
Synthesis and Strong Solvatochromism of Push-Pull Thienylthiazole Boron Complexes


Abstract
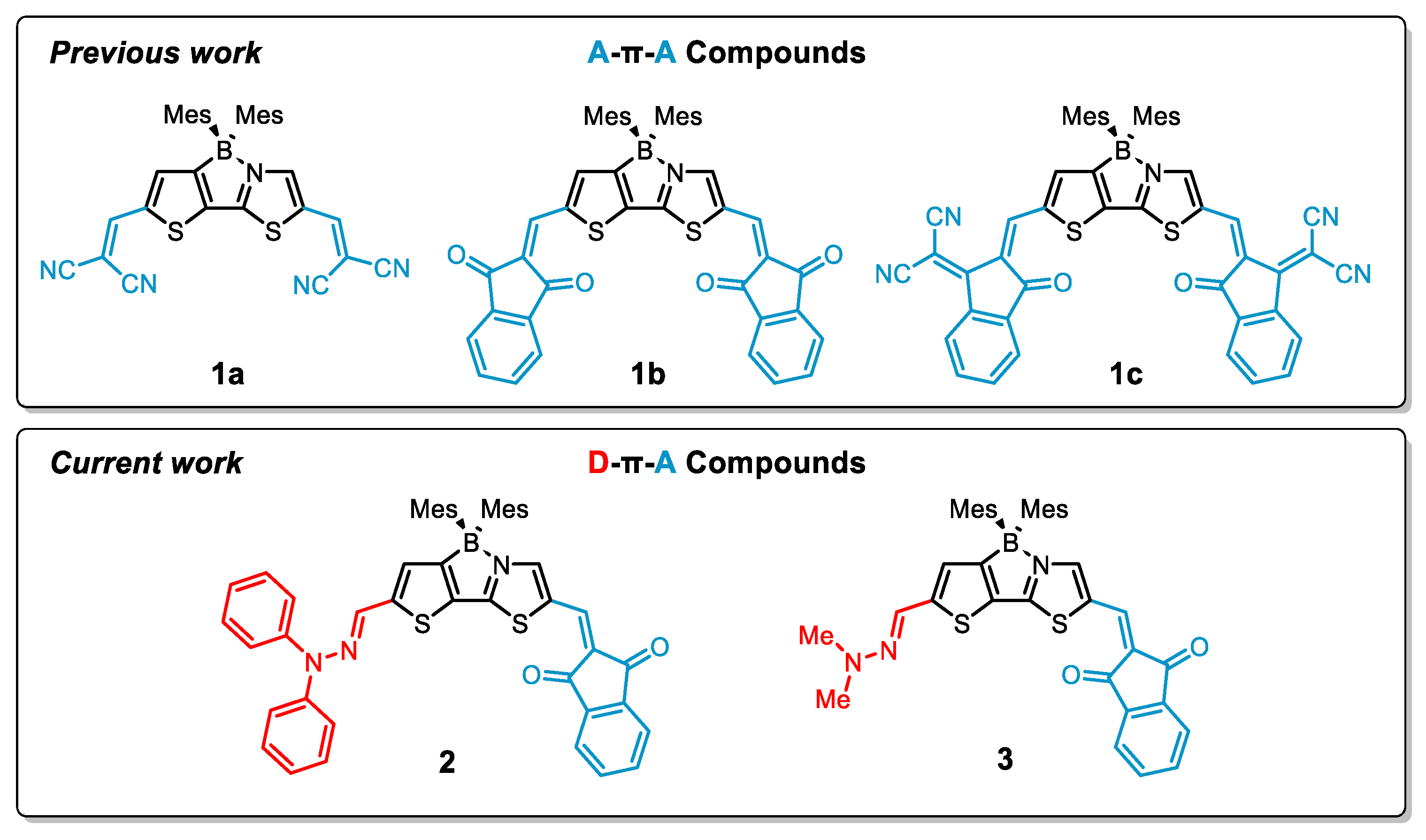
:1. Introduction
2. Results and Discussion
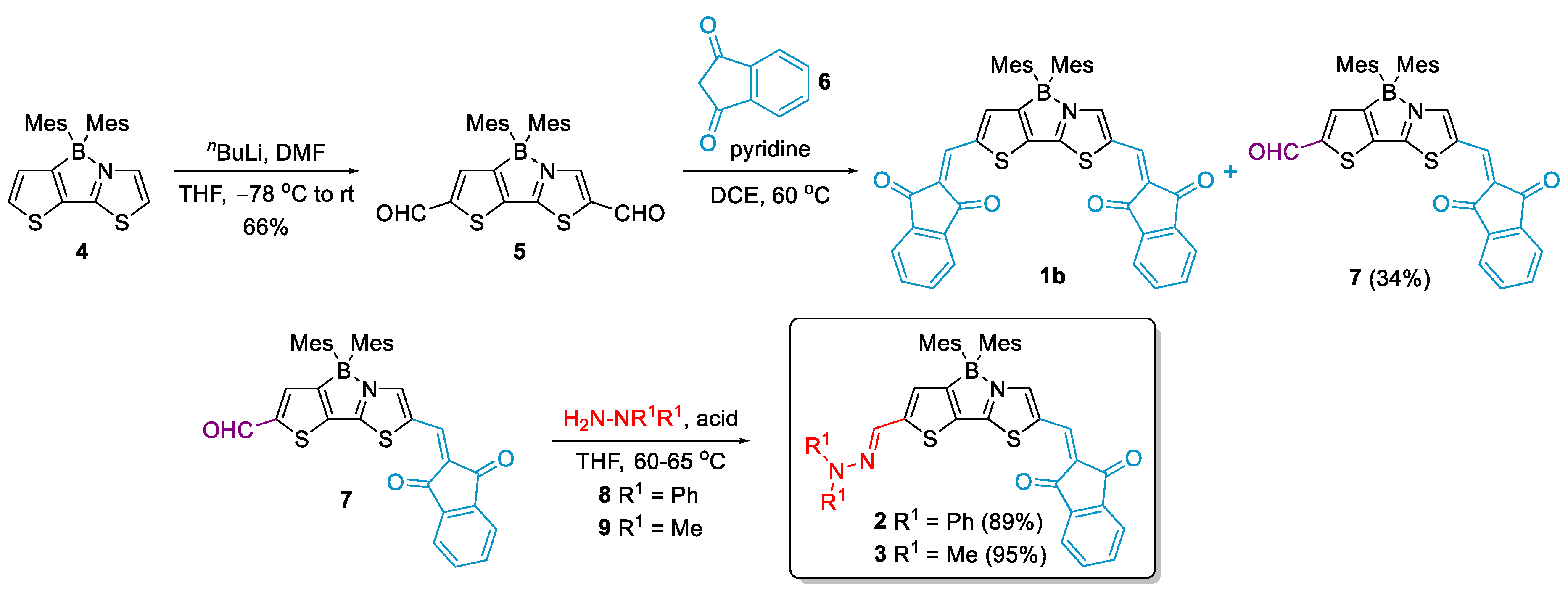
2.1. Synthesis
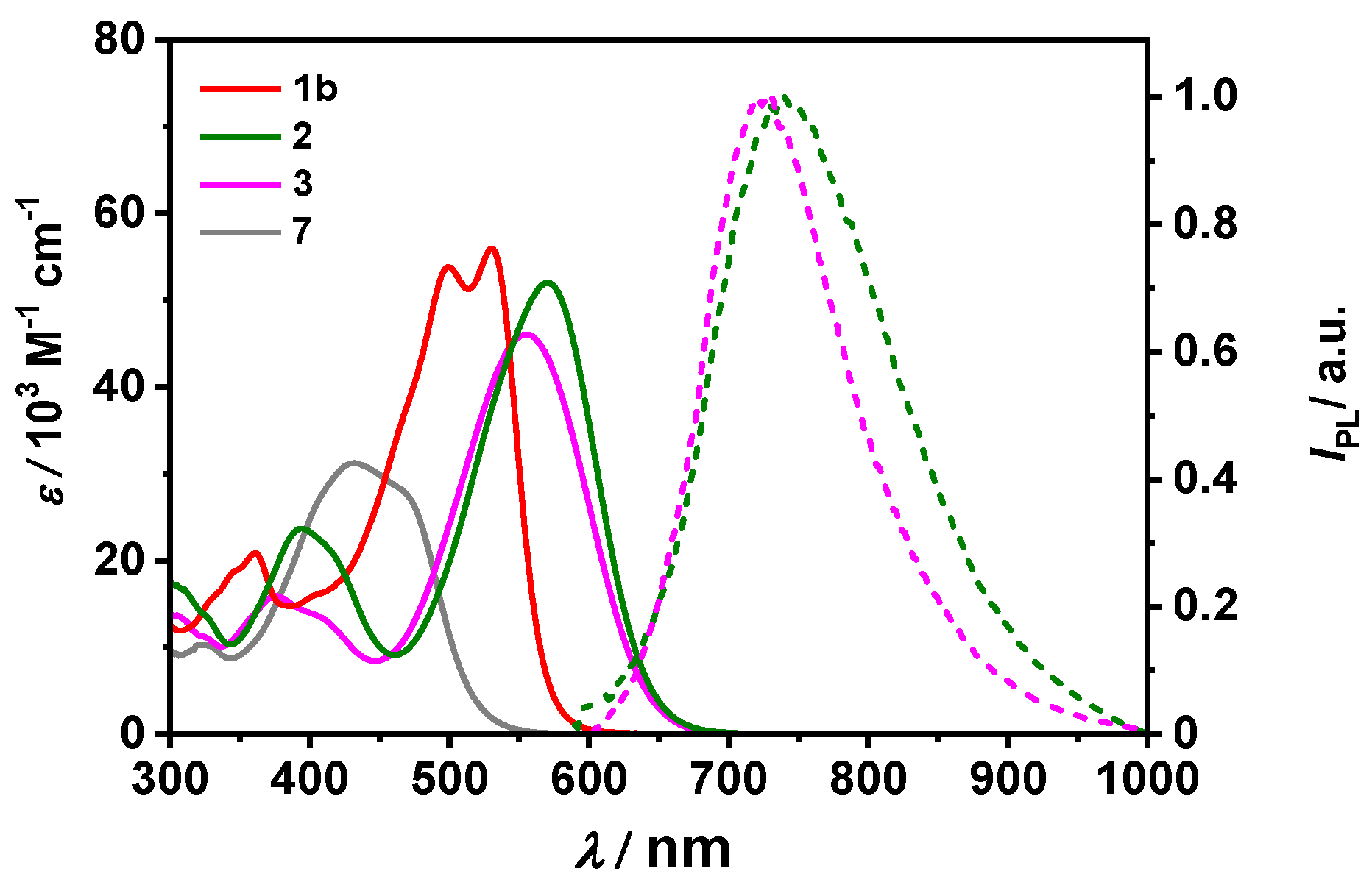
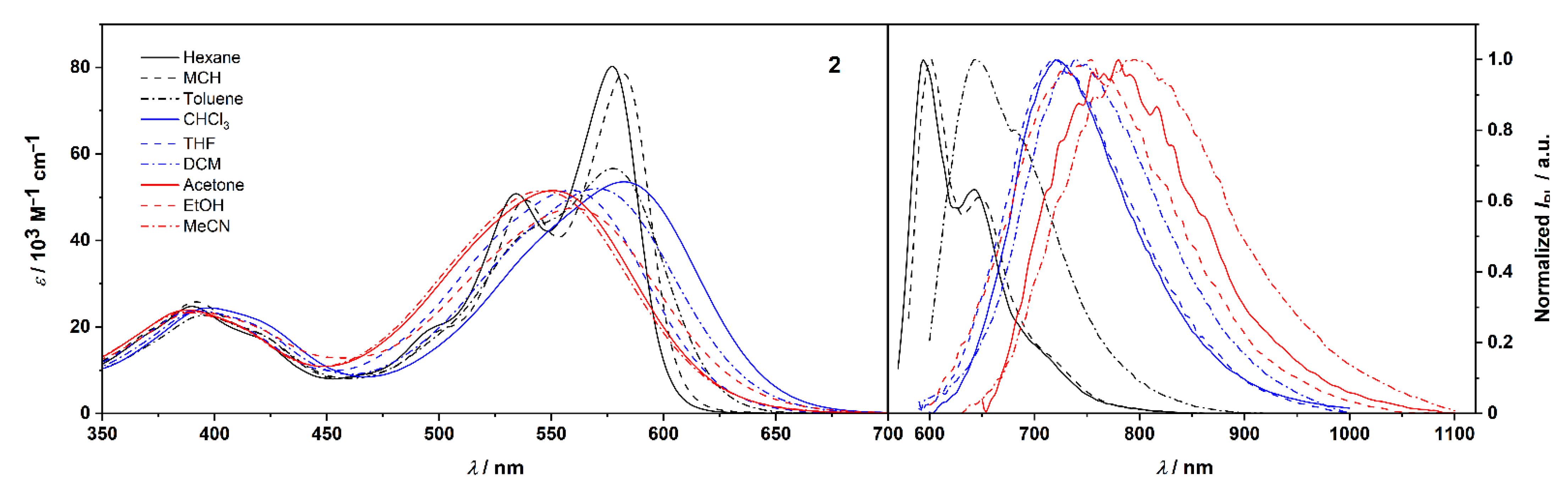
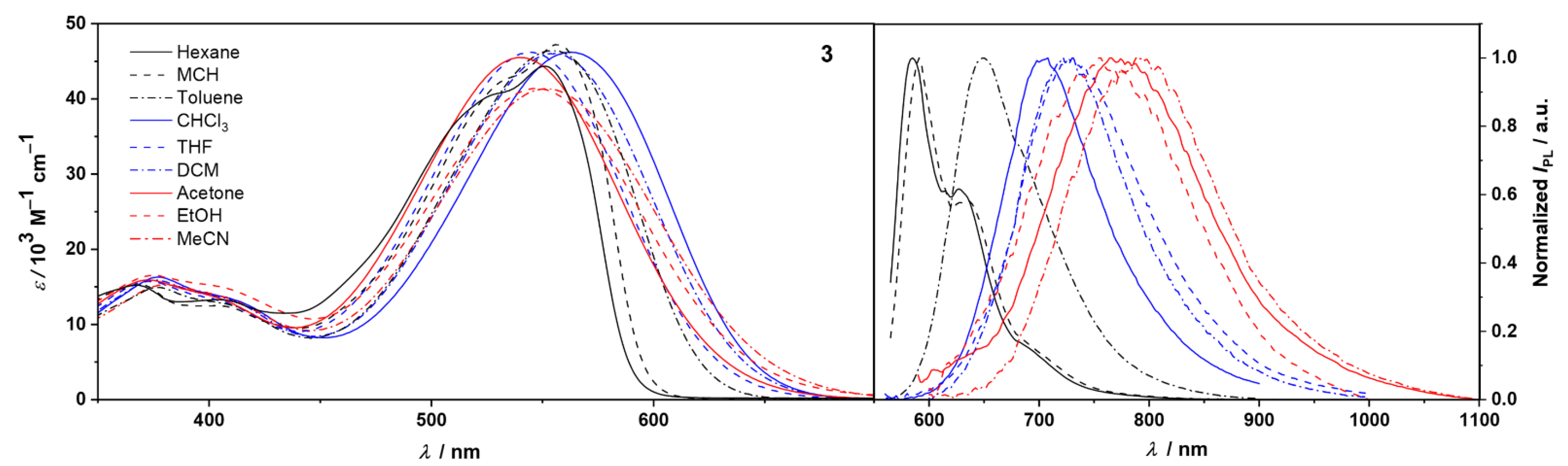
2.2. Photophysical and Solvatochromic Properties
2.3. Calculation of Ground and Excited State Dipole Moments
2.4. Quantum Chemical Calculations
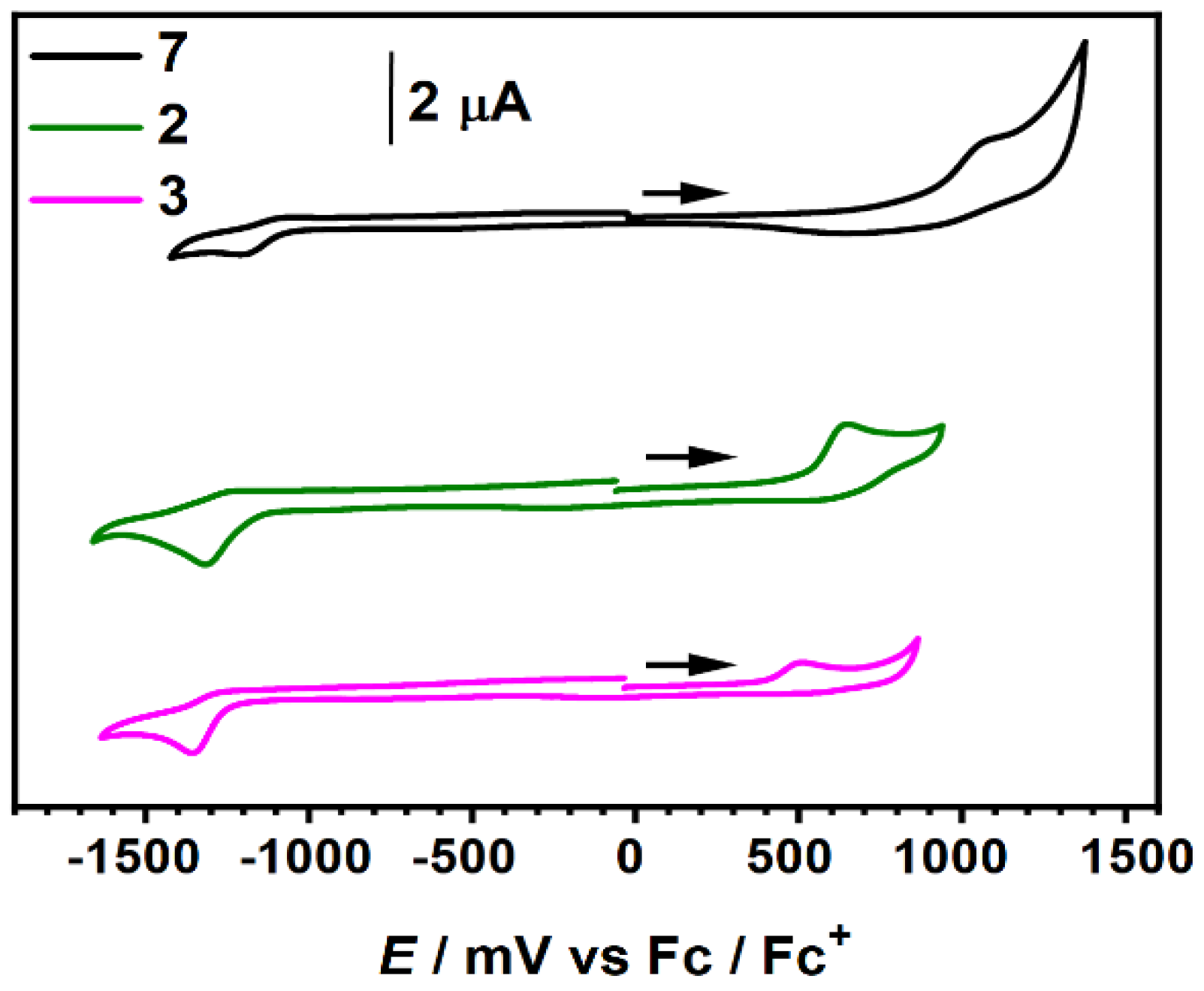
2.5. Electrochemistry
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedures
3.3. Computational Details
3.4. Absorption Spectroscopy
3.5. Fluorescence Spectroscopy
3.6. Electrochemical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, B.; Zhao, M.; Zhang, F. Rational Design of Near-Infrared-II Organic Molecular Dyes for Bioimaging and Biosensing. ACS Mater. Lett. 2020, 2, 905–917. [Google Scholar] [CrossRef]
- Fernandes, S.S.; Belsley, M.; Pereira, A.I.; Ivanou, D.; Mendes, A.; Justino, L.L.G.; Burrows, H.D.; Raposo, M.M.M. Push-Pull N,N-Diphenylhydrazones Bearing Bithiophene or Thienothiophene Spacers as Nonlinear Optical Second Harmonic Generators and as Photosensitizers for Nanocrystalline TiO2 Dye-Sensitized Solar Cells. ACS Omega 2018, 3, 12893–12904. [Google Scholar] [CrossRef]
- Dang, D.; Yu, D.; Wang, E. Conjugated Donor–Acceptor Terpolymers Toward High-Efficiency Polymer Solar Cells. Adv. Mater. 2019, 31, 1807019. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.K.; Kotresh, M.G.; Inamdar, S.R. A Combined Solvatochromic Shift and TDDFT Study Probing Solute-Solvent Interactions of Blue Fluorescent Alexa Fluor 350 Dye: Evaluation of Ground and Excited State Dipole Moments. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 215, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Issa, Y.M.; Abdel-Latif, S.A.; El-Ansary, A.L.; Hassib, H.B. The Synthesis, Spectroscopic Characterization, DFT/TD-DFT/PCM Calculations of the Molecular Structure and NBO of the Novel Charge-Transfer Complexes of Pyrazine Schiff Base Derivatives with Aromatic Nitro Compounds. New J. Chem. 2021, 45, 1482–1499. [Google Scholar] [CrossRef]
- Wei, Z.; Sharma, S.; Philip, A.M.; Sengupta, S.; Grozema, F.C. Excited State Dynamics of BODIPY-Based Acceptor-Donor-Acceptor Systems: A Combined Experimental and Computational Study. Phys. Chem. Chem. Phys. 2021, 23, 8900–8907. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Irfan, A. Computational Analysis to Understand the Performance Difference between Two Small-Molecule Acceptors Differing in Their Terminal Electron-Deficient Group. J. Comput. Electron. 2020, 19, 931–939. [Google Scholar] [CrossRef]
- Köse, M.E. Theoretical Estimation of Donor Strength of Common Conjugated Units for Organic Electronics. J. Phys. Chem. A 2019, 123, 5566–5573. [Google Scholar] [CrossRef]
- Bureš, F. Fundamental Aspects of Property Tuning in Push-Pull Molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Gandhimathi, S.; Balakrishnan, C.; Venkataraman, R.; Neelakantan, M.A. Crystal Structure, Solvatochromism and Estimation of Ground and Excited State Dipole Moments of an Allyl Arm Containing Schiff Base: Experimental and Theoretical Calculations. J. Mol. Liq. 2016, 219, 239–250. [Google Scholar] [CrossRef]
- Klikar, M.; Kityk, I.V.; Kulwas, D.; Mikysek, T.; Pytela, O.; Bureš, F. Multipodal Arrangement of Push-Pull Chromophores: A Fundamental Parameter Affecting Their Electronic and Optical Properties. New J. Chem. 2017, 41, 1459–1472. [Google Scholar] [CrossRef]
- Effenberger, F.; Würthner, F.; Steybe, F. Synthesis and Solvatochromic Properties of Donor-Acceptor-Substituted Oligothiophenes. J. Org. Chem. 1995, 60, 2082–2091. [Google Scholar] [CrossRef]
- Liu, J.; Gao, W.; Kityk, I.V.; Liu, X.; Zhen, Z. Optimization of Polycyclic Electron-Donors Based on Julolidinyl Structure in Push-Pull Chromophores for Second Order NLO Effects. Dyes Pigments 2015, 122, 74–84. [Google Scholar] [CrossRef]
- He, G.S.; Tan, L.S.; Zheng, Q.; Prasad, P.N. Multiphoton Absorbing Materials: Molecular Designs, Characterizations, and Applications. Chem. Rev. 2008, 108, 1245–1330. [Google Scholar] [CrossRef]
- Beckmann, S.; Etzbach, K.H.; Krämer, P.; Lukaszuk, K.; Matschiner, R.; Schmidt, A.J.; Schuhmacher, P.; Sens, R.; Seybold, G.; Wortmann, R.; et al. Electrooptical Chromophores for Nonlinear Optical and Photorefractive Applications. Adv. Mater. 1999, 11, 536–541. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, J.; Wan, X. Design and Synthesis of Piezochromic Materials Based on Push-Pull Chromophores: A Mechanistic Perspective. Chem. Eur. J. 2012, 18, 4558–4567. [Google Scholar] [CrossRef]
- Qian, F.; Zhang, C.; Zhang, Y.; He, W.; Gao, X.; Hu, P.; Guo, Z. Visible Light Excitable Zn2+ Fluorescent Sensor Derived from an Intramolecular Charge Transfer Fluorophore and Its In Vitro and In Vivo Application. J. Am. Chem. Soc. 2009, 131, 1460–1468. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Ji, L.; Edkins, R.M.; Sewell, L.J.; Beeby, A.; Batsanov, A.S.; Fucke, K.; Drafz, M.; Howard, J.A.K.; Moutounet, O.; Ibersiene, F.; et al. Experimental and Theoretical Studies of Quadrupolar Oligothiophene-Cored Chromophores Containing Dimesitylboryl Moieties as π-Accepting End-Groups: Syntheses, Structures, Fluorescence, and One- and Two-Photon Absorption. Chem. Eur. J. 2014, 20, 13618–13635. [Google Scholar] [CrossRef]
- Zhang, Z.; Edkins, R.M.; Nitsch, J.; Fucke, K.; Eichhorn, A.; Steffen, A.; Wang, Y.; Marder, T.B. D-π-A Triarylboron Compounds with Tunable Push-Pull Character Achieved by Modification of Both the Donor and Acceptor Moieties. Chem. Eur. J. 2015, 21, 177–190. [Google Scholar] [CrossRef]
- Zhang, Z.; Edkins, R.M.; Nitsch, J.; Fucke, K.; Steffen, A.; Longobardi, L.E.; Stephan, D.W.; Lambert, C.; Marder, T.B. Optical and Electronic Properties of Air-Stable Organoboron Compounds with Strongly Electron-Accepting Bis(fluoromesityl)boryl Groups. Chem. Sci. 2015, 6, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Shirasaka, T.; Akiyama, S.; Tamao, K. Dibenzoborole-Containing π-Electron Systems: Remarkable Fluorescence Change Based on the “on/off” Control of the pπ–π* Conjugation. J. Am. Chem. Soc. 2002, 124, 8816–8817. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.Y.W.; Wong, H.L.; Wong, Y.C.; Chan, M.Y.; Yam, V.W.W. Air-Stable Spirofluorene-Containing Ladder-Type Bis(alkynyl)borane Compounds with Readily Tunable Full Color Emission Properties. Chem. Eur. J. 2016, 22, 15095–15106. [Google Scholar] [CrossRef] [PubMed]
- Pais, V.F.; Alcaide, M.M.; Lõpez-Rodríguez, R.; Collado, D.; Nájera, F.; Pérez-Inestrosa, E.; Álvarez, E.; Lassaletta, J.M.; Fernández, R.; Ros, A.; et al. Strongly Emissive and Photostable Four-Coordinate Organoboron N, C Chelates and Their Use in Fluorescence Microscopy. Chem. Eur. J. 2015, 21, 15369–15376. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Kasatani, K.; Niwa, T.; Sato, H.; Funabiki, K.; Matsui, M. Synthesis and Fluorescence Properties of Pyrimidine-Based Diboron Complexes with Donor-π-Acceptor Structures. Chem. Eur. J. 2016, 22, 1816–1824. [Google Scholar] [CrossRef]
- Poon, C.-T.; Wu, D.; Yam, V.W.-W. Boron(III)-Containing Donor-Acceptor Compound with Goldlike Reflective Behavior for Organic Resistive Memory Devices. Angew. Chem. Int. Ed. 2016, 128, 3711–3715. [Google Scholar] [CrossRef]
- Dhanunjayarao, K.; Sa, S.; Aradhyula, B.P.R.; Venkatasubbaiah, K. Synthesis of Phenanthroimidazole-Based Four Coordinate Organoboron Compounds. Tetrahedron 2018, 74, 5819–5825. [Google Scholar] [CrossRef]
- Ruelas-Álvarez, G.Y.; Cárdenas-Valenzuela, A.J.; Galaviz-Moreno, L.L.; Cruz-Enríquez, A.; Campos-Gaxiola, J.J.; Höpf, H.; Baldenebro-López, J.; Vargas-Olvera, E.C.; Miranda-Soto, V.; García Grajeda, B.A.; et al. Four-Coordinate Monoboron Complexes with 8-Hydroxyquinolin-5-Sulfonate: Synthesis, Crystal Structures, Theoretical Studies, and Luminescence Properties. Crystals 2022, 12, 783. [Google Scholar] [CrossRef]
- Hecht, R.; Kade, J.; Schmidt, D.; Nowak-Król, A. n-Channel Organic Semiconductors Derived from Air-Stable Four-Coordinate Boron Complexes of Substituted Thienylthiazoles. Chem. Eur. J. 2017, 23, 11620–11628. [Google Scholar] [CrossRef]
- Aprahamian, I. Hydrazone Switches and Things in Between. Chem. Commun. 2017, 53, 6674–6684. [Google Scholar] [CrossRef]
- Li, X.; Zhang, C.; Wang, Y.; Liu, J.; Liu, J. Hydrazone Organic Compound with R2C=N–NR Substructure for Ultrafast Photonics. J. Phys. Chem. C 2020, 124, 22638–22645. [Google Scholar] [CrossRef]
- Abdelrahman, M.S.; Khattab, T.A.; Kamel, S. Development of a Novel Colorimetric Thermometer Based on Poly(N-Vinylcaprolactam) with Push-π-Pull Tricyanofuran Hydrazone Anion Dye. New J. Chem. 2021, 45, 5382–5390. [Google Scholar] [CrossRef]
- Beverina, L.; Crippa, M.; Landenna, M.; Ruffo, R.; Salice, P.; Silvestri, F.; Versari, S.; Villa, A.; Ciaffoni, L.; Collini, E.; et al. Assessment of Water-Soluble π-Extended Squaraines as One- and Two-Photon Singlet Oxygen Photosensitizers: Design, Synthesis, and Characterization. J. Am. Chem. Soc. 2008, 130, 1894–1902. [Google Scholar] [CrossRef]
- Chung, S.J.; Zheng, S.; Odani, T.; Beverina, L.; Fu, J.; Padilha, L.A.; Biesso, A.; Hales, J.M.; Zhan, X.; Schmidt, K.; et al. Extended Squaraine Dyes with Large Two-Photon Absorption Cross-Sections. J. Am. Chem. Soc. 2006, 128, 14444–14445. [Google Scholar] [CrossRef]
- Abbotto, A.; Beverina, L.; Manfredi, N.; Pagani, G.A.; Archetti, G.; Kuball, H.G.; Wittenburg, C.; Heck, J.; Holtmann, J. Second-Order Nonlinear Optical Activity of Dipolar Chromophores Ased on Pyrrole-Hydrazono Donor Moieties. Chem. Eur. J. 2009, 15, 6175–6185. [Google Scholar] [CrossRef]
- Wakamiya, A.; Taniguchi, T.; Yamaguchi, S. Intramolecular B–N Coordination as a Scaffold for Electron-Transporting Materials: Synthesis and Properties of Boryl-Substituted Thienylthiazoles. Angew. Chem. Int. Ed. 2006, 45, 3170–3173. [Google Scholar] [CrossRef]
- Kubota, Y.; Ozaki, Y.; Funabiki, K.; Matsui, M. Synthesis and Fluorescence Properties of Pyrimidine Mono- and Bisboron Complexes. J. Org. Chem. 2013, 78, 7058–7067. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, H.; Wang, S.; Li, Z.; Shen, Z. Asymmetric Boron-Complexes Containing Keto-Isoindolinyl and Pyridyl Groups: Solvatochromic Fluorescence, Efficient Solid-State Emission and DFT Calculations. J. Mater. Chem. C 2015, 3, 12281–12289. [Google Scholar] [CrossRef]
- Effenberger, F.; Würthner, F. 5-Dimethylamino-5′-nitro-2, 2′-bithiophene—A New Dye with Pronounced Positive Solvatochromism. Angew. Chem. Int. Ed. 1993, 32, 719–721. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4031. [Google Scholar] [CrossRef]
- Lee, S.C.; Heo, J.; Woo, H.C.; Lee, J.A.; Seo, Y.H.; Lee, C.L.; Kim, S.; Kwon, O.P. Fluorescent Molecular Rotors for Viscosity Sensors. Chem. Eur. J. 2018, 24, 13706–13718. [Google Scholar] [CrossRef]
- Haidekker, M.A.; Theodorakis, E.A. Environment-Sensitive Behavior of Fluorescent Molecular Rotors. J. Biol. Eng. 2010, 4, 11. [Google Scholar] [CrossRef]
- Karpenko, I.A.; Niko, Y.; Yakubovskyi, V.P.; Gerasov, A.O.; Bonnet, D.; Kovtun, Y.P.; Klymchenko, A.S. Push-Pull Dioxaborine as Fluorescent Molecular Rotor: Far-Red Fluorogenic Probe for Ligand-Receptor Interactions. J. Mater. Chem. C 2016, 4, 3002–3009. [Google Scholar] [CrossRef]
- Lippert, E. Dipolmoment Und Elektronenstruktur von Angeregten Molekülen. Z. Naturforsch. A 1955, 10, 541–545. [Google Scholar] [CrossRef]
- Mataga, N.; Kaifu, Y.; Koizumi, M. Solvent Effects upon Fluorescence Spectra and the Dipolemoments of Excited Molecules. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. [Google Scholar] [CrossRef]
- Onsager, L. Electric Moments of Molecular Liquids. J. Am. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlatin-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Apporximation with Correct Asymptotic Behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Sıdır, İ.; Sarı, T.; Gülseven Sıdır, Y.; Berber, H. Synthesis, Solvatochromism and Dipole Moment in the Ground and Excited States of Substitute Phenol Derivative Fluorescent Schiff Base Compounds. J. Mol. Liq. 2021, 346, 117075. [Google Scholar] [CrossRef]
- Korsaye, F.A. Following the Density Evolution Using Real Time Density Functional Theory and Density Based Indexes: Application to Model Push–Pull Molecules. J. Comput. Chem. 2022, 43, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Hickey, A.L.; Rowley, C.N. Benchmarking Quantum Chemical Methods for the Calculation of Molecular Dipole Moments and Polarizabilities. J. Phys. Chem. A 2014, 118, 3678–3687. [Google Scholar] [CrossRef]
- Hait, D.; Head-Gordon, M. How Accurate Is Density Functional Theory at Predicting Dipole Moments? An Assessment Using a New Database of 200 Benchmark Values. J. Chem. Theory Comput. 2018, 14, 1969–1981. [Google Scholar] [CrossRef]
- Lokshin, V.; Sigalov, M.; Larina, N.; Khodorkovsky, V. Dipole Moments of Conjugated Donor-Acceptor Substituted Systems: Calculations vs. Experiments. RSC Adv. 2021, 11, 934–945. [Google Scholar] [CrossRef]
- Brand, C.; Meerts, W.L.; Schmitt, M. How and Why Do Transition Dipole Moment Orientations Depend on Conformer Structure? J. Phys. Chem. A 2011, 115, 9612–9619. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999. [Google Scholar]
- Sens, R.; Drexhage, K.H. Fluorescence Quantum Yield of Oxazine and Carbazine Laser Dyes. J. Lumin. 1981, 24-25, 709–712. [Google Scholar] [CrossRef]
- Seybold, G.; Wagenblast, G. New Perylene and Violanthrone Dyestuffs for Fluorescent Collectors. Dyes Pigments 1989, 11, 303–317. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Hansen, W.N.; Hansen, G.J. Absolute half-cell potential: A simple direct measurement. Phys. Rev. A 1987, 36, 1396–1402. [Google Scholar] [CrossRef]
- Cardona, C.M.; Li, W.; Kaifer, A.E.; Stockdale, D.; Bazan, G.C. Electrochemical considerations for determining absolute frontier orbital energy levels of conjugated polymers for solar cell applications. Adv. Mater. 2011, 23, 2367–2371. [Google Scholar] [CrossRef]
- Wong, M.W.; Wiberg, K.B.; Frisch, M.J. Ab initio calculation of molar volumes: Comparison with experiment and use in solvation models. J. Comp. Chem. 1995, 16, 385–394. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | λabsa [nm] | εmaxb [103 M−1 cm−1] | μ2egc [D2] | [D2 mol g−1] | [V] | [V] |
|---|---|---|---|---|---|---|
| 2 | 571 | 52.0 | 91 | 0.12 | −1.32 f | +0.65f |
| 3 | 556 | 46.0 | 88 | 0.14 | −1.36 f | +0.51 f |
| 7 | 432 | 31.2 | 79 | 0.13 | −1.21 f | +1.08 f |
| 1b [29] | 530 | 55.9 | 118 | 0.16 | −1.12, −1.49 f | +0.89 f, +1.01 f, +1.17 f |
| 499 | 53.8 |
| Cpd | Solvent | εra | λabsb [nm] | εmaxc [103 M−1cm−1] | µ2egd [D2] | λeme [nm] | [cm−1] | τ1g [ns] (% amp) | τ2h [ns] (% amp) | Φfli [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | n-hexane | 1.9 | 577 | 80.3 | 89 | 595 | 520 | 0.5 (96.4) | 2.12 (3.6) | 11 ± 1 |
| MCH | 2.02 | 582 | 78.6 | 89 | 601 | 540 | 0.91 | - | 19 ± 2 | |
| toluene | 2.38 | 577 | 56.7 | 86 | 644 | 1800 | 0.52 (96.3) | 2.31 (3.7) | 8 ± 1 | |
| CHCl3 | 4.8 | 583 | 53.6 | 91 | 722 | 3300 | 0.44 (93.8) | 1.61 (6.2) | 3 ± 1 | |
| THF | 7.6 | 558 | 51.6 | 90 | 720 | 4030 | 0.3 (90.5) | 1.38 (9.5) | 2 ± 1 | |
| CH2Cl2 | 9.1 | 571 | 52.0 | 91 | 740 | 4000 | 0.19 (81.6) | 1.32 (18.4) | 1 ± 1 | |
| acetone | 20.6 | 551 | 51.6 | 93 | 772 | 5200 | 0.23 (78.1) | 1.02 (21.9) | <1 | |
| EtOH | 22.4 | 560 | 47.4 | 89 | 748 | 4490 | - | - | 5 ± 1 | |
| MeCN | 37.5 | 548 | 49.8 | 93 | 800 | 5750 | - | - | 1 ± 1 | |
| 3 | n-hexane | 1.9 | 551 | 44.4 | 78 | 585 | 1060 | 2.73 | - | 75 ± 1 |
| MCH | 2.02 | 557 | 47.2 | 78 | 591 | 1030 | 2.77 | - | 73 ± 1 | |
| toluene | 2.38 | 554 | 46.4 | 82 | 648 | 2620 | 2.88 | - | 41 ± 1 | |
| CHCl3 | 4.8 | 562 | 46.2 | 87 | 698 | 3470 | 1.57 | - | 15 ± 1 | |
| THF | 7.6 | 545 | 46.2 | 88 | 729 | 4630 | 0.69 (93.3) | 2.21 (6.7) | 5 ± 1 | |
| CH2Cl2 | 9.1 | 556 | 46.0 | 88 | 726 | 4210 | 1.02 | - | 7 ± 1 | |
| acetone | 20.6 | 539 | 45.5 | 90 | 776 | 5670 | 0.31 (75.6) | 1.18 (24.4) | <1 | |
| EtOH | 22.4 | 547 | 41.4 | 84 | 756 | 5050 | 1.57 | - | 1± 1 | |
| MeCN | 37.5 | 537 | 44.0 | 89 | 774 | 5700 | - | - | <1 |
| Cpd | Solvent | μg | μe a | Δμeg |
|---|---|---|---|---|
| 2 | n-hexane | 5.08 | 12.57 | 7.49 |
| MCH | 5.11 | 12.71 | 7.6 | |
| toluene | 5.18 | 13.00 | 7.82 | |
| CHCl3 | 5.36 | 13.81 | 8.45 | |
| THF | 5.41 | 14.14 | 8.73 | |
| CH2Cl2 | 5.43 | 14.23 | 8.8 | |
| acetone | 5.45 | 14.59 | 9.14 | |
| EtOH | 5.46 | 14.54 | 9.08 | |
| MeCN | 5.46 | 14.60 | 9.14 | |
| 3 | n-hexane | 8.05 | 13.57 | 5.52 |
| MCH | 8.16 | 13.81 | 5.65 | |
| toluene | 8.39 | 14.30 | 5.91 | |
| CHCl3 | 9.16 | 15.97 | 6.81 | |
| THF | 9.49 | 16.72 | 7.23 | |
| CH2Cl2 | 9.59 | 16.96 | 7.37 | |
| acetone | 9.89 | 17.87 | 7.98 | |
| EtOH | 9.92 | 17.76 | 7.84 | |
| MeCN | 9.98 | 17.91 | 7.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wildervanck, M.J.; Hecht, R.; Nowak-Król, A. Synthesis and Strong Solvatochromism of Push-Pull Thienylthiazole Boron Complexes. Molecules 2022, 27, 5510. https://doi.org/10.3390/molecules27175510
Wildervanck MJ, Hecht R, Nowak-Król A. Synthesis and Strong Solvatochromism of Push-Pull Thienylthiazole Boron Complexes. Molecules. 2022; 27(17):5510. https://doi.org/10.3390/molecules27175510
Chicago/Turabian StyleWildervanck, Martijn J., Reinhard Hecht, and Agnieszka Nowak-Król. 2022. "Synthesis and Strong Solvatochromism of Push-Pull Thienylthiazole Boron Complexes" Molecules 27, no. 17: 5510. https://doi.org/10.3390/molecules27175510
APA StyleWildervanck, M. J., Hecht, R., & Nowak-Król, A. (2022). Synthesis and Strong Solvatochromism of Push-Pull Thienylthiazole Boron Complexes. Molecules, 27(17), 5510. https://doi.org/10.3390/molecules27175510