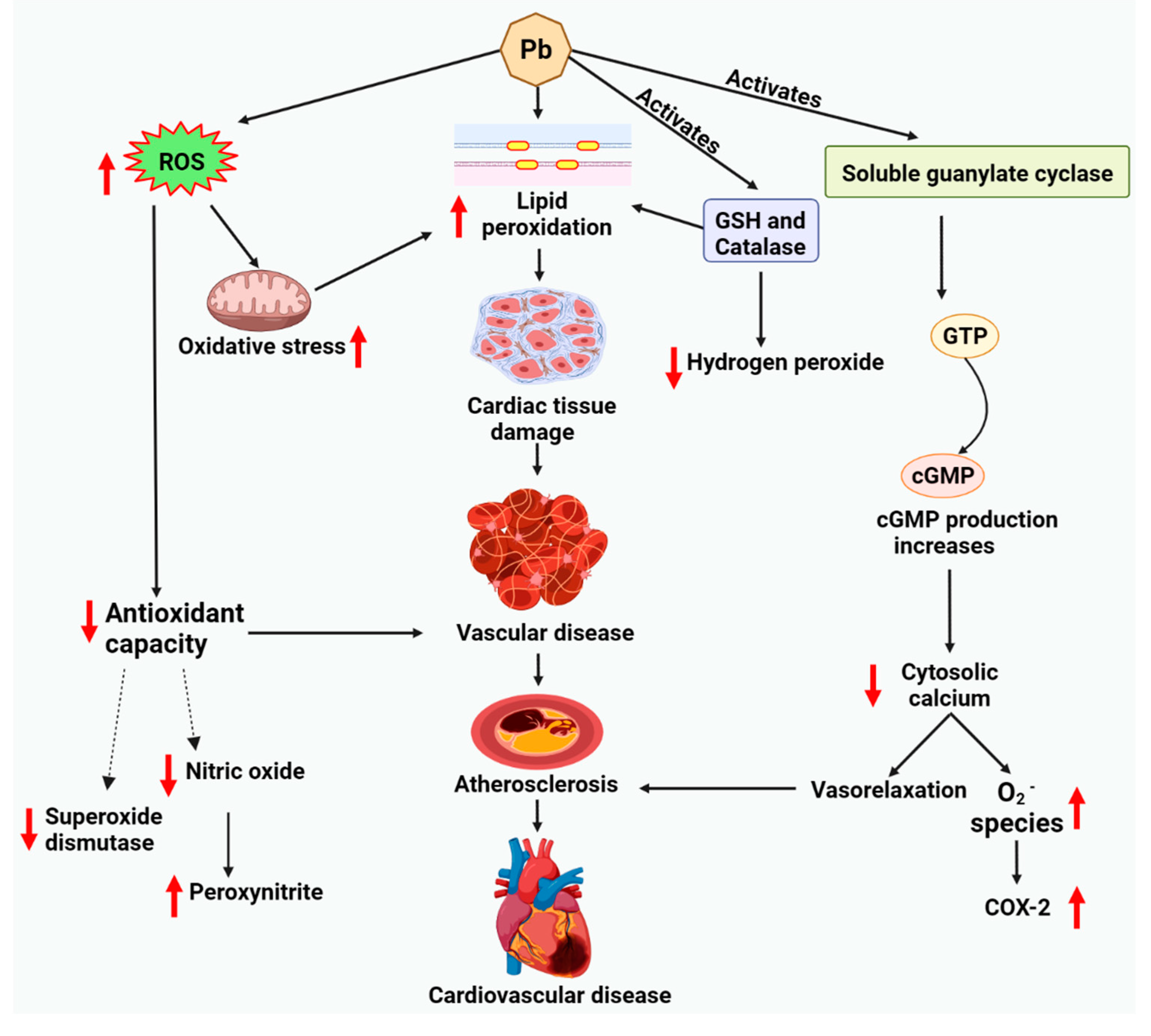
8. Lead-Associated Cardiotoxicity via Lipid Metabolism
8.1. Cardiac Lipotoxicity Evidence upon Lead Exposure
8.2. Lipid-Driven Signaling Pathways upon Lead Exposure
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duruibe, J.O.; Ogwuegbu, M.; Egwurugwu, J. Heavy metal pollution and human biotoxic effects. Int. J. Phys. Sci. 2007, 2, 112–118. [Google Scholar]
- Mukherjee, A.G.; Wanjari, U.R.; Renu, K.; Vellingiri, B.; Gopalakrishnan, A.V. Heavy metal and metalloid-induced reproductive toxicity. Environ. Toxicol. Pharmacol. 2022, 92, 103859. [Google Scholar] [CrossRef]
- Goutam Mukherjee, A.; Ramesh Wanjari, U.; Eladl, M.A.; El-Sherbiny, M.; Elsherbini, D.M.A.; Sukumar, A.; Kannampuzha, S.; Ravichandran, M.; Renu, K.; Vellingiri, B. Mixed Contaminants: Occurrence, Interactions, Toxicity, Detection, and Remediation. Molecules 2022, 27, 2577. [Google Scholar] [CrossRef]
- Famurewa, A.C.; Renu, K.; Eladl, M.A.; Chakraborty, R.; Myakala, H.; El-Sherbiny, M.; Elsherbini, D.M.A.; Vellingiri, B.; Madhyastha, H.; Wanjari, U.R. Hesperidin and hesperetin against heavy metal toxicity: Insight on the molecular mechanism of mitigation. Biomed. Pharmacother. 2022, 149, 112914. [Google Scholar] [CrossRef]
- Bradu, P.; Biswas, A.; Nair, C.; Sreevalsakumar, S.; Patil, M.; Kannampuzha, S.; Mukherjee, A.G.; Wanjari, U.R.; Renu, K.; Vellingiri, B. Recent advances in green technology and Industrial Revolution 4.0 for a sustainable future. Environ. Sci. Pollut. Res. 2022, 1–32. [Google Scholar] [CrossRef]
- Alissa, E.M.; Ferns, G.A. Heavy metal poisoning and cardiovascular disease. J. Toxicol. 2011, 2011, 870125. [Google Scholar] [CrossRef]
- Abu-Elfotuh, K.; Ragab, G.M.; Salahuddin, A.; Jamil, L.; Abd Al Haleem, E.N. Attenuative Effects of Fluoxetine and Triticum aestivum against Aluminum-Induced Alzheimer’s Disease in Rats: The Possible Consequences on Hepatotoxicity and Nephrotoxicity. Molecules 2021, 26, 6752. [Google Scholar] [CrossRef]
- El-Said, K.S.; Hussein, S.; Alrashdi, B.M.; Mahmoud, H.A.; Ibrahim, M.A.; Elbakry, M.; El-Tantawy, H.; Kabil, D.I.; El-Naggar, S.A. Musa sp. Leaves Extract Ameliorates the Hepato-Renal Toxicities Induced by Cadmium in Mice. Molecules 2022, 27, 559. [Google Scholar] [CrossRef]
- Rettig, P. ATSDR (Agency for Toxic Substances and Disease Registry): Medical waste poses no threat to public. Health Facil. Manag. 1990, 3, 60, 62, 64–66. [Google Scholar]
- Valko, M.; Morris, H.; Cronin, M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Lamas, G.A.; Goertz, C.; Boineau, R.; Mark, D.B.; Rozema, T.; Nahin, R.L.; Drisko, J.A.; Lee, K.L. Design of the trial to assess chelation therapy (TACT). Am. Heart J. 2012, 163, 7–12. [Google Scholar] [CrossRef]
- Messner, B.; Bernhard, D. Cadmium and cardiovascular diseases: Cell biology, pathophysiology, and epidemiological relevance. Biometals 2010, 23, 811–822. [Google Scholar] [CrossRef]
- Washington, B.; Williams, S.; Armstrong, P.; Mtshali, C.; Robinson, J.T.; Myles, E.L. Cadmium toxicity on arterioles vascular smooth muscle cells of spontaneously hypertensive rats. Int. J. Environ. Res. Public Health 2006, 3, 323–328. [Google Scholar] [CrossRef]
- Oluranti, O.I.; Agboola, E.A.; Fubara, N.E.; Ajayi, M.O.; Michael, O.S. Cadmium exposure induces cardiac glucometabolic dysregulation and lipid accumulation independent of pyruvate dehydrogenase activity. Ann. Med. 2021, 53, 1109–1118. [Google Scholar] [CrossRef]
- Vineetha, V.P.; Raghu, K.G. An overview on arsenic trioxide-induced cardiotoxicity. Cardiovasc. Toxicol. 2019, 19, 105–119. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Carocci, A.; Lauria, G.; Catalano, A. Mercury exposure and heart diseases. Int. J. Environ. Res. Public Health 2017, 14, 74. [Google Scholar] [CrossRef]
- Ali, S.; Awan, Z.; Mumtaz, S.; Shakir, H.A.; Ahmad, F.; Ulhaq, M.; Tahir, H.M.; Awan, M.S.; Sharif, S.; Irfan, M. Cardiac toxicity of heavy metals (cadmium and mercury) and pharmacological intervention by vitamin C in rabbits. Environ. Sci. Pollut. Res. 2020, 27, 29266–29279. [Google Scholar] [CrossRef]
- Attri, J.; Dhawan, V.; Mahmood, S.; Pandhi, P.; Parwana, H.; Nath, R. Effect of vitamin C supplementation on oxidative DNA damage in an experimental model of lead-induced hypertension. Ann. Nutr. Metab. 2003, 47, 294–301. [Google Scholar] [CrossRef]
- Sharifi, A.M.; Darabi, R.; Akbarloo, N.; Larijani, B.; Khoshbaten, A. Investigation of circulatory and tissue ACE activity during development of lead-induced hypertension. Toxicol. Lett. 2004, 153, 233–238. [Google Scholar] [CrossRef]
- Vaziri, N.D. Mechanisms of lead-induced hypertension and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H454–H465. [Google Scholar] [CrossRef]
- Buhari, O.A.; Atanda, A.C.; Medhane, F.; Faillace, R.T. Exposure to Heavy Metals Increases Cardiovascular Risk by Elevation of Serum Lipid Levels-a National Health and Nutrition Examination Survey Based Study. Circulation 2018, 138, A16885. [Google Scholar] [CrossRef]
- Buhari, O.; Dayyab, F.; Igbinoba, O.; Atanda, A.; Medhane, F.; Faillace, R. The association between heavy metal and serum cholesterol levels in the US population: National Health and Nutrition Examination Survey 2009–2012. Hum. Exp. Toxicol. 2020, 39, 355–364. [Google Scholar] [CrossRef]
- Antonowicz, J.; Andrzejak, R.; Lepetow, T. Influence of heavy metals, especially lead, on lipid metabolism, serum alpha-tocopherol level, total antioxidant status, and erythrocyte redox status of copper smelter workers. Fresenius’ J. Anal. Chem. 1998, 361, 365–367. [Google Scholar] [CrossRef]
- Samarghandian, S.; Azimi-Nezhad, M.; Shabestari, M.M.; Azad, F.J.; Farkhondeh, T.; Bafandeh, F. Effect of chronic exposure to cadmium on serum lipid, lipoprotein and oxidative stress indices in male rats. Interdiscip. Toxicol. 2015, 8, 151–154. [Google Scholar] [CrossRef]
- Waghe, P.; Sarkar, S.N.; Sarath, T.S.; Kandasamy, K.; Choudhury, S.; Gupta, P.; Harikumar, S.; Mishra, S.K. Subchronic arsenic exposure through drinking water alters lipid profile and electrolyte status in rats. Biol. Trace Elem. Res. 2017, 176, 350–354. [Google Scholar] [CrossRef]
- Park, H.; Kim, K. Comparisons among machine learning models for the prediction of hypercholestrolemia associated with exposure to lead, mercury, and cadmium. Int. J. Environ. Res. Public Health 2019, 16, 2666. [Google Scholar] [CrossRef]
- Kristal-Boneh, E.; Coller, D.; Froom, P.; Harari, G.; Ribak, J. The association between occupational lead exposure and serum cholesterol and lipoprotein levels. Am. J. Public Health 1999, 89, 1083–1087. [Google Scholar] [CrossRef]
- Kim, C.; Ashrap, P.; Watkins, D.J.; Mukherjee, B.; Rosario-Pabón, Z.Y.; Vélez-Vega, C.M.; Alshawabkeh, A.N.; Cordero, J.F.; Meeker, J.D. Maternal Metals/Metalloid Blood Levels Are Associated With Lipidomic Profiles Among Pregnant Women in Puerto Rico. Front. Public Health 2021, 9, 4706. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid use and misuse by the heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Unger, R.H. Lipotoxic diseases. Annu. Rev. Med. 2002, 53, 319–336. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Oh, H.; Hoang, N.H.M.; Kim, M.-S. Association between heavy metals, high-sensitivity C-reaction protein and 10-year risk of cardiovascular diseases among adult Korean population. Sci. Rep. 2021, 11, 14664. [Google Scholar] [CrossRef]
- Zhou, Z.; Lu, Y.-h.; Pi, H.-f.; Gao, P.; Li, M.; Zhang, L.; Pei, L.-p.; Mei, X.; Liu, L.; Zhao, Q. Cadmium exposure is associated with the prevalence of dyslipidemia. Cell. Physiol. Biochem. 2016, 40, 633–643. [Google Scholar] [CrossRef]
- Tellez-Plaza, M.; Guallar, E.; Howard, B.V.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Silbergeld, E.K.; Devereux, R.B.; Navas-Acien, A. Cadmium exposure and incident cardiovascular disease. Epidemiology 2013, 24, 421. [Google Scholar] [CrossRef]
- Ledda, C.; Iavicoli, I.; Bracci, M.; Avola, R.; Senia, P.; Santarelli, L.; Pomara, C.; Rapisarda, V. Serum lipid, lipoprotein and apolipoprotein profiles in workers exposed to low arsenic levels: Lipid profiles and occupational arsenic exposure. Toxicol. Lett. 2018, 282, 49–56. [Google Scholar] [CrossRef]
- Sohn, S.H.; Heo, H.C.; Jo, S.; Park, C.; Sakong, J. The association between mercury concentrations and lipid profiles in the Korean National Environmental Health Survey (KoNEHS) cycle 3. Ann. Occup. Environ. Med. 2020, 32, e19. [Google Scholar] [CrossRef]
- Sharma, S.V.; Kumar, P.; Atam, V.; Verma, A.; Murthy, R. Lipid profiles with increase blood lead level: Risk of cardiovascular disease in battery workers of Lucknow City. J. Indian Acad. Forensic. Med. 2012, 34, 328–331. [Google Scholar]
- Ademuyiwa, O.; Ugbaja, R.N.; Idumebor, F.; Adebawo, O. Plasma lipid profiles and risk of cardiovascular disease in occupational lead exposure in Abeokuta, Nigeria. Lipids Health Dis. 2005, 4, 19. [Google Scholar] [CrossRef]
- Vlad, M.; Caseanu, E.; Uza, G.; Petrescu, M. Concentration of copper, zinc, chromium, iron and nickel in the abdominal aorta of patients deceased with coronary heart disease. J. Trace Elem. Electrolytes Health Dis. 1994, 8, 111–114. [Google Scholar]
- Janik, A. Effect of cadmium on certain factors of lipid metabolism in the aorta and myocardium of rats. Folia Med. Crac. 1992, 33, 53–58. [Google Scholar]
- Prentice, R.C.; Hawley, P.L.; Glonek, T.; Kopp, S.J. Calcium-dependent effects of cadmium on energy metabolism and function of perfused rat heart. Toxicol. Appl. Pharmacol. 1984, 75, 198–210. [Google Scholar] [CrossRef]
- Unsal, V.; Dalkıran, T.; Çiçek, M.; Kölükçü, E. The role of natural antioxidants against reactive oxygen species produced by cadmium toxicity: A review. Adv. Pharm. Bull. 2020, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Lagoa, R.; Marques-da-Silva, D.; Diniz, M.; Daglia, M.; Bishayee, A. Molecular mechanisms linking environmental toxicants to cancer development: Significance for protective interventions with polyphenols. Semin. Cancer Biol. 2022, 80, 118–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-y.; Zhang, S.-l.; Liu, Z.-y.; Tian, Y.; Sun, Q. Cadmium toxicity induces ER stress and apoptosis via impairing energy homoeostasis in cardiomyocytes. Biosci. Rep. 2015, 35, e00214. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-Induced cytotoxicity: Effects on mitochondrial electron transport chain. Front. Cell Dev. Biol. 2020, 8, 4377. [Google Scholar] [CrossRef]
- Cai, H.; Chen, S.; Liu, J.; He, Y. An attempt to reverse cardiac lipotoxicity by aerobic interval training in a high-fat diet-and streptozotocin-induced type 2 diabetes rat model. Diabetol. Metab. Syndr. 2019, 11, 43. [Google Scholar] [CrossRef]
- Schrey, P.; Wittsiepe, J.; Budde, U.; Heinzow, B.; Idel, H.; Wilhelm, M. Dietary intake of lead, cadmium, copper and zinc by children from the German North Sea island Amrum. Int. J. Hyg. Environ. Health 2000, 203, 1–9. [Google Scholar] [CrossRef]
- Rogalska, J.; Brzóska, M.M.; Roszczenko, A.; Moniuszko-Jakoniuk, J. Enhanced zinc consumption prevents cadmium-induced alterations in lipid metabolism in male rats. Chem. Biol. Interact. 2009, 177, 142–152. [Google Scholar] [CrossRef]
- Beloucif, A.; Kechrid, Z.; Bekada, A.M.A. Effect of zinc deficiency on blood glucose, lipid profile, and antioxidant status in streptozotocin diabetic rats and the potential role of sesame oil. Biol. Trace Elem. Res. 2021, 200, 3236–3247. [Google Scholar] [CrossRef]
- MacDiarmid, C.W.; Gaither, L.A.; Eide, D. Zinc transporters that regulate vacuolar zinc storage in Saccharomyces cerevisiae. EMBO J. 2000, 19, 2845–2855. [Google Scholar] [CrossRef]
- Rajakumar, S.; Abhishek, A.; Selvam, G.S.; Nachiappan, V. Effect of cadmium on essential metals and their impact on lipid metabolism in Saccharomyces cerevisiae. Cell Stress Chaperones 2020, 25, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Limaye, D.A.; Shaikh, Z.A. Cytotoxicity of cadmium and characteristics of its transport in cardiomyocytes. Toxicol. Appl. Pharmacol. 1999, 154, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ruta, L.L.; Popa, V.C.; Nicolau, I.; Danet, A.F.; Iordache, V.; Neagoe, A.D.; Farcasanu, I.C. Calcium signaling mediates the response to cadmium toxicity in Saccharomyces cerevisiae cells. FEBS Lett. 2014, 588, 3202–3212. [Google Scholar] [CrossRef]
- Muthukumar, K.; Nachiappan, V. Phosphatidylethanolamine from phosphatidylserine decarboxylase2 is essential for autophagy under cadmium stress in Saccharomyces cerevisiae. Cell Biochem. Biophys. 2013, 67, 1353–1363. [Google Scholar] [CrossRef]
- Badin, J.K. Coronary Smooth Muscle Cell Cytodifferentiation and Intracellular Ca2+ Handling in Coronary Artery Disease; Indiana University-Purdue University Indianapolis: Indianapolis, IN, USA, 2019. [Google Scholar]
- Rajak, C.; Singh, N.; Parashar, P. Metal toxicity and natural antidotes: Prevention is better than cure. Environ. Sci. Pollut. Res. 2020, 27, 43582–43598. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Miao, S.; Zhou, W.; Elnesr, S.S.; Dong, X.; Zou, X. MAPK, AKT/FoxO3a and mTOR pathways are involved in cadmium regulating the cell cycle, proliferation and apoptosis of chicken follicular granulosa cells. Ecotoxicol. Environ. Saf. 2021, 214, 112091. [Google Scholar] [CrossRef]
- Hughes, M.F.; Edwards, B.C.; Herbin-Davis, K.M.; Saunders, J.; Styblo, M.; Thomas, D.J. Arsenic (+3 oxidation state) methyltransferase genotype affects steady-state distribution and clearance of arsenic in arsenate-treated mice. Toxicol. Appl. Pharmacol. 2010, 249, 217–223. [Google Scholar] [CrossRef]
- Bozack, A.K.; Saxena, R.; Gamble, M.V. Nutritional influences on one-carbon metabolism: Effects on arsenic methylation and toxicity. Annu. Rev. Nutr. 2018, 38, 401–429. [Google Scholar] [CrossRef]
- Thomas, D.J. Arsenic methylation–Lessons from three decades of research. Toxicol. Appl. Pharmacol. 2021, 457, 152800. [Google Scholar] [CrossRef]
- Stýblo, M.; Venkatratnam, A.; Fry, R.C.; Thomas, D.J. Origins, fate, and actions of methylated trivalent metabolites of inorganic arsenic: Progress and prospects. Arch. Toxicol. 2021, 95, 1547–1572. [Google Scholar] [CrossRef]
- Roy, N.K.; Murphy, A.; Costa, M. Arsenic methyltransferase and methylation of inorganic arsenic. Biomolecules 2020, 10, 1351. [Google Scholar] [CrossRef]
- Ratnaike, R.N. Acute and chronic arsenic toxicity. Postgrad. Med. J. 2003, 79, 391–396. [Google Scholar] [CrossRef]
- Alamolhodaei, N.S.; Shirani, K.; Karimi, G. Arsenic cardiotoxicity: An overview. Environ. Toxicol. Pharmacol. 2015, 40, 1005–1014. [Google Scholar] [CrossRef]
- Balakumar, P.; Kaur, J. Arsenic exposure and cardiovascular disorders: An overview. Cardiovasc. Toxicol. 2009, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-J.; Chuu, J.-J.; Chang, C.-Y.; Tsai, W.-C.; Chen, K.-J.; Guo, H.-R. Atherosclerosis induced by arsenic in drinking water in rats through altering lipid metabolism. Toxicol. Appl. Pharmacol. 2011, 256, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Bae, O.-N.; Chung, S.-M.; Kang, K.-T.; Lee, J.-Y.; Chung, J.-H. Enhancement of platelet aggregation and thrombus formation by arsenic in drinking water: A contributing factor to cardiovascular disease. Toxicol. Appl. Pharmacol. 2002, 179, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, P.P.; Hulderman, T.; Harki, D.; Luster, M.I. Arsenic exposure accelerates atherogenesis in apolipoprotein E (-/-) mice. Environ. Health Perspect. 2003, 111, 1744–1748. [Google Scholar] [CrossRef]
- Peng, D.; Hiipakka, R.A.; Xie, J.-T.; Reardon, C.A.; Getz, G.S.; Liao, S. Differential effects of activation of liver X receptor on plasma lipid homeostasis in wild-type and lipoprotein clearance-deficient mice. Atherosclerosis 2010, 208, 126–133. [Google Scholar] [CrossRef]
- Groot, P.H.E.; Pearce, N.J.; Yates, J.W.; Stocker, C.; Sauermelch, C.; Doe, C.P.; Willette, R.N.; Olzinski, A.; Peters, T.; D'Epagnier, D.; et al. Synthetic LXR agonists increase LDL in CETP species. J. Lipid Res. 2005, 46, 2182–2191. [Google Scholar] [CrossRef]
- States, J.C.; Srivastava, S.; Chen, Y.; Barchowsky, A. Arsenic and cardiovascular disease. Toxicol. Sci. 2009, 107, 312–323. [Google Scholar] [CrossRef]
- Muthumani, M.; Prabu, S.M. Silibinin potentially attenuates arsenic-induced oxidative stress mediated cardiotoxicity and dyslipidemia in rats. Cardiovasc. Toxicol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Guallar, E.; Navas-Acien, A. Arsenic exposure and cardiovascular disease: An updated systematic review. Curr. Atheroscler. Rep. 2012, 14, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The role of reactive oxygen species in arsenic toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef]
- Balarastaghi, S.; Rezaee, R.; Hayes, A.W.; Yarmohammadi, F.; Karimi, G. Mechanisms of Arsenic Exposure-Induced Hypertension and Atherosclerosis: An Updated Overview. Biol. Trace Elem. Res. 2022, 1–16. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, F.; Graziano, J.H.; Parvez, F.; Liu, M.; Paul, R.R.; Shaheen, I.; Sarwar, G.; Ahmed, A.; Islam, T. Arsenic exposure from drinking water, arsenic methylation capacity, and carotid intima-media thickness in Bangladesh. Am. J. Epidemiol. 2013, 178, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, M.; Lemarie, C.A.; Flores Molina, M.; Guilbert, C.; Lehoux, S.; Mann, K.K. Genetic deletion of LXRα prevents arsenic-enhanced atherosclerosis, but not arsenic-altered plaque composition. Toxicol. Sci. 2014, 142, 477–488. [Google Scholar] [CrossRef]
- Ye, G.; Lu, W.; Zhang, L.; Gao, H.; Liao, X.; Zhang, X.; Zhang, H.; Chen, J.; Huang, Q. Integrated metabolomic and transcriptomic analysis identifies benzo [a] pyrene-induced characteristic metabolic reprogramming during accumulation of lipids and reactive oxygen species in macrophages. Sci. Total Environ. 2022, 829, 154685. [Google Scholar] [CrossRef]
- Garciafigueroa, D.Y.; Klei, L.R.; Ambrosio, F.; Barchowsky, A. Arsenic-stimulated lipolysis and adipose remodeling is mediated by G-protein-coupled receptors. Toxicol. Sci. 2013, 134, 335–344. [Google Scholar] [CrossRef]
- Vigouroux, C.; Caron-Debarle, M.; Le Dour, C.; Magré, J.; Capeau, J. Molecular mechanisms of human lipodystrophies: From adipocyte lipid droplet to oxidative stress and lipotoxicity. Int. J. Biochem. Cell Biol. 2011, 43, 862–876. [Google Scholar] [CrossRef]
- Turer, A.T.; Hill, J.A.; Elmquist, J.K.; Scherer, P.E. Adipose tissue biology and cardiomyopathy: Translational implications. Circ. Res. 2012, 111, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Kolditz, C.-I.; Langin, D. Adipose tissue lipolysis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Bézaire, V.; Mairal, A.; Anesia, R.; Lefort, C.; Langin, D. Chronic TNFα and cAMP pre-treatment of human adipocytes alter HSL, ATGL and perilipin to regulate basal and stimulated lipolysis. FEBS Lett. 2009, 583, 3045–3049. [Google Scholar] [CrossRef]
- He, X.; H’ng, S.-C.; Leong, D.T.; Hutmacher, D.W.; Melendez, A.J. Sphingosine-1-phosphate mediates proliferation maintaining the multipotency of human adult bone marrow and adipose tissue-derived stem cells. J. Mol. Cell Biol. 2010, 2, 199–208. [Google Scholar] [CrossRef]
- Straub, A.C.; Klei, L.R.; Stolz, D.B.; Barchowsky, A. Arsenic requires sphingosine-1-phosphate type 1 receptors to induce angiogenic genes and endothelial cell remodeling. Am. J. Pathol. 2009, 174, 1949–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branco, V.; Coppo, L.; Aschner, M.; Carvalho, C. N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II). Toxics 2022, 10, 433. [Google Scholar] [CrossRef]
- Houston, M.C. Role of mercury toxicity in hypertension, cardiovascular disease, and stroke. J. Clin. Hypertens. 2011, 13, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Houston, M.C. The role of mercury in cardiovascular disease. J. Cardiovasc. Dis. Diagn. 2014, 2, 1–8. [Google Scholar] [CrossRef]
- Kim, S.H.; Johnson, V.J.; Sharma, R.P. Mercury inhibits nitric oxide production but activates proinflammatory cytokine expression in murine macrophage: Differential modulation of NF-κB and p38 MAPK signaling pathways. Nitric Oxide 2002, 7, 67–74. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Afshari, R.; Mehrpour, O.; Samarghandian, S. Mercury and atherosclerosis: Cell biology, pathophysiology, and epidemiological studies. Biol. Trace Elem. Res. 2020, 196, 27–36. [Google Scholar] [CrossRef]
- Marques-da-Silva, D.; Videira, P.; Lagoa, R. Registered human trials addressing environmental and occupational toxicant exposures: Scoping review of immunological markers and protective strategies. Environ. Toxicol. Pharmacol. 2022, 93, 103886. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Lee, S.; Park, Y.; Min, T.; Bai, S.C. Evaluation of dietary selenium, vitamin C and E as the multi-antioxidants on the methylmercury intoxicated mice based on mercury bioaccumulation, antioxidant enzyme activity, lipid peroxidation and mitochondrial oxidative stress. Chemosphere 2021, 273, 129673. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, J.K.; Rissanen, T.H.; Voutilainen, S.; Tuomainen, T.-P. Mercury as a risk factor for cardiovascular diseases. J. Nutr. Biochem. 2007, 18, 75–85. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Chen, W.; Xu, S.; Feng, X.; Zhang, L. Natural products: The role and mechanism in low-density lipoprotein oxidation and atherosclerosis. Phytother. Res. 2021, 35, 2945–2967. [Google Scholar] [CrossRef]
- Pollard, K.M.; Cauvi, D.M.; Toomey, C.B.; Hultman, P.; Kono, D.H. Mercury-induced inflammation and autoimmunity. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 129299. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, F.; Niaz, K.; Hassan, F.I.; Khan, F.; Abdollahi, M. Immunotoxicity of mercury: Pathological and toxicological effects. J. Environ. Sci. Health Part C 2017, 35, 29–46. [Google Scholar] [CrossRef]
- Shanmugam, G.; Narasimhan, M.; Sakthivel, R.; Kumar, R.; Davidson, C.; Palaniappan, S.; Claycomb, W.W.; Hoidal, J.R.; Darley-Usmar, V.M.; Rajasekaran, N.S. A biphasic effect of TNF-α in regulation of the Keap1/Nrf2 pathway in cardiomyocytes. Redox Biol. 2016, 9, 77–89. [Google Scholar] [CrossRef]
- Turillazzi, E.; Neri, M.; Cerretani, D.; Cantatore, S.; Frati, P.; Moltoni, L.; Busardo, F.P.; Pomara, C.; Riezzo, I.; Fineschi, V. Lipid peroxidation and apoptotic response in rat brain areas induced by long-term administration of nandrolone: The mutual crosstalk between ROS and NF-kB. J. Cell. Mol. Med. 2016, 20, 601–612. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, R.; Xiong, J.; Yan, D.; Li, A.; Wang, S.; Xu, J.; Zhou, J. JWA antagonizes paraquat-induced neurotoxicity via activation of Nrf2. Toxicol. Lett. 2017, 277, 32–40. [Google Scholar] [CrossRef]
- Uruno, A.; Motohashi, H. The Keap1–Nrf2 system as an in vivo sensor for electrophiles. Nitric Oxide 2011, 25, 153–160. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, X.; Yang, D.; Lu, J.; Liu, B.; Baiyun, R.; Zhang, Z. Dietary luteolin attenuates chronic liver injury induced by mercuric chloride via the Nrf2/NF-κB/P53 signaling pathway in rats. Oncotarget 2017, 8, 40982. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Bennett, A.M. Mitogen-activated protein kinase regulation in hepatic metabolism. Trends Endocrinol. Metab. 2017, 28, 868–878. [Google Scholar] [CrossRef]
- Jennrich, P. The influence of arsenic, lead, and mercury on the development of cardiovascular diseases. Int. Sch. Res. Not. 2013, 2013, 1–16. [Google Scholar] [CrossRef]
- Pall, M.L. Nitric oxide synthase partial uncoupling as a key switching mechanism for the NO/ONOO–cycle. Med. Hypotheses 2007, 69, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Sica, D.A. Lead-induced hypertension: Role of oxidative stress. Curr. Hypertens. Rep. 2004, 6, 314–320. [Google Scholar] [CrossRef]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Haskó, G.; Schmidt, H.H.; Stasch, J.-P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef]
- Evora, R.B.; M Evora, P.; C Celotto, A.; J Rodrigues, A.; E Joviliano, E. Cardiovascular therapeutics targets on the NO–sGC–cGMP signaling pathway: A critical overview. Curr. Drug Targets 2012, 13, 1207–1214. [Google Scholar] [CrossRef]
- Aje, T.O.; Miller, M. Cardiovascular disease: A global problem extending into the developing world. World J. Cardiol. 2009, 1, 3. [Google Scholar] [CrossRef]
- Van der Sande, M.; Inskip, H.M.; Jaiteh, K.O.; Maine, N.P.; Walraven, G.E.; Hall, A.J.; McAdam, K. Changing causes of death in the West African town of Banjul, 1942–1997. Bull. World Health Organ. 2001, 79, 133. [Google Scholar]
- Brotons, C.; Ribera, A.; Perich, R.M.; Abrodos, D.; Magaña, P.; Pablo, S.; Terradas, D.; Fernández, F.; Permanyer, G. Worldwide distribution of blood lipids and lipoproteins in childhood and adolescence: A review study. Atherosclerosis 1998, 139, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Pletcher, M.J.; Vittinghoff, E.; Clemons, A.M.; Jacobs, D.R.; Allen, N.B.; Alonso, A.; Bellows, B.K.; Oelsner, E.C.; Al Hazzouri, A.Z. Association between cumulative low-density lipoprotein cholesterol exposure during young adulthood and middle age and risk of cardiovascular events. JAMA Cardiol. 2021, 6, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Kosma, K.; Barbetseas, J.; Karagiorga, M.; Ladis, I.; Stefanadis, C. Distribution of serum lipids and lipoproteins in patients with beta thalassaemia major; an epidemiological study in young adults from Greece. Lipids Health Dis. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, G.J.; Halliwell, B.; Moorhouse, C.P.; Gutteridge, J.M. Action of lead (II) and aluminium (III) ions on iron-stimulated lipid peroxidation in liposomes, erythrocytes and rat liver microsomal fractions. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1988, 962, 196–200. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Rodríguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef]
- Lee, J.-C.; Son, Y.-O.; Pratheeshkumar, P.; Shi, X. Oxidative stress and metal carcinogenesis. Free Radic. Biol. Med. 2012, 53, 742–757. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renu, K.; Mukherjee, A.G.; Wanjari, U.R.; Vinayagam, S.; Veeraraghavan, V.P.; Vellingiri, B.; George, A.; Lagoa, R.; Sattu, K.; Dey, A.; et al. Misuse of Cardiac Lipid upon Exposure to Toxic Trace Elements—A Focused Review. Molecules 2022, 27, 5657. https://doi.org/10.3390/molecules27175657
Renu K, Mukherjee AG, Wanjari UR, Vinayagam S, Veeraraghavan VP, Vellingiri B, George A, Lagoa R, Sattu K, Dey A, et al. Misuse of Cardiac Lipid upon Exposure to Toxic Trace Elements—A Focused Review. Molecules. 2022; 27(17):5657. https://doi.org/10.3390/molecules27175657
Chicago/Turabian StyleRenu, Kaviyarasi, Anirban Goutam Mukherjee, Uddesh Ramesh Wanjari, Sathishkumar Vinayagam, Vishnu Priya Veeraraghavan, Balachandar Vellingiri, Alex George, Ricardo Lagoa, Kamaraj Sattu, Abhijit Dey, and et al. 2022. "Misuse of Cardiac Lipid upon Exposure to Toxic Trace Elements—A Focused Review" Molecules 27, no. 17: 5657. https://doi.org/10.3390/molecules27175657
APA StyleRenu, K., Mukherjee, A. G., Wanjari, U. R., Vinayagam, S., Veeraraghavan, V. P., Vellingiri, B., George, A., Lagoa, R., Sattu, K., Dey, A., & Gopalakrishnan, A. V. (2022). Misuse of Cardiac Lipid upon Exposure to Toxic Trace Elements—A Focused Review. Molecules, 27(17), 5657. https://doi.org/10.3390/molecules27175657