
Volumetric Absorptive Microsampling (VAMS) for Targeted LC-MS/MS Determination of Tryptophan-Related Biomarkers

 ,
, 
 , , and
, , and 
Abstract
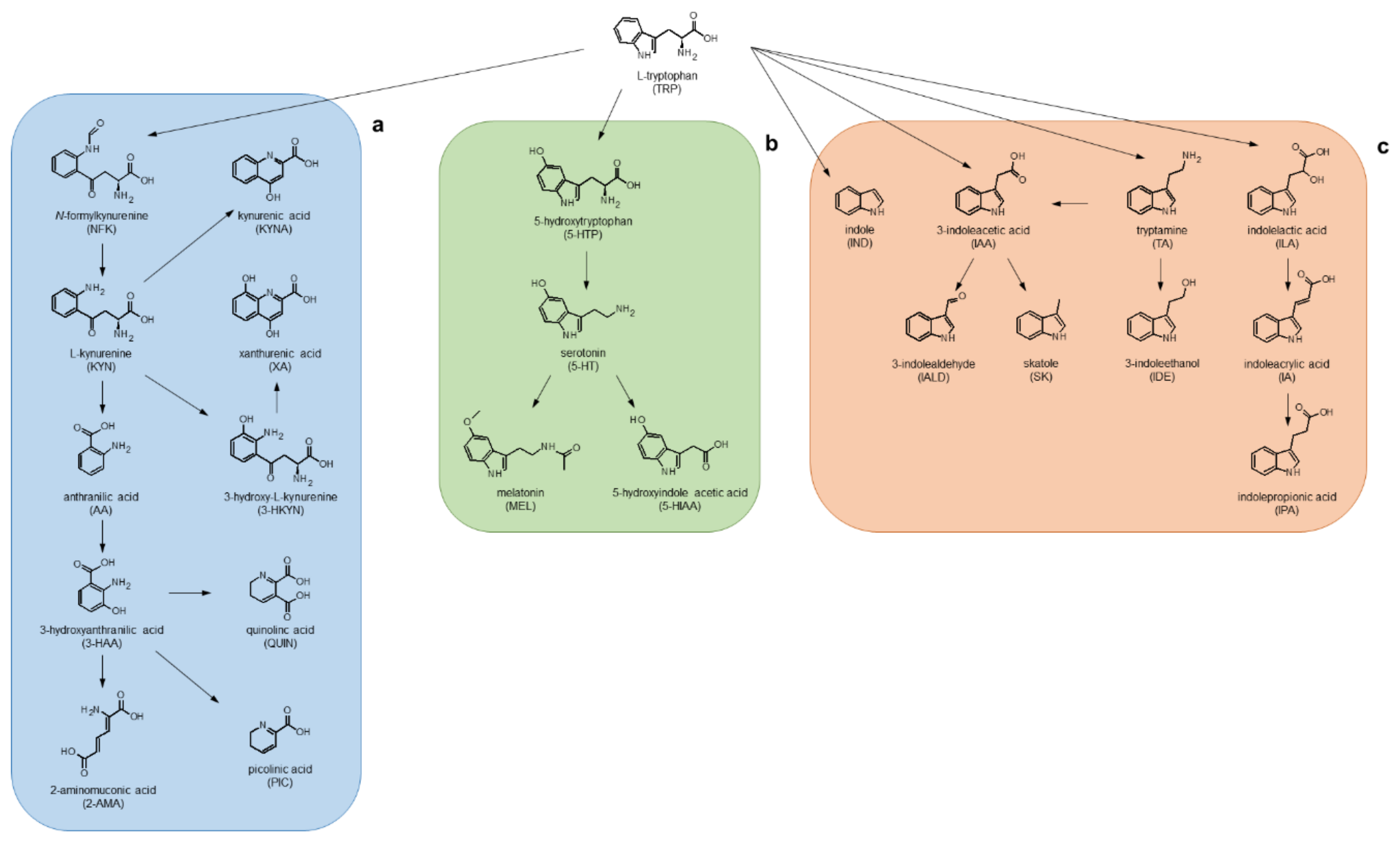
:1. Introduction
2. Results and Discussion
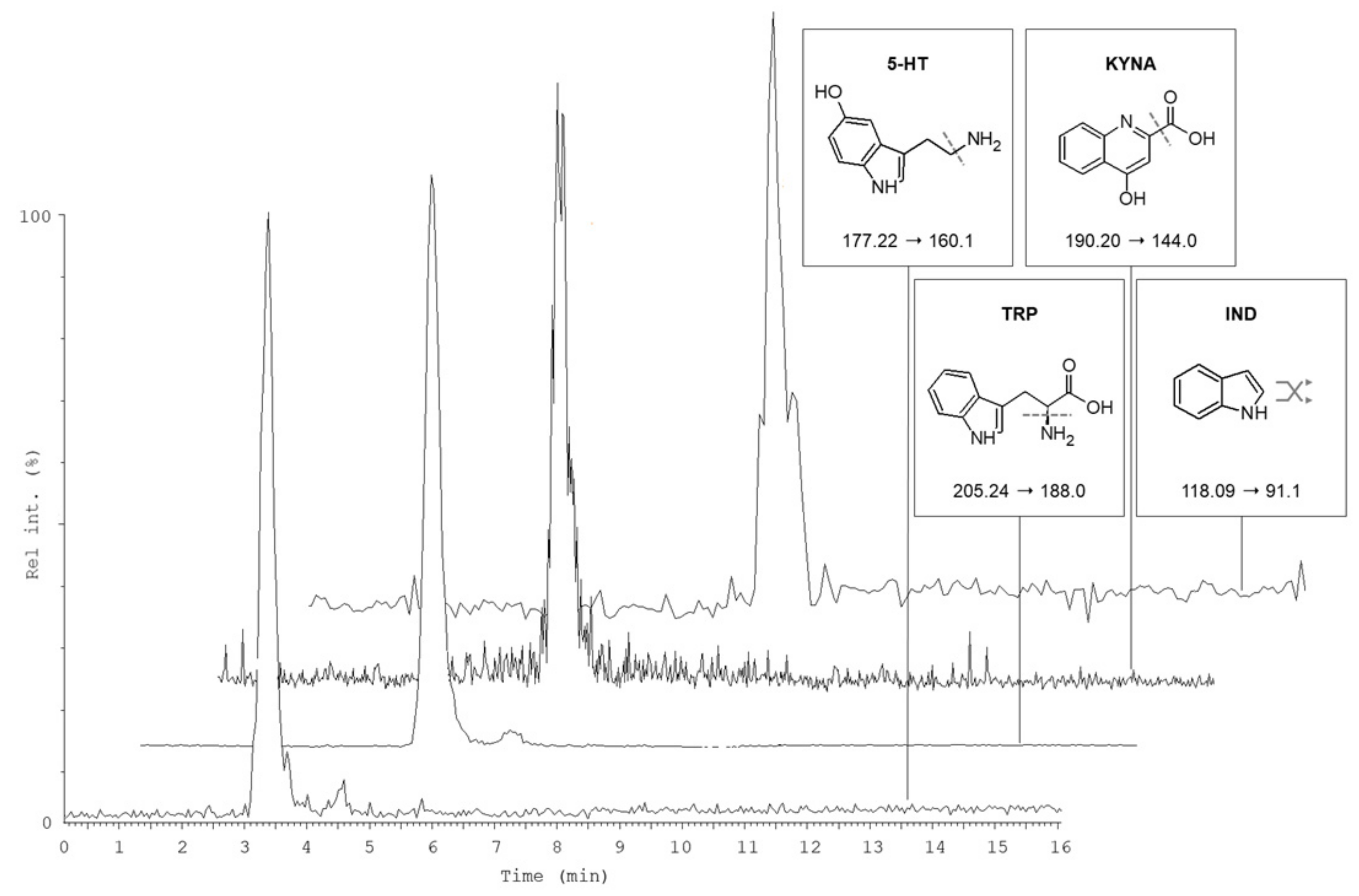
2.1. Liquid Chromatography and Mass Spectrometry
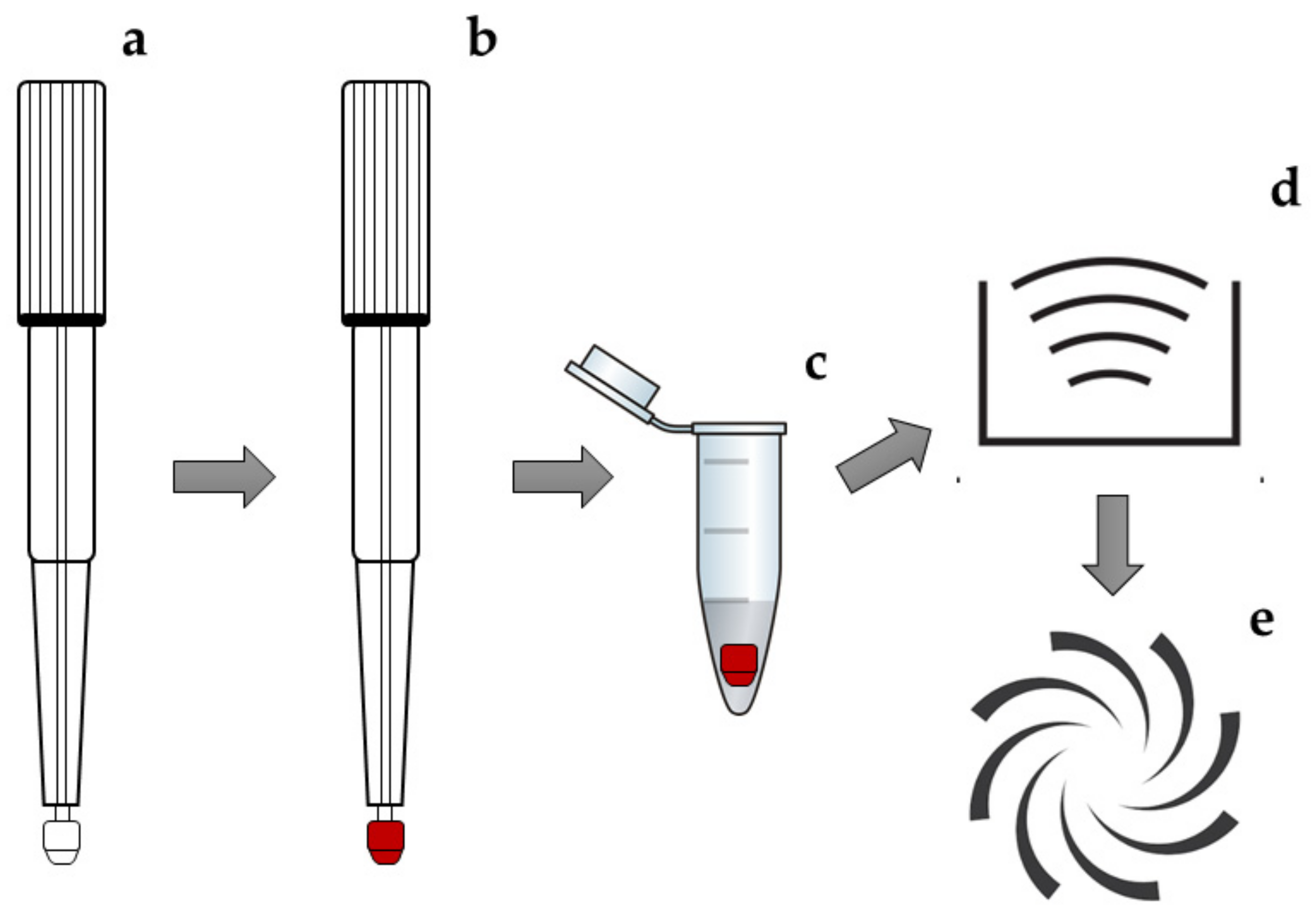
2.2. VAMS Microsample Pretreatment
2.3. VAMS-LC-MS/MS Method Qualification for Targeted Biomarker Assessment
2.3.1. Absolute Recovery, Precision, Matrix Effect and Carryover
2.3.2. Selectivity, Linearity and Sensitivity
2.3.3. Stability
2.4. Analysis of Real Samples and Accuracy
3. Materials and Methods
3.1. Chemicals and Standard Solutions
3.2. LC-MS/MS Instrumentation and Conditions
3.3. Microsampling: VAMS Collection and Pretreatment
3.4. Method Qualification
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Amaral, M.; Outeiro, T.F.; Scrutton, N.S.; Giorgini, F. The causative role and therapeutic potential of the kynurenine pathway in neurodegenerative disease. J. Mol. Med. 2013, 91, 705–713. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nakahama, T.; Le, D.H.; Van Son, L.; Chu, H.H.; Kishimoto, T. Aryl hydrocarbon receptor and kynurenine: Recent advances in autoimmune disease research. Front. Immunol. 2014, 5, 551. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan metabolic pathways and brain serotonergic activity: A comparative review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef]
- Li, X.; Zhang, B.; Hu, Y.; Zhao, Y. New insights into gut-bacteria-derived indole and its derivatives in intestinal and liver diseases. Front. Pharmacol. 2021, 12, 769501. [Google Scholar] [CrossRef]
- Jacob, M.; Malkawi, A.; Albast, N.; Al Bougha, S.; Lopata, A.; Dasouki, M.; Abdel Rahman, A.M. A targeted metabolomics approach for clinical diagnosis of inborn errors of metabolism. Anal. Chim. Acta 2018, 1025, 141–153. [Google Scholar] [CrossRef]
- Dittakavi, S.; Jat, R.K.; Mullangi, R. Quantitative analysis of enasidenib in dried blood spots of mouse blood using an increased-sensitivity LC-MS/MS method: Application to a pharmacokinetic study. Biomed. Chromatogr. 2019, 33, e4491. [Google Scholar] [CrossRef]
- Mercolini, L.; Mandrioli, R.; Protti, M.; Conca, A.; Albers, L.J.; Raggi, M.A. Dried blood spot testing: A novel approach for the therapeutic drug monitoring of ziprasidone-treated patients. Bioanalysis 2014, 6, 1487–1495. [Google Scholar] [CrossRef]
- Protti, M.; Vignali, A.; Sanchez Blanco, T.; Rudge, J.; Bugamelli, F.; Ferranti, A.; Mandrioli, R.; Mercolini, L. Enantioseparation and determination of asenapine in biological fluid micromatrices by HPLC with diode array detection. J. Sep. Sci. 2018, 41, 1257–1265. [Google Scholar] [CrossRef]
- Björkesten, J.; Enroth, S.; Shen, Q.; Wik, L.; Hougaard, D.M.; Cohen, A.S.; Sörensen, L.; Giedraitis, V.; Ingelsson, M.; Larsson, A.; et al. Stability of proteins in dried blood spot biobanks. Mol. Cell. Proteom. 2017, 16, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Marasca, C.; Arana, M.E.B.; Protti, M.; Cavalli, A.; Mercolini, L.; Armirotti, A. Volumetric absorptive microsampling of blood for untargeted lipidomics. Molecules 2021, 26, 262. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S.; Cooper, J.D.; Lago, S.G.; Kenny, D.; Rustogi, N.; Stocki, P.; Bahn, S. Towards reproducible MRM based biomarker discovery using dried blood spots. Sci. Rep. 2017, 7, 45178. [Google Scholar] [CrossRef] [PubMed]
- Eshghi, A.; Pistawka, A.J.; Liu, J.; Chen, M.; Sinclair, N.J.T.; Hardie, D.B.; Elliott, M.; Chen, L.; Newman, R.; Mohammed, Y.; et al. concentration determination of >200 proteins in dried blood spots for biomarker discovery and validation. Mol. Cell. Proteom. 2020, 19, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Denniff, P.; Spooner, N. Volumetric absorptive microsampling: A dried sample collection technique for quantitative bioanalysis. Anal. Chem. 2014, 86, 8489–8495. [Google Scholar] [CrossRef]
- Kok, M.G.M.; Fillet, M. Volumetric absorptive microsampling: Current advances and applications. J. Pharm. Biomed. Anal. 2018, 147, 288–296. [Google Scholar] [CrossRef]
- Verougstraete, N.; Lapauw, B.; Van Aken, S.; Delanghe, J.; Stove, C.; Stove, V. Volumetric Absorptive Microsampling at Home as an Alternative Tool for the Monitoring of HbA1c in Diabetes Patients. Clin. Chem. Lab. Med. 2017, 55, 462–469. [Google Scholar] [CrossRef]
- Luo, Y.; Korfmacher, W.; Ho, S.; Shen, L.; Wang, J.; Wu, Z.; Guo, Y.; Snow, G.; O’Shea, T. Evaluation of two blood microsampling approaches for drug discovery PK studies in rats. Bioanalysis 2015, 7, 2345–2359. [Google Scholar] [CrossRef]
- Protti, M.; Mandrioli, R.; Mercolini, L. Tutorial: Volumetric absorptive microsampling (VAMS). Anal. Chim. Acta 2019, 1046, 32–47. [Google Scholar] [CrossRef]
- Kita, K.; Noritake, K.; Mano, Y. Application of a volumetric absorptive microsampling device to a pharmacokinetic study of tacrolimus in rats: Comparison with wet blood and plasma. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 91–102. [Google Scholar] [CrossRef]
- Protti, M.; Catapano, M.C.; Samolsky Dekel, B.G.; Rudge, J.; Gerra, G.; Somaini, L.; Mandrioli, R.; Mercolini, L. Determination of oxycodone and its major metabolites in haematic and urinary matrices: Comparison of traditional and miniaturised sampling approaches. J. Pharm. Biomed. Anal. 2018, 152, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Marasca, C.; Mandrioli, R.; Sardella, R.; Vovk, T.; Armirotti, A.; Cavalli, A.; Serretti, A.; Protti, M.; Mercolini, L. Dried volumetric microsampling approaches for the therapeutic drug monitoring of psychiatric patients undergoing clozapine treatment. Front. Psychiatry 2022, 13, 794609. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Rudge, J.; Sberna, A.E.; Gerra, G.; Mercolini, L. Dried haematic microsamples and LC-MS/MS for the analysis of natural and synthetic cannabinoids. J. Chromatogr. B 2017, 1044, 77–86. [Google Scholar] [CrossRef]
- Mandrioli, R.; Mercolini, L.; Protti, M. Blood and plasma volumetric absorptive microsampling (VAMS) coupled to LC-MS/MS for the forensic assessment of cocaine consumption. Molecules 2020, 25, 1046. [Google Scholar] [CrossRef]
- Mercolini, L.; Protti, M.; Catapano, M.C.; Rudge, J.; Sberna, A.E. LC-MS/MS and volumetric absorptive microsampling for quantitative bioanalysis of cathinone analogues in dried urine, plasma and oral fluid samples. J. Pharm. Biomed. Anal. 2016, 123, 186–194. [Google Scholar] [CrossRef]
- Protti, M.; Marasca, C.; Cirrincione, M.; Sberna, A.E.; Mandrioli, R.; Mercolini, L. Dried urine microsampling coupled to liquid chromatography-tandem mass spectrometry (LC-MS/MS) for the analysis of unconjugated anabolic androgenic steroids. Molecules 2020, 25, 3210. [Google Scholar] [CrossRef]
- Marasca, C.; Protti, M.; Mandrioli, R.; Atti, A.R.; Armirotti, A.; Cavalli, A.; De Ronchi, D.; Mercolini, L. whole blood and oral fluid microsampling for the monitoring of patients under treatment with antidepressant drugs. J. Pharm. Biomed. Anal. 2020, 188, 113384. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Sberna, P.M.; Sberna, A.E.; Ferrante, R.; Mandrioli, R.; Mercolini, L. Enhanced urinary stability of peptide hormones and growth factors by dried urine microsampling. J. Pharm. Biomed. Anal. 2021, 204, 114234. [Google Scholar] [CrossRef]
- Protti, M.; Sberna, P.M.; Sardella, R.; Vovk, T.; Mercolini, L.; Mandrioli, R. VAMS and StAGE as innovative tools for the enantioselective determination of clenbuterol in urine by LC-MS/MS. J. Pharm. Biomed. Anal. 2021, 195, 113873. [Google Scholar] [CrossRef]
- Protti, M.; Mandrioli, R.; Mercolini, L. Microsampling and LC-MS/MS for antidoping testing of glucocorticoids in urine. Bioanalysis 2020, 12, 769–782. [Google Scholar] [CrossRef]
- Van den Broek, I.; Fu, Q.; Kushon, S.; Kowalski, M.P.; Millis, K.; Percy, A.; Holewinski, R.J.; Venkatraman, V.; Van Eyk, J.E. Application of volumetric absorptive microsampling for robust, high-throughput mass spectrometric quantification of circulating protein biomarkers. Clin. Mass Spectrom. 2017, 4, 25–33. [Google Scholar] [CrossRef]
- Kok, M.G.M.; Nix, C.; Nys, G.; Fillet, M. Targeted metabolomics of whole blood using volumetric absorptive microsampling. Talanta 2019, 197, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Houghton, R.; Horro Pita, C.; Ward, I.; Macarthur, R. Generic approach to validation of small-molecule LC-MS/MS biomarker assays. Bioanalysis 2009, 1, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Devanarayan, V.; Barrett, Y.C.; Weiner, R.; Allinson, J.; Fountain, S.; Keller, S.; Weinryb, I.; Green, M.; Duan, L.; et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm. Res. 2006, 23, 312–328. [Google Scholar] [CrossRef]
- European Medicines Agency, Guideline on Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 26 July 2022).

{kind=link}
{kind=link}
{kind=link}
| Analyte | MW (g/mol) | Q1 (m/z) | Q3 (m/z) | Collision Energy (eV) |
|---|---|---|---|---|
| TRP | 204.22 | 205.24 | 188.0 | 15 |
| NFK | 236.22 | 237.24 | 142.2 | 10 |
| KYN | 208.21 | 209.30 | 192.0 | 10 |
| AA | 137.14 | 138.16 | 120.0 | 10 |
| KYNA | 189.17 | 190.20 | 144.0 | 20 |
| 3-HKYN | 224.21 | 225.19 | 208.1 | 10 |
| XA | 205.17 | 206.21 | 160.1 | 15 |
| 3-HAA | 153.14 | 154.20 | 136.1 | 15 |
| QUIN | 167.12 | 168.14 | 150.1 | 10 |
| PIC | 123.11 | 122.09 | 78.1 | 15 |
| 2-AMA | 157.12 | 156.15 | 66.0 | 15 |
| 5-HTP | 220.22 | 221.21 | 204.0 | 10 |
| 5-HT | 176.21 | 177.22 | 160.1 | 10 |
| 5-HIAA | 191.18 | 192.21 | 160.2 | 15 |
| MEL | 232.28 | 233.31 | 173.9 | 15 |
| IND | 117.15 | 118.09 | 91.1 | 20 |
| TA | 160.22 | 161.27 | 144.1 | 10 |
| IDE | 161.20 | 162.17 | 144.1 | 10 |
| IPA | 189.21 | 190.23 | 130.2 | 10 |
| ILA | 205.21 | 204.22 | 158.1 | 15 |
| IAA | 175.18 | 176.18 | 130.2 | 15 |
| SK | 131.17 | 132.20 | 117.1 | 20 |
| IALD | 145.16 | 146.18 | 118.1 | 15 |
| IA | 187.19 | 188.19 | 115.1 | 10 |
| IS1 | 209.26 | 210.31 | 192.0 | 15 |
| IS2 | 194.20 | 195.23 | 149.1 | 20 |
| IS3 | 180.24 | 181.22 | 160.1 | 10 |
| IS4 | 124.19 | 125.09 | 98.2 | 20 |
| Compound | Concentration Level a | Intraday Precision (%RSD) b | Interday Precision (%RSD) b | Absolute Recovery (%) c | Matrix Effect (%) c |
|---|---|---|---|---|---|
| TRP | Low | 7.9 | 9.5 | 86 | 88 |
| Medium | 6.8 | 8.3 | 88 | 90 | |
| High | 6.3 | 7.8 | 93 | 92 | |
| NFK | Low | 6.6 | 7.4 | 91 | 88 |
| Medium | 6.2 | 6.8 | 93 | 92 | |
| High | 5.7 | 6.2 | 96 | 93 | |
| KYN | Low | 7.7 | 8.9 | 88 | 89 |
| Medium | 6.6 | 7.8 | 91 | 93 | |
| High | 6.1 | 7.3 | 94 | 93 | |
| AA | Low | 6.3 | 7.2 | 88 | 87 |
| Medium | 6.0 | 6.5 | 92 | 90 | |
| High | 5.5 | 5.9 | 95 | 94 | |
| KYNA | Low | 6.4 | 7.1 | 87 | 89 |
| Medium | 5.9 | 6.6 | 92 | 93 | |
| High | 5.6 | 5.8 | 93 | 95 | |
| 3-HKYN | Low | 6.0 | 7.0 | 90 | 90 |
| Medium | 5.8 | 6.2 | 94 | 93 | |
| High | 5.3 | 5.7 | 94 | 96 | |
| XA | Low | 6.3 | 6.8 | 87 | 91 |
| Medium | 5.9 | 6.3 | 89 | 92 | |
| High | 5.2 | 5.6 | 93 | 95 | |
| 3-HAA | Low | 6.0 | 6.8 | 88 | 91 |
| Medium | 5.6 | 6.1 | 91 | 91 | |
| High | 5.3 | 5.5 | 95 | 93 | |
| QUIN | Low | 6.4 | 7.4 | 90 | 89 |
| Medium | 5.8 | 6.7 | 91 | 94 | |
| High | 5.2 | 6.1 | 94 | 94 | |
| PIC | Low | 6.7 | 7.6 | 89 | 92 |
| Medium | 5.9 | 7.0 | 93 | 93 | |
| High | 5.7 | 6.3 | 94 | 94 | |
| 2-AMA | Low | 5.5 | 6.3 | 88 | 90 |
| Medium | 5.1 | 5.6 | 92 | 93 | |
| High | 4.8 | 5.0 | 96 | 95 | |
| 5-HTP | Low | 5.6 | 6.2 | 89 | 92 |
| Medium | 5.2 | 5.5 | 93 | 95 | |
| High | 4.8 | 4.9 | 94 | 96 | |
| 5-HT | Low | 6.9 | 7.7 | 90 | 88 |
| Medium | 6.5 | 7.1 | 95 | 91 | |
| High | 6.0 | 6.5 | 96 | 92 | |
| 5-HIAA | Low | 5.8 | 6.1 | 88 | 90 |
| Medium | 5.1 | 5.7 | 89 | 94 | |
| High | 4.9 | 4.8 | 92 | 94 | |
| MEL | Low | 6.1 | 6.5 | 90 | 92 |
| Medium | 5.5 | 6.0 | 90 | 95 | |
| High | 5.1 | 5.6 | 93 | 95 | |
| IND | Low | 5.7 | 6.3 | 87 | 91 |
| Medium | 5.3 | 5.6 | 88 | 92 | |
| High | 4.9 | 5.0 | 89 | 94 | |
| TA | Low | 5.6 | 6.4 | 89 | 92 |
| Medium | 5.3 | 5.6 | 92 | 95 | |
| High | 5.0 | 4.9 | 92 | 96 | |
| IDE | Low | 5.5 | 6.3 | 88 | 93 |
| Medium | 5.3 | 5.4 | 90 | 93 | |
| High | 4.8 | 4.9 | 92 | 95 | |
| IPA | Low | 6.8 | 7.4 | 89 | 91 |
| Medium | 6.3 | 6.8 | 91 | 94 | |
| High | 5.8 | 6.2 | 93 | 96 | |
| ILA | Low | 6.6 | 7.1 | 90 | 88 |
| Medium | 6.2 | 6.5 | 92 | 91 | |
| High | 5.5 | 6.0 | 94 | 95 | |
| IAA | Low | 6.8 | 7.4 | 89 | 89 |
| Medium | 6.3 | 6.8 | 93 | 92 | |
| High | 5.8 | 6.2 | 95 | 96 | |
| SK | Low | 5.3 | 6.0 | 87 | 90 |
| Medium | 5.0 | 5.2 | 90 | 93 | |
| High | 4.5 | 4.7 | 92 | 96 | |
| IALD | Low | 6.0 | 6.5 | 90 | 92 |
| Medium | 5.5 | 6.0 | 90 | 92 | |
| High | 5.0 | 5.6 | 93 | 94 | |
| IA | Low | 6.0 | 6.8 | 90 | 90 |
| Medium | 5.6 | 6.1 | 93 | 93 | |
| High | 5.3 | 5.5 | 96 | 96 | |
| IS1 | \ | 3.2 | 3.9 | 95 | 93 |
| IS2 | \ | 3.5 | 4.0 | 97 | 95 |
| IS3 | \ | 3.8 | 4.2 | 93 | 98 |
| IS4 | \ | 3.3 | 3.9 | 93 | 96 |
| Analyte | Linearity Range (ng/mL) | r2 | LOQ (ng/mL) |
|---|---|---|---|
| TRP | 25–10,000 | 0.9988 | 25 |
| NFK | 0.3–100 | 0.9991 | 0.3 |
| KYN | 5–1000 | 0.9989 | 5 |
| AA | 0.2–100 | 0.9993 | 0.2 |
| KYNA | 0.2–100 | 0.9992 | 0.2 |
| 3-HKYN | 0.2–100 | 0.9993 | 0.2 |
| XA | 0.2–100 | 0.9994 | 0.2 |
| 3-HAA | 0.2–100 | 0.9992 | 0.2 |
| QUIN | 1–500 | 0.9991 | 1 |
| PIC | 1–500 | 0.9990 | 1 |
| 2-AMA | 0.1–50 | 0.9995 | 0.1 |
| 5-HTP | 0.1–50 | 0.9994 | 0.1 |
| 5-HT | 1–500 | 0.9991 | 1 |
| 5-HIAA | 0.1–50 | 0.9996 | 0.1 |
| MEL | 0.2–100 | 0.9994 | 0.2 |
| IND | 0.1–50 | 0.9994 | 0.1 |
| TA | 0.1–50 | 0.9992 | 0.1 |
| IDE | 0.1–50 | 0.9993 | 0.1 |
| IPA | 1–500 | 0.9989 | 1 |
| ILA | 1–500 | 0.9991 | 1 |
| IAA | 1–500 | 0.9991 | 1 |
| SK | 0.1–50 | 0.9994 | 0.1 |
| IALD | 0.2–100 | 0.9992 | 0.2 |
| IA | 0.2–100 | 0.9993 | 0.2 |
| Individual | 1 | 2 | 3 |
|---|---|---|---|
| Sex | F | M | F |
| Blood VAMS Concentrations (ng/mL) | |||
| TRP | 6544 | 7620 | 7481 |
| NFK | 17.3 | 17.2 | 17.0 |
| KYN | 359 | 336 | 342 |
| AA | 8.2 | 7.5 | 7.4 |
| KYNA | 9.4 | 9.0 | 8.6 |
| 3-HKYN | 9.0 | 9.7 | 12.2 |
| XA | 7.4 | 7.7 | 7.0 |
| 3-HAA | 7.8 | 8.1 | 8.3 |
| QUIN | 49 | 52 | 55 |
| PIC | 55 | 51 | 51 |
| 2-AMA | 5.0 | 5.5 | 6.1 |
| 5-HTP | 1.4 | 3.2 | 2.3 |
| 5-HT | 189 | 159 | 175 |
| 5-HIAA | 3.1 | 2.7 | 2.7 |
| MEL | 15.0 | 12.9 | 13.2 |
| IND | 1.2 | 2.1 | 1.8 |
| TA | 1.3 | 1.5 | 1.7 |
| IDE | 0.3 | 0.3 | 0.3 |
| IPA | 166 | 168 | 187 |
| ILA | 226 | 230 | 255 |
| IAA | 300 | 311 | 296 |
| SK | 3.5 | 3.5 | 5.1 |
| IALD | 27.4 | 27.2 | 31.9 |
| IA | 11.6 | 11.6 | 16.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Protti, M.; Cirrincione, M.; Mandrioli, R.; Rudge, J.; Regazzoni, L.; Valsecchi, V.; Volpi, C.; Mercolini, L. Volumetric Absorptive Microsampling (VAMS) for Targeted LC-MS/MS Determination of Tryptophan-Related Biomarkers. Molecules 2022, 27, 5652. https://doi.org/10.3390/molecules27175652
Protti M, Cirrincione M, Mandrioli R, Rudge J, Regazzoni L, Valsecchi V, Volpi C, Mercolini L. Volumetric Absorptive Microsampling (VAMS) for Targeted LC-MS/MS Determination of Tryptophan-Related Biomarkers. Molecules. 2022; 27(17):5652. https://doi.org/10.3390/molecules27175652
Chicago/Turabian StyleProtti, Michele, Marco Cirrincione, Roberto Mandrioli, James Rudge, Luca Regazzoni, Valeria Valsecchi, Claudia Volpi, and Laura Mercolini. 2022. "Volumetric Absorptive Microsampling (VAMS) for Targeted LC-MS/MS Determination of Tryptophan-Related Biomarkers" Molecules 27, no. 17: 5652. https://doi.org/10.3390/molecules27175652
APA StyleProtti, M., Cirrincione, M., Mandrioli, R., Rudge, J., Regazzoni, L., Valsecchi, V., Volpi, C., & Mercolini, L. (2022). Volumetric Absorptive Microsampling (VAMS) for Targeted LC-MS/MS Determination of Tryptophan-Related Biomarkers. Molecules, 27(17), 5652. https://doi.org/10.3390/molecules27175652