Assessment of a Computational Approach to Predict Drug Resistance Mutations for HIV, HBV and SARS-CoV-2

 ,
,
Abstract
:1. Introduction
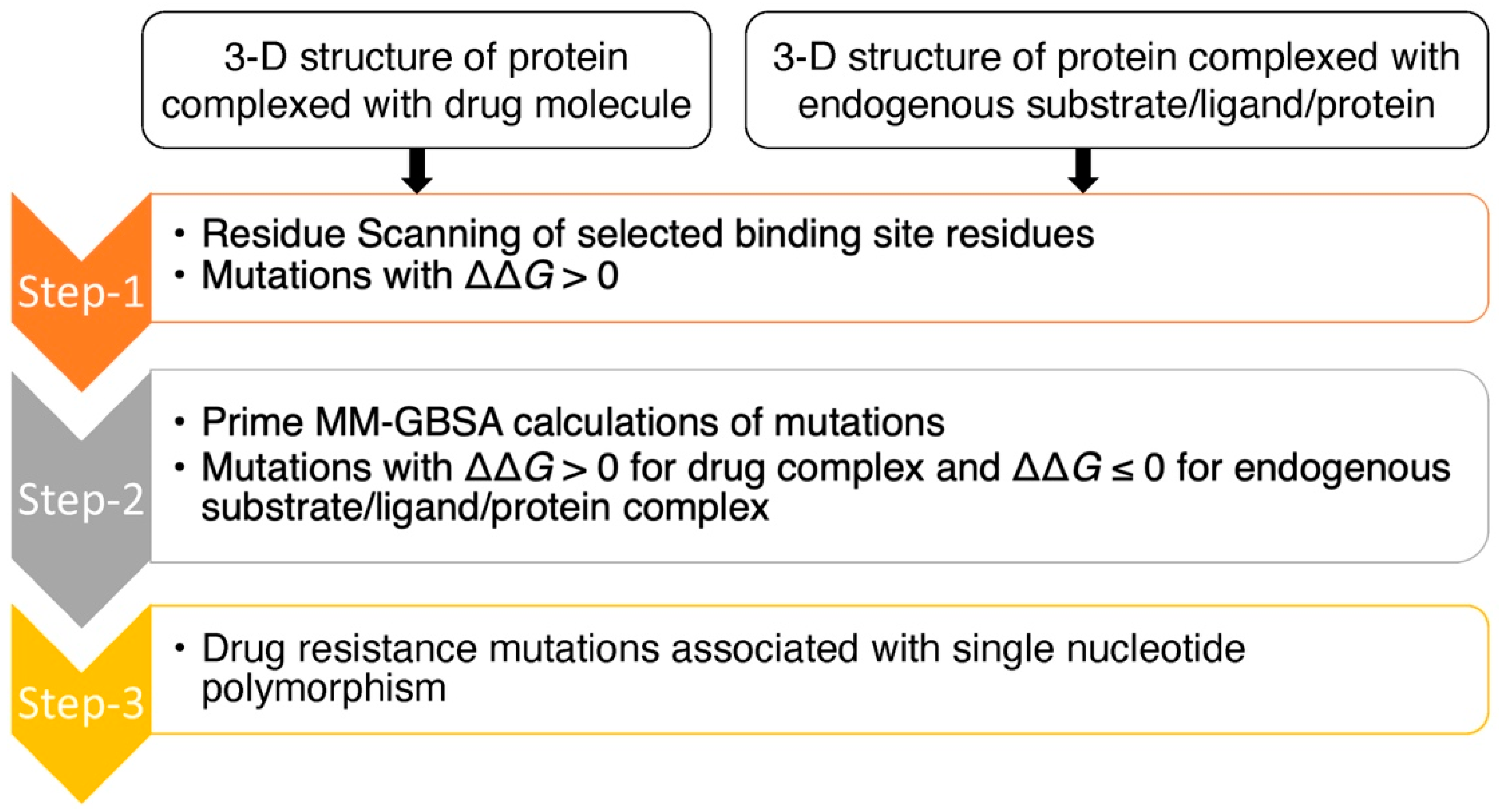
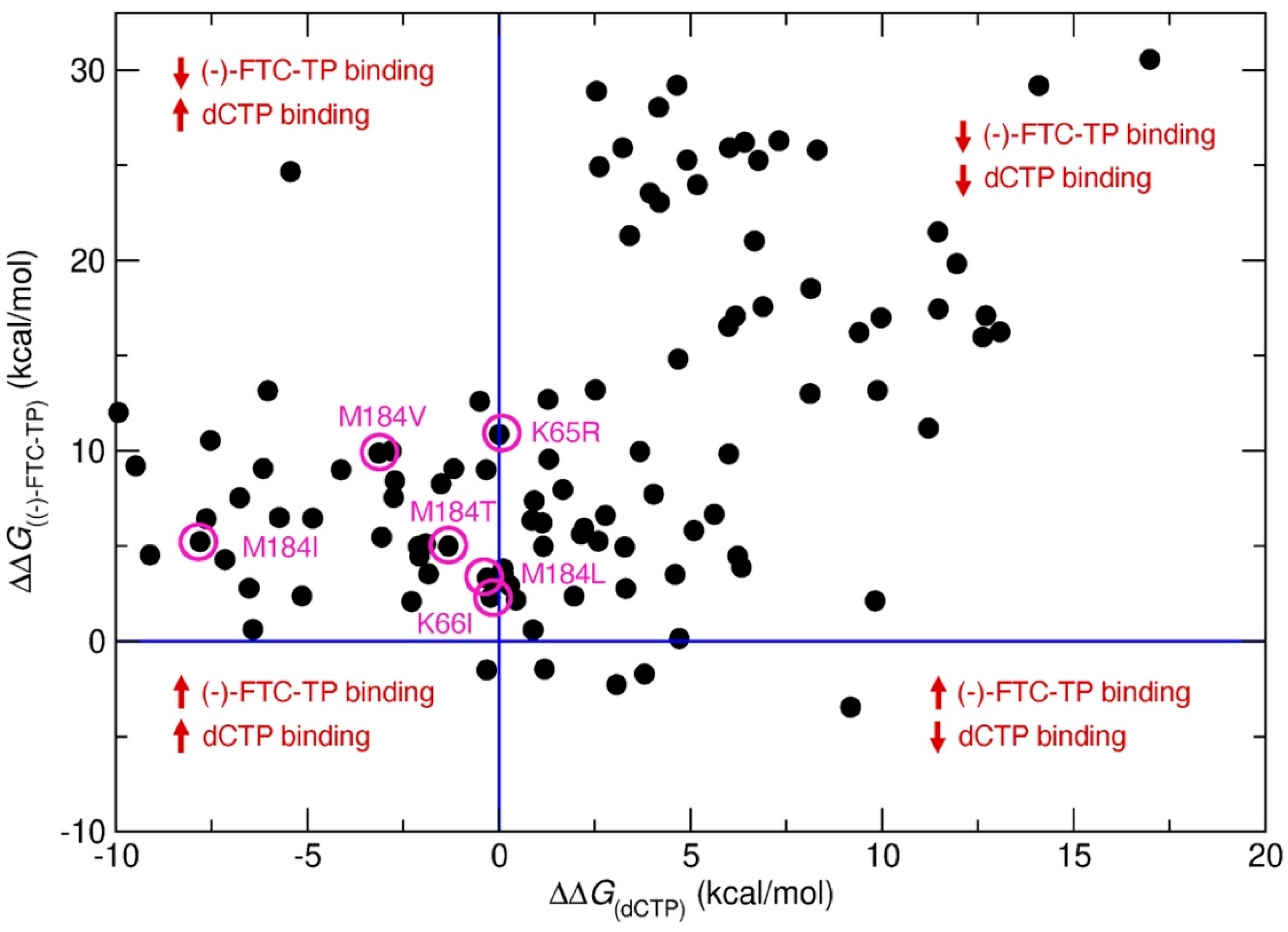
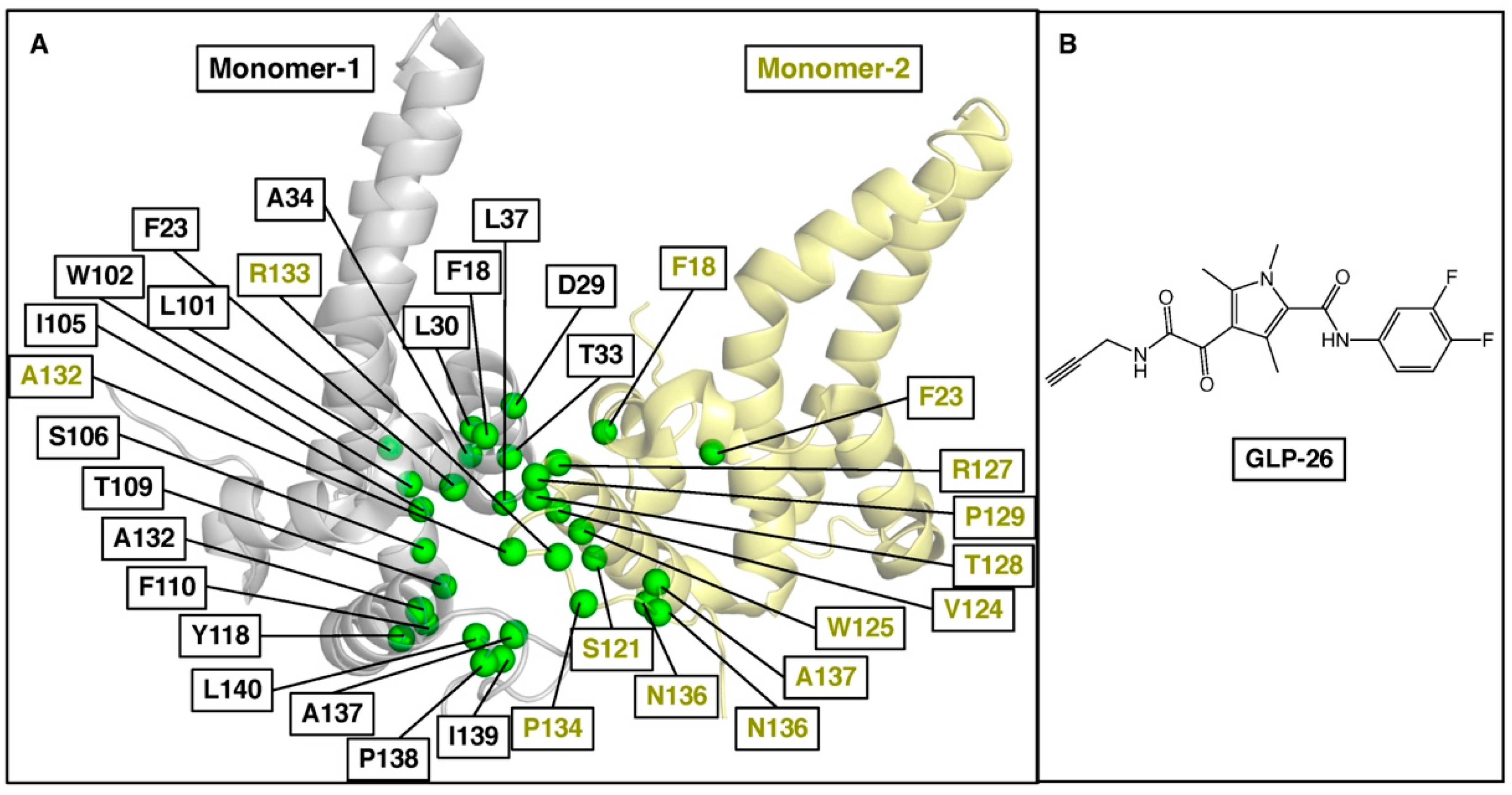
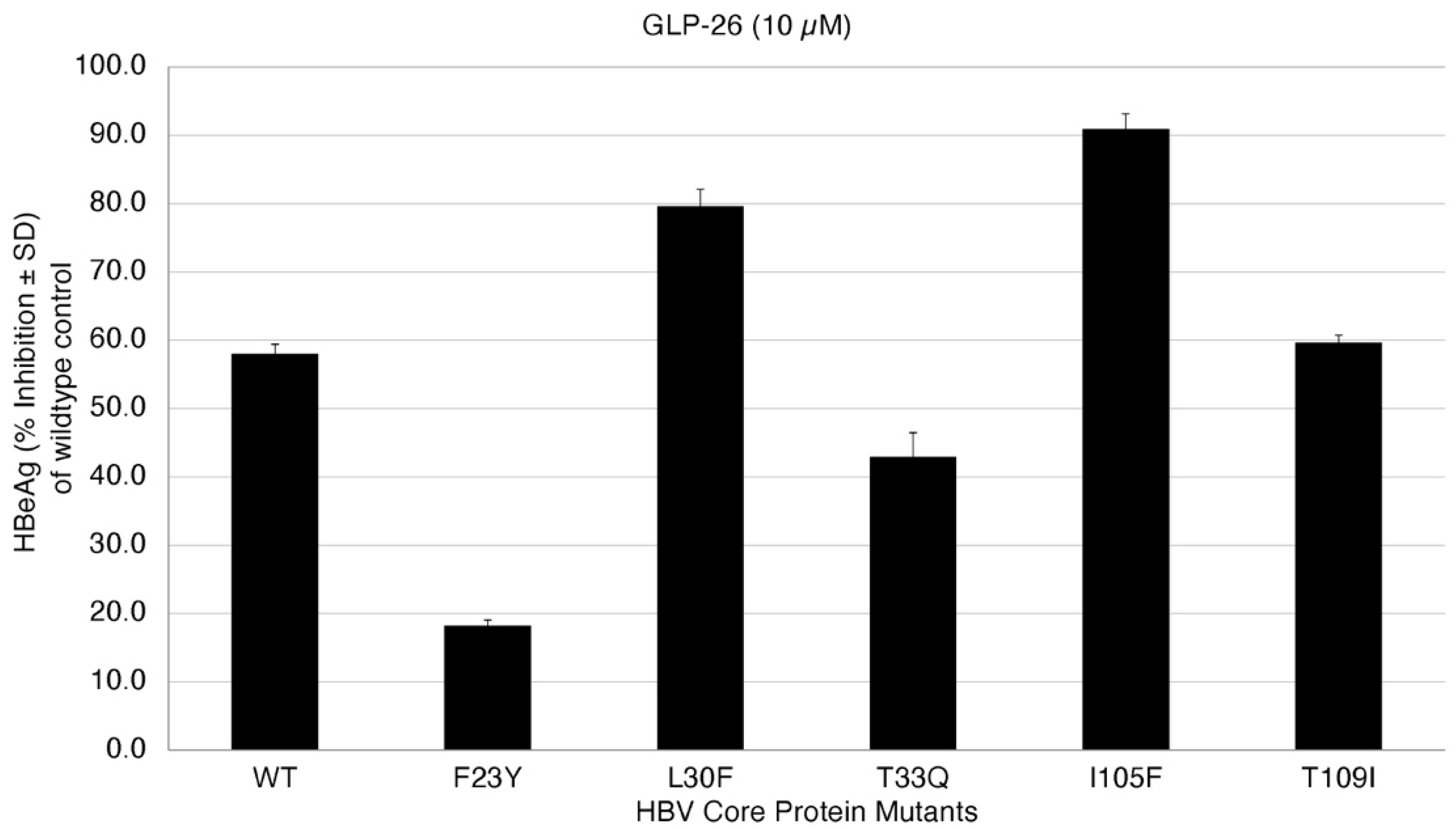
2. Results and Discussion
3. Materials and Methods
3.1. Test System Selection and Preparation
3.2. Residue Scanning
3.3. Prime MM-GBSA Calculations
3.4. Cell Lines
3.5. Compound Synthesis
3.6. Transfection of Full-Length HBV DNA into HepNTCP-DL Cells
3.7. Analysis of HBV HBeAg Production

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pair Name | Sequence Information |
|---|---|
| Core T109I Mutant c326t | 5′-ctataactgtttctcttccaaaaatgagacaagaaatgtgaaaccac-3’ |
| 5′-tgtgtttcacatttcttgtctcatttttggaagagaaacagttatag-3’ | |
| Core I105F Mutant a313t | 5’-ccaaaagtgagacaagaaaagtgaaaccacaagagttgc-3’ |
| 5’-gcaactcttgtggtttcacttttcttgtctcacttttgg-3’ | |
| Core T33Q Mutant | 5’-atacagagctgaggcctgatctagaagatctcgtactgaaggaaaga-3’ |
| a97c_c98a_c99g | 5’-tctttccttcagtacgagatcttctagatcaggcctcagctctgtat-3’ |
| Core L30F Mutant c88t | 5’-gcggtatctagaaaatctcgtactgaaggaaagaagtc-3’ |
| 5’-gacttctttccttcagtacgagattttctagataccgc-3’ | |
| Core F23Y Mutant t68a | 5’-tctcgtactgaaggaaagtagtcagaaggcaaaaacg-3’ |
| 5’-cgtttttgccttctgactactttccttcagtacgaga-3’ |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perrin, L.; Telenti, A. Hiv treatment failure: Testing for HIV resistance in clinical practice. Science 1998, 280, 1871–1873. [Google Scholar] [CrossRef] [PubMed]
- Elhence, A.; Singh, A.; Anand, A.; Kumar, R.; Ashraf, A.; Kumar, S.; Pradhan, D.; Pathak, P.; Vaishnav, M.; Rajput, M.S.; et al. Real-world re-treatment outcomes of direct-acting antiviral therapy failure in patients with chronic hepatitis C. J. Med. Virol. 2021, 93, 4982–4991. [Google Scholar] [CrossRef] [PubMed]
- Stuyver, L.J.; Locarnini, S.A.; Lok, A.; Richman, D.D.; Carman, W.F.; Dienstag, J.L.; Schinazi, R.F. Nomenclature for antiviral-resistant human hepatitis B virus mutations in the polymerase region. Hepatology 2001, 33, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Burrel, S.; Topalis, D.; Boutolleau, D. Herpes simplex virus resistance to antivirals. Virologie 2020, 24, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Borst, P. Genetic mechanisms of drug resistance. A review. Acta Oncol. 1991, 30, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.W.; Burt, S.K. Structural mechanisms of HIV drug resistance. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 545–571. [Google Scholar] [CrossRef]
- Mason, S.; Devincenzo, J.P.; Toovey, S.; Wu, J.Z.; Whitley, R.J. Comparison of antiviral resistance across acute and chronic viral infections. Antivir. Res. 2018, 158, 103–112. [Google Scholar] [CrossRef]
- Vere Hodge, A.; Field, H.J. General mechanisms of antiviral resistance. In Genetics and Evolution of Infectious Disease; Elsevier: Amsterdam, The Netherlands, 2011; pp. 339–362. [Google Scholar] [CrossRef]
- Amblard, F.; Patel, D.; Michailidis, E.; Coats, S.J.; Kasthuri, M.; Biteau, N.; Tber, Z.; Ehteshami, M.; Schinazi, R.F. HIV nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2022, 240, 114554. [Google Scholar] [CrossRef]
- Schinazi, R. Combined chemotherapeutic modalities for viral infections: Rationale and clinical potential. In Synergism and Antagonism in Chemotherapy; Chou, T.C., Rideout, D.C., Eds.; Academic Press: Orlando, FL, USA, 1991. [Google Scholar]
- Schinazi, R.; Halfon, P.; Marcellin, P.; Asselah, T. HCV direct-acting antiviral agents: The best interferon-free combinations. Liver Int. 2014, 34, 69–78. [Google Scholar] [CrossRef]
- Vacca, J.P.; Dorsey, B.D.; Schleif, W.A.; Levin, R.B.; McDaniel, S.L.; Darke, P.L.; Zugay, J.; Quintero, J.C.; Blahy, O.M.; Roth, E.; et al. L-735,524: An orally bioavailable human immunodeficiency virus type 1 protease inhibitor. Proc. Natl. Acad. Sci. USA 1994, 91, 4096–4100. [Google Scholar] [CrossRef] [Green Version]
- Larder, B.A.; Kemp, S.D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (azt). Science 1989, 246, 1155–1158. [Google Scholar] [CrossRef]
- Cao, Z.W.; Han, L.Y.; Zheng, C.J.; Ji, Z.L.; Chen, X.; Lin, H.H.; Chen, Y.Z. Computer prediction of drug resistance mutations in proteins. Drug Discov. Today 2005, 10, 521–529. [Google Scholar] [CrossRef]
- Steiner, M.C.; Gibson, K.M.; Crandall, K.A. Drug resistance prediction using deep learning techniques on HIV-1 sequence data. Viruses 2020, 12, 560. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Däumer, M.; Oette, M.; Korn, K.; Hoffmann, D.; Kaiser, R.; Lengauer, T.; Selbig, J.; Walter, H. Geno2pheno: Estimating phenotypic drug resistance from hiv-1 genotypes. Nucleic Acids Res. 2003, 31, 3850–3855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemenschneider, M.; Hummel, T.; Heider, D. Shiva—A web application for drug resistance and tropism testing in hiv. BMC Bioinform. 2016, 17, 314. [Google Scholar] [CrossRef] [Green Version]
- Bonet, I. Machine learning for prediction of hiv drug resistance: A review. Curr. Bioinform. 2015, 10, 579–585. [Google Scholar] [CrossRef]
- Khalid, Z.; Sezerman, O.U. Prediction of hiv drug resistance by combining sequence and structural properties. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 966–973. [Google Scholar] [CrossRef]
- Weber, I.T.; Harrison, R.W. Tackling the problem of HIV drug resistance. Postep. Biochem. 2016, 62, 273–279. [Google Scholar] [CrossRef]
- Jenwitheesuk, E.; Samudrala, R. Prediction of HIV-1 protease inhibitor resistance using a protein-inhibitor flexible docking approach. Antivir. Ther. 2005, 10, 157–166. [Google Scholar] [CrossRef]
- Toor, J.S.; Sharma, A.; Kumar, R.; Gupta, P.; Garg, P.; Arora, S.K. Prediction of drug-resistance in HIV-1 subtype c based on protease sequences from art naive and first-line treatment failures in north india using genotypic and docking analysis. Antivir. Res. 2011, 92, 213–218. [Google Scholar] [CrossRef]
- Hou, T.; Yu, R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J. Med. Chem. 2007, 50, 1177–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agniswamy, J.; Louis, J.M.; Roche, J.; Harrison, R.W.; Weber, I.T. Structural studies of a rationally selected multi-drug resistant HIV-1 protease reveal synergistic effect of distal mutations on flap dynamics. PLoS ONE 2016, 11, e0168616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, G.F.; Yang, G.F.; Zhan, C.G. Structure-based methods for predicting target mutation-induced drug resistance and rational drug design to overcome the problem. Drug Discov. Today 2012, 17, 1121–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Lupyan, D.; Wang, L. Improving the accuracy of protein thermostability predictions for single point mutations. Biophys. J. 2020, 119, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Pucci, F.; Bernaerts, K.; Teheux, F.; Gilis, D.; Rooman, M. Symmetry principles in optimization problems: An application to protein stability prediction. IFAC-Pap. 2015, 48, 458–463. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011, 79, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Smit, E. Antiviral resistance testing. Curr. Opin. Infect. Dis. 2014, 27, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Irwin, K.K.; Renzette, N.; Kowalik, T.F.; Jensen, J.D. Antiviral drug resistance as an adaptive process. Virus Evol. 2016, 2, vew014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, E.; Das, K.; Ding, J.; Yadav, P.N.; Hsiou, Y.; Boyer, P.L.; Hughes, S.H. Targeting HIV reverse transcriptase for anti-aids drug design: Structural and biological considerations for chemotherapeutic strategies. Drug Des. Discov. 1996, 13, 29–47. [Google Scholar] [PubMed]
- Schinazi, R.F. Emtricitabine: A viewpoint by Raymond F. Schinazi. Drugs 2003, 63, 2425–2426. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Lloyd, R.M., Jr.; Nguyen, M.H.; Cannon, D.L.; McMillan, A.; Ilksoy, N.; Chu, C.K.; Liotta, D.C.; Bazmi, H.Z.; Mellors, J.W. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob. Agents Chemother. 1993, 37, 875–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinazi, R.F.; McMillan, A.; Cannon, D.; Mathis, R.; Lloyd, R.M.; Peck, A.; Sommadossi, J.P.; St Clair, M.; Wilson, J.; Furman, P.A.; et al. Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob. Agents Chemother. 1992, 36, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.S.; Murakami, E.; Peterson, C.N.; Shi, J.; Schinazi, R.F.; Anderson, K.S. Interactions of enantiomers of 2′,3′-didehydro-2′,3′-dideoxy-fluorocytidine with wild type and m184v mutant hiv-1 reverse transcriptase. Antivir. Res. 2002, 56, 189–205. [Google Scholar] [CrossRef]
- Tisdale, M.; Kemp, S.D.; Parry, N.R.; Larder, B.A. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3′-thiacytidine inhibitors due to a mutation in the ymdd region of reverse transcriptase. Proc. Natl. Acad. Sci. USA 1993, 90, 5653–5656. [Google Scholar] [CrossRef] [Green Version]
- Hiv Drug Resistance Database. Available online: https://hivdb.stanford.edu/dr-summary/resistance-notes/NRTI/ (accessed on 14 July 2022).
- Feng, J.Y.; Myrick, F.T.; Margot, N.A.; Mulamba, G.B.; Rimsky, L.; Borroto-Esoda, K.; Selmi, B.; Canard, B. Virologic and enzymatic studies revealing the mechanism of K65R- and Q151M-associated HIV-1 drug resistance towards emtricitabine and lamivudine. Nucleosides Nucleotides Nucleic Acids 2006, 25, 89–107. [Google Scholar] [CrossRef]
- Seitz, S.; Habjanic, J.; Schutz, A.K.; Bartenschlager, R. The hepatitis B virus envelope proteins: Molecular gymnastics throughout the viral life cycle. Annu. Rev. Virol. 2020, 7, 263–288. [Google Scholar] [CrossRef]
- Amblard, F.; Boucle, S.; Bassit, L.; Chen, Z.; Sari, O.; Cox, B.; Verma, K.; Ozturk, T.; Ollinger-Russell, O.; Schinazi, R.F. Discovery and structure activity relationship of glyoxamide derivatives as anti-hepatitis B virus agents. Bioorg. Med. Chem. 2021, 31, 115952. [Google Scholar] [CrossRef] [PubMed]
- Amblard, F.; Boucle, S.; Bassit, L.; Cox, B.; Sari, O.; Tao, S.; Chen, Z.; Ozturk, T.; Verma, K.; Russell, O.; et al. Novel hepatitis B virus capsid assembly modulator induces potent antiviral responses in vitro and in humanized mice. Antimicrob. Agents Chemother. 2020, 64, e01701-19. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.J.; McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Tao, S.; Bassit, L.; Chen, Z.; Kohler, J.J.; Amblard, F.; Feitelson, M.A.; et al. Studies on the efficacy, potential cardiotoxicity and monkey pharmacokinetics of GLP-26 as a potent hepatitis B virus capsid assembly modulator. Viruses 2021, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Verbinnen, T.; Tan, Y.; Wang, G.; Dehertogh, P.; Vergauwen, K.; Neefs, J.M.; Jacoby, E.; Lenz, O.; Berke, J.M. Anti-HBV activity of the HBV capsid assembly modulator jnj-56136379 across full-length genotype a-h clinical isolates and core site-directed mutants in vitro. J. Antimicrob. Chemother. 2020, 75, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Hadden, J.A.; Zlotnick, A. Assembly properties of hepatitis B virus core protein mutants correlate with their resistance to assembly-directed antivirals. J. Virol. 2018, 92, e01018–e01082. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Wu, D.; Hu, H.; Zeng, J.; Yu, X.; Xu, Z.; Zhou, Z.; Zhou, X.; Yang, G.; Young, J.A.T.; et al. Direct inhibition of hepatitis B e antigen by core protein allosteric modulator. Hepatology 2019, 70, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Guo, H.; Guo, J.T.; Cuconati, A.; Mehta, A.; Block, T.M. Hepatitis b virus e antigen production is dependent upon covalently closed circular (ccc) DNA in hepad38 cell cultures and may serve as a cccdna surrogate in antiviral screening assays. Antivir. Res. 2006, 72, 116–124. [Google Scholar] [CrossRef]
- Patel, D.; Patel, J.S.; Ytreberg, F.M. Implementing and assessing an alchemical method for calculating protein-protein binding free energy. J. Chem. Theory Comput. 2021, 17, 2457–2464. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. Pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 2015, 36, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pkaprediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Liu, X.; Luo, Z.; Li, J.; Wang, C.; Goldmann, S.; Zhang, J.; Zhang, Y. Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (gls4). Bioorg. Med. Chem. 2017, 25, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Li, B.C.; Miska, S.; Krüger, D.H.; Meisel, H.; Will, H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J. Virol. 1995, 69, 5437–5444. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; NY. 16.56–16.67; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]


| WT | Predicted Resistance Mutations |
|---|---|
| M184 | V, I, A, R, N, D, C, Q, G, L, K, P, S, T, W, Y |
| I63 | D, E |
| K65 | R |
| K66 | I, P, T, G |
| D113 | E |
| Y115 | A, R, N, D, C, Q, E, G, H, I, L, K, P, S, T, V |
| Q151 | E, I, C, D, P |
| G152 | D, E |
| Y183 | D, E |
| WT | Predicted Resistance Mutations |
|---|---|
| A132 | Y, W, V, S |
| S106 | W, Y, F, L, R, C |
| L140 | P, C, I, V, H, M, Y |
| T128 | K, R, P, I, M |
| L30 | W, H, R |
| W102 | G, C, L, Y, F, S, R |
| Y118 | Q, D, C, W, E, H, L, K, R, F |
| P134 | L, H, S |
| R133 | I, C, L, T, H, M, G |
| W125 | G |
| T33 | R, M, Q, I |
| F110 | I, L, M |
| P25 | L, Q, R |
| F23 | M, L, Y |
| I105 | M, F, S, V, T |
| P138 | H, A |
| P129 | H, T, S, Q |
| F122 | V, L, C |
| N136 | R |
| V124 | G, A |
| L37 | P, Y, Q |
| R127 | Q |
| A34 | E |
| HBV Core Protein Mutants | GLP26 | Predictions for GLP26 |
|---|---|---|
| WT | Sensitive | - |
| F23Y | Resistant | Resistant |
| L30F | Sensitive | Sensitive |
| T33Q | Resistant | Resistant |
| I105F | Sensitive | Sensitive |
| T109I | Sensitive | Sensitive |
| WT | Predicted Resistance Mutations |
|---|---|
| S144 | Y, W |
| H163 | Y, Q |
| L167 | Q, P, F, R, I, M, H |
| T190 | N, I, R, K, A |
| R188 | H, K, N, L |
| M49 | N, R, K, F |
| P168 | S, Q, R, A, H |
| Q192 | H, K, Y, L, P |
| E166 | G, A, K, V, N |
| F140 | S, I, V, L |
| G143 | A, C |
| Y54 | D, C, E, Q, N, F, G, H, M, S, W, K |
| P52 | R, S |
| H172 | Q, N |
| H164 | N, L, D, Q, K, T, E |
| Q189 | H |
| A191 | G, D |
| D187 | G, Q, A |
| L141 | Q, H |
| M165 | L |
| F181 | L, I, V, S, C |
| N142 | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, D.; Ono, S.K.; Bassit, L.; Verma, K.; Amblard, F.; Schinazi, R.F. Assessment of a Computational Approach to Predict Drug Resistance Mutations for HIV, HBV and SARS-CoV-2. Molecules 2022, 27, 5413. https://doi.org/10.3390/molecules27175413
Patel D, Ono SK, Bassit L, Verma K, Amblard F, Schinazi RF. Assessment of a Computational Approach to Predict Drug Resistance Mutations for HIV, HBV and SARS-CoV-2. Molecules. 2022; 27(17):5413. https://doi.org/10.3390/molecules27175413
Chicago/Turabian StylePatel, Dharmeshkumar, Suzane K. Ono, Leda Bassit, Kiran Verma, Franck Amblard, and Raymond F. Schinazi. 2022. "Assessment of a Computational Approach to Predict Drug Resistance Mutations for HIV, HBV and SARS-CoV-2" Molecules 27, no. 17: 5413. https://doi.org/10.3390/molecules27175413
APA StylePatel, D., Ono, S. K., Bassit, L., Verma, K., Amblard, F., & Schinazi, R. F. (2022). Assessment of a Computational Approach to Predict Drug Resistance Mutations for HIV, HBV and SARS-CoV-2. Molecules, 27(17), 5413. https://doi.org/10.3390/molecules27175413