
Para-Substituted O-Benzyl Sulfohydroxamic Acid Derivatives as Redox-Triggered Nitroxyl (HNO) Sources


Abstract
:
1. Introduction
2. Results
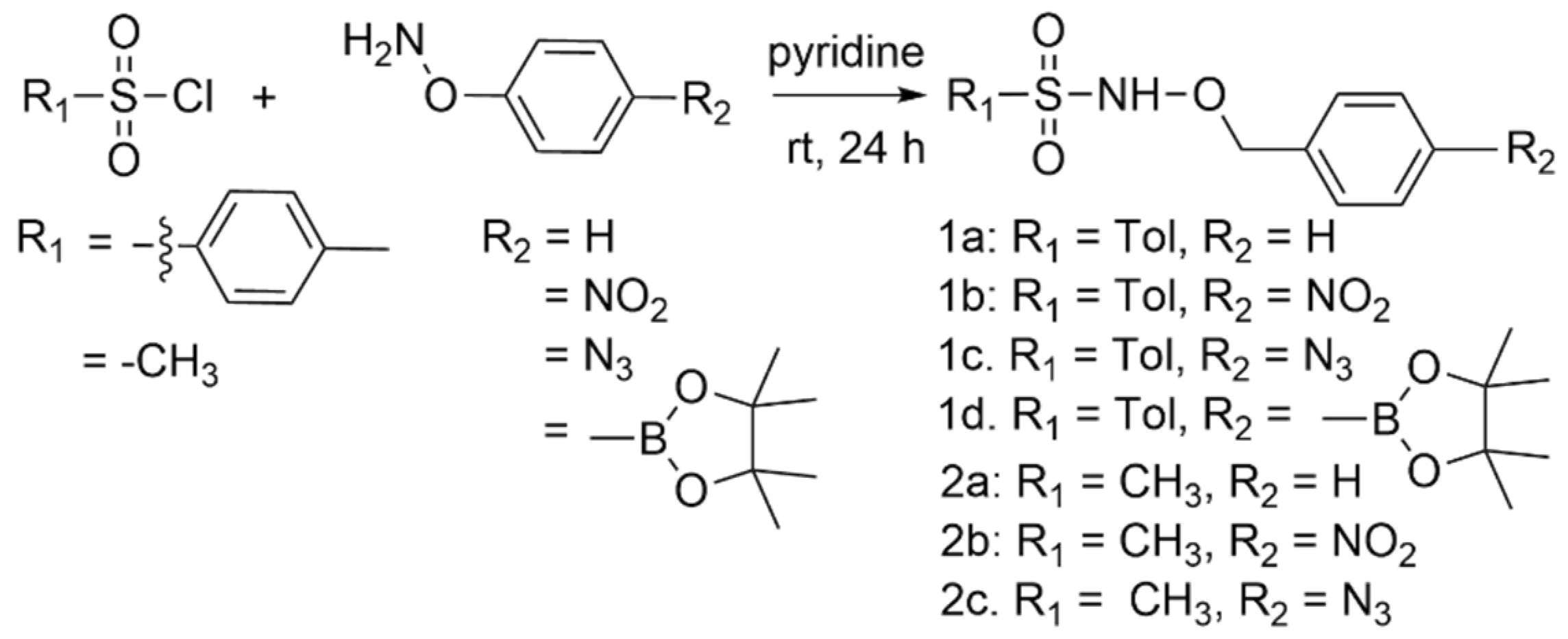
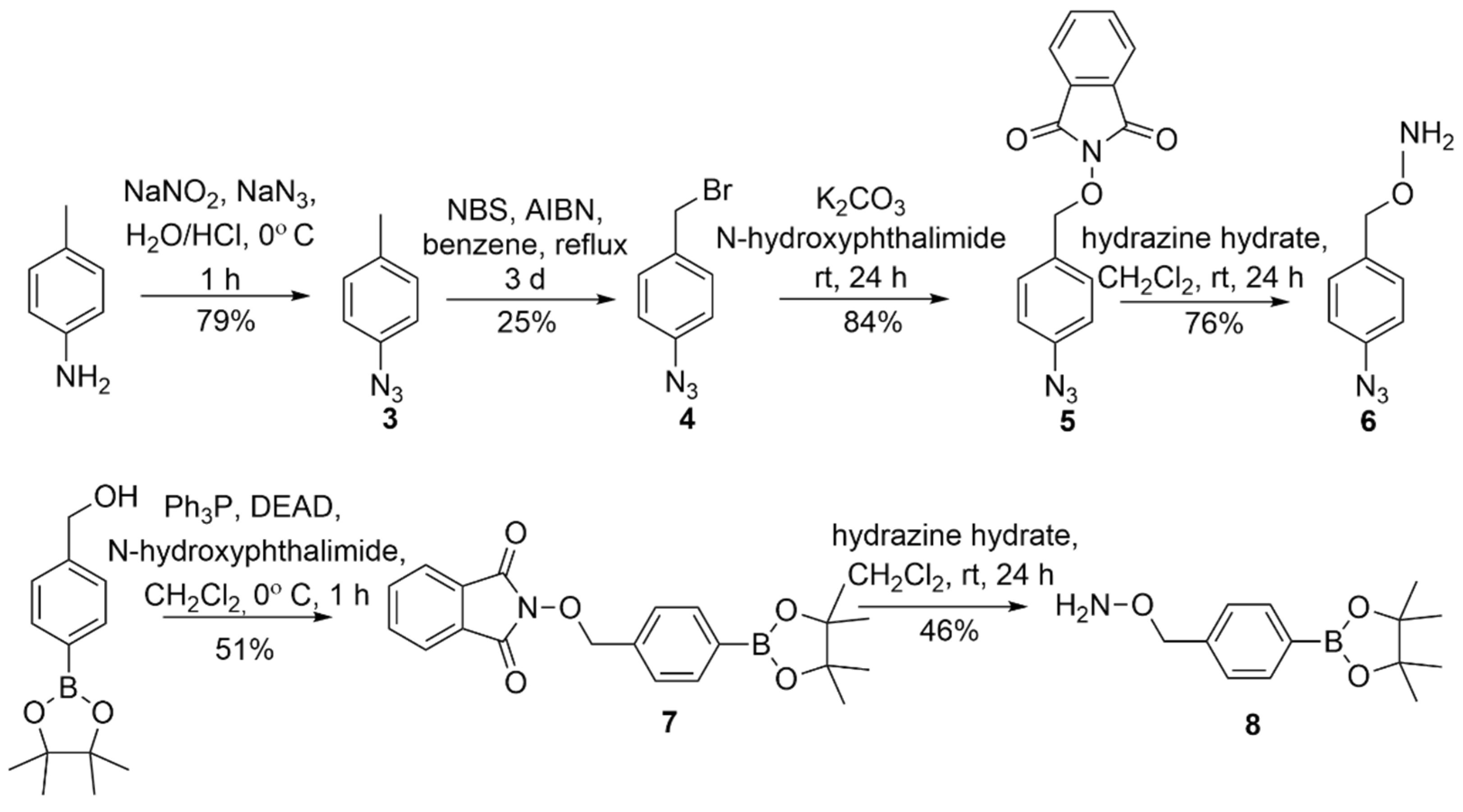
2.1. Synthesis
2.2. HNO Donation Ability
3. Materials and Methods
3.1. Synthesis of Piloty’s Acid Derivatives
3.2. Gas Chromatographic N2O Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, G.; Sener, A.; Ji, Y.; Pei, Y.; Pluth, M.D. Gasotransmitters in Biology and Medicine: Molecular Mechanisms and Drug Targets. Oxid. Med. Cell. Longev. 2016, 2016, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Bonner, F.T.; Hughes, M.N. The Aqueous Solution Chemistry of Nitrogen in Low Positive Oxidation States. Comments Inorg. Chem. 1988, 7, 215–234. [Google Scholar] [CrossRef]
- Dumond, J.F.; King, S.B. The chemistry of nitroxyl-releasing compounds. Antioxid. Redox Signal. 2011, 14, 1637–1648. [Google Scholar] [CrossRef] [Green Version]
- Miranda, K.M.; Paolocci, N.; Katori, T.; Thomas, D.D.; Ford, E.; Bartberger, M.D.; Espey, M.G.; Kass, D.A.; Feelisch, M.; Fukuto, J.M.; et al. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2003, 100, 9196–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuto, J.M.; Chiang, K.; Hszieh, R.; Wong, P.; Chaudhuri, G. The pharmacological activity of nitroxyl: A potent vasodilator with activity similar to nitric oxide and/or endothelium-derived relaxing factor. J. Pharmacol. Exp. Ther. 1992, 263, 546–551, PMID: 1331403. [Google Scholar] [PubMed]
- King, S.B. The Nitric Oxide Producing Reactions of Hydroxyurea. Curr. Med. Chem. 2003, 10, 437–452. [Google Scholar] [CrossRef]
- Nagasawa, H.T.; DeMaster, E.G.; Redfern, B.; Shirota, F.N.; Goon, D.J.W. Evidence for Nitroxyl in the Catalase-Mediated Bioactivation of the Alcohol Deterrent Agent Cyanamide. J. Med. Chem. 1990, 33, 3122–3124. [Google Scholar] [CrossRef]
- Lang, N.N.; Ahmad, F.A.; Cleland, J.G.; O’Connor, C.M.; Teerlink, J.R.; Voors, A.A.; Taubel, J.; Hodes, A.R.; Anwar, M.; Karra, R.; et al. Haemodynamic effects of the nitroxyl donor cimlanod (BMS-986231) in chronic heart failure: A randomized trial. Eur. J. Heart Fail. 2021, 23, 1147–1155. [Google Scholar] [CrossRef]
- Switzer, C.H.; Flores-Santana, W.; Mancardi, D.; Donzelli, S.; Basudhar, D.; Ridnour, L.A.; Miranda, K.M.; Fukuto, J.M.; Paolocci, N.; Wink, D.A. The emergence of nitroxyl (HNO) as a pharmacological agent. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Miranda, K.M. The chemistry of nitroxyl (HNO) and implications in biology. Coord. Chem. Rev. 2005, 249, 433–455. [Google Scholar] [CrossRef]
- Zarenkiewicz, J.; Khodade, V.S.; Toscano, J.P. Reaction of Nitroxyl (HNO) with Hydrogen Sulfide and Hydropersulfides. J. Org. Chem. 2021, 86, 868–877. [Google Scholar] [CrossRef] [PubMed]
- DeMaster, E.G.; Redfern, B.; Nagasawa, H.T. Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem. Pharmacol. 1998, 55, 2007–2015. [Google Scholar] [CrossRef]
- Lopez, B.E.; Wink, D.A.; Fukuto, J.M. The inhibition of glyceraldehyde-3-phosphate dehydrogenase by nitroxyl (HNO). Arch. Biochem. Biophys. 2007, 465, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Tian, Y.; Tocchetti, C.G.; Katori, T.; Murphy, A.M.; Kass, D.A.; Paolocci, N.; Gao, W.D. Nitroxyl increases force development in rat cardiac muscle. J. Physiol. 2007, 580, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Moss, R.L. HNO, Redox Switches, Cardiac Myofilaments, and Heart Failure: A Prequel to Novel Therapeutics. Circ. Res. 2012, 23, 954–956. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, M.; Dux, M.; Namer, B.; Miljkovic, J.; Cordasic, N.; Will, C.; Kichko, T.I.; De La Roche, J.; Fischer, M.; Suárez, S.A.; et al. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 2014, 5, 4381. [Google Scholar] [CrossRef]
- Peng, H.; Shen, J.; Edmonds, K.A.; Luebke, J.L.; Hickey, A.K.; Palmer, L.D.; Chang, F.-M.J.; Bruce, K.A.; Kehl-Fie, T.E.; Skaar, E.P.; et al. Sulfide Homeostasis and Nitroxyl Intersect via Formation of Reactive Sulfur Species in Staphylococcus aureus. mSphere 2017, 2, e00082-17. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Walsh, B.J.C.; Flores-Mireles, A.L.; Peng, H.; Zhang, Y.; Zhang, Y.; Trinidad, J.C.; Hultgren, S.J.; Giedroc, D.P. Hydrogen Sulfide Sensing through Reactive Sulfur Species (RSS) and Nitroxyl (HNO) in Enterococcus faecalis. ACS Chem. Biol. 2018, 13, 1610–1620. [Google Scholar] [CrossRef]
- Yadav, R.; Goldstein, S.; Nasef, M.O.; Lee, W.; Samuni, U. Synergistic activity of acetohydroxamic acid on prokaryotes under oxidative stress: The role of reactive nitrogen species. Free Radic. Biol. Med. 2014, 77, 291–297. [Google Scholar] [CrossRef]
- Sirsalmath, K.; Suárez, S.A.; Bikiel, D.E.; Doctorovich, F. The pH of HNO donation is modulated by ring substituents in Piloty’s acid derivatives: Azanone donors at biological pH. J. Inorg. Biochem. 2013, 118, 134–139. [Google Scholar] [CrossRef]
- Chauhan, P.; Bora, P.; Ravikumar, G.; Jos, S.; Chakrapani, H. Esterase activated carbonyl sulfide/hydrogen sulfide (H2S) donors. Org. Lett. 2017, 19, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Xu, L.; Ou, S.; Edwards, H.; Luedtke, D.; Ge, Y.; Qin, Z. H2O2/Peroxynitrite-Activated Hydroxamic Acid HDAC Inhibitor Prodrugs Show Antileukemic Activities against AML Cells. ACS Med. Chem. Lett. 2018, 9, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Kazuyuki, A.; Hidehiko, N.; Kazuya, M.; Kodai, K.; Naoya, I.; Takayoshi, S.; Naoki, M. Piloty’s acid derivative with improved nitroxyl-releasing characteristics. Bioorg. Med. Chem. Lett. 2013, 23, 2340–2343. [Google Scholar] [CrossRef]
- Porcheddu, A.; De Luca, L.; Giacomelli, G. A Straightforward Route to Piloty’s Acid Derivatives: A Class of Potential Nitroxyl-Generating Prodrugs. Synlett 2009, 13, 2149–2153. [Google Scholar] [CrossRef]
- Gjonaj, L.; Roelfes, G. Selective chemical modification of DNA with alkoxy- and benzyloxyamines. Org. Biomol. Chem. 2015, 13, 6059–6065. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Sommers, E.M.; Kim-Shapiro, D.B.; King, S.B. Horseradish peroxidase catalyzed nitric oxide formation from hydroxyurea. J. Am. Chem. Soc. 2002, 124, 3473–3480. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.M.; Faig, A.; Wolff, D.E.; King, S.B. Sodium borohydride and thiol mediated nitrite release from nitroaromatic antibiotics. Bioorganic Med. Chem. Lett. 2021, 48, 128245. [Google Scholar] [CrossRef]
- Tomaszewska, J.; Koroniak-Szejn, K.; Koroniak, H. Fluorinated organic azides—their preparation and synthetic applications. Arkivoc 2016, 2017, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Montoya, L.A.; Pluth, M.D. Selective turn-on fluorescent probes for imaging hydrogen sulfide in living cells. Chem. Commun. 2012, 48, 4767–4769. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, Y.; Zhang, N.; Xu, S.; Zhang, L.; Wang, Q.; Zhang, Q.; Hu, H.Y. Cell-Permeable Fluorogenic Probes for Identification and Imaging Nitroreductases in Live Bacterial Cells. J. Org. Chem. 2019, 84, 1299–1309. [Google Scholar] [CrossRef]
- Dillon, K.M.; Morrison, H.A.; Powell, C.R.; Carrazzone, R.J.; Ringel-Scaia, V.M.; Winckler, E.W.; Council-Troche, R.M.; Allen, I.C.; Matson, J.B. Targeted Delivery of Persulfides to the Gut: Effects on the Microbiome. Angew. Chemie 2021, 133, 6126–6132. [Google Scholar] [CrossRef]
- Jackson, M.I.; Han, T.H.; Serbulea, L.; Dutton, A.; Ford, E.; Miranda, K.M.; Houk, K.N.; Wink, D.A.; Fukuto, J.M. Kinetic feasibility of nitroxyl reduction by physiological reductants and biological implications. Free Radic. Biol. Med. 2009, 47, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Toscano, J.P.; Brookfield, F.A.; Cohen, A.D.; Courtney, S.M.; Frost, L.M.; Kalish, V.J. N-Hydroxylsulfonamide Derivatives as New Physiologically Useful Nitroxyl Donors. World Intellect. Prop. Organ. Int. Bur. 2007, 1–75. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % N2O | +GSH (5 equiv.) |
|---|---|---|
| pCH3C4H6SO2NHOH | 76 | 0 |
| 1a | 1 | |
| 1b | 1 | |
| 1c | 1 | |
| 1d | 1 | |
| 2a | 1 | |
| 2b | 1 | |
| 2c | 1 |
| Treatment (% N2O) | ||||||
|---|---|---|---|---|---|---|
| Compound | NaBH4 | NaBH4/Cu(II)SO4 | GSH | H2S 2.5 equiv. | H2S 5 equiv. | H2O2 |
| 1a | 0 | 1 | - | - | - | 0 |
| 1b | 25(7) | - | - | - | - | - |
| 1c | - | 89(14) | 0 | 6 | 0 | |
| 1d | - | - | - | - | 23(5) | |
| 2a | 1 | 1 | - | - | -- | 0 |
| 2b | 51(2) | - | - | - | - | - |
| 2c | - | 92 | 0 | 5 | 0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, Y.; Xia, Z.; Rice, A.M.; King, S.B. Para-Substituted O-Benzyl Sulfohydroxamic Acid Derivatives as Redox-Triggered Nitroxyl (HNO) Sources. Molecules 2022, 27, 5305. https://doi.org/10.3390/molecules27165305
Long Y, Xia Z, Rice AM, King SB. Para-Substituted O-Benzyl Sulfohydroxamic Acid Derivatives as Redox-Triggered Nitroxyl (HNO) Sources. Molecules. 2022; 27(16):5305. https://doi.org/10.3390/molecules27165305
Chicago/Turabian StyleLong, Yueming, Zijun Xia, Allison M. Rice, and S. Bruce King. 2022. "Para-Substituted O-Benzyl Sulfohydroxamic Acid Derivatives as Redox-Triggered Nitroxyl (HNO) Sources" Molecules 27, no. 16: 5305. https://doi.org/10.3390/molecules27165305
APA StyleLong, Y., Xia, Z., Rice, A. M., & King, S. B. (2022). Para-Substituted O-Benzyl Sulfohydroxamic Acid Derivatives as Redox-Triggered Nitroxyl (HNO) Sources. Molecules, 27(16), 5305. https://doi.org/10.3390/molecules27165305