
Why Ortho- and Para-Hydroxy Metabolites Can Scavenge Free Radicals That the Parent Atorvastatin Cannot? Important Pharmacologic Insight from Quantum Chemistry


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
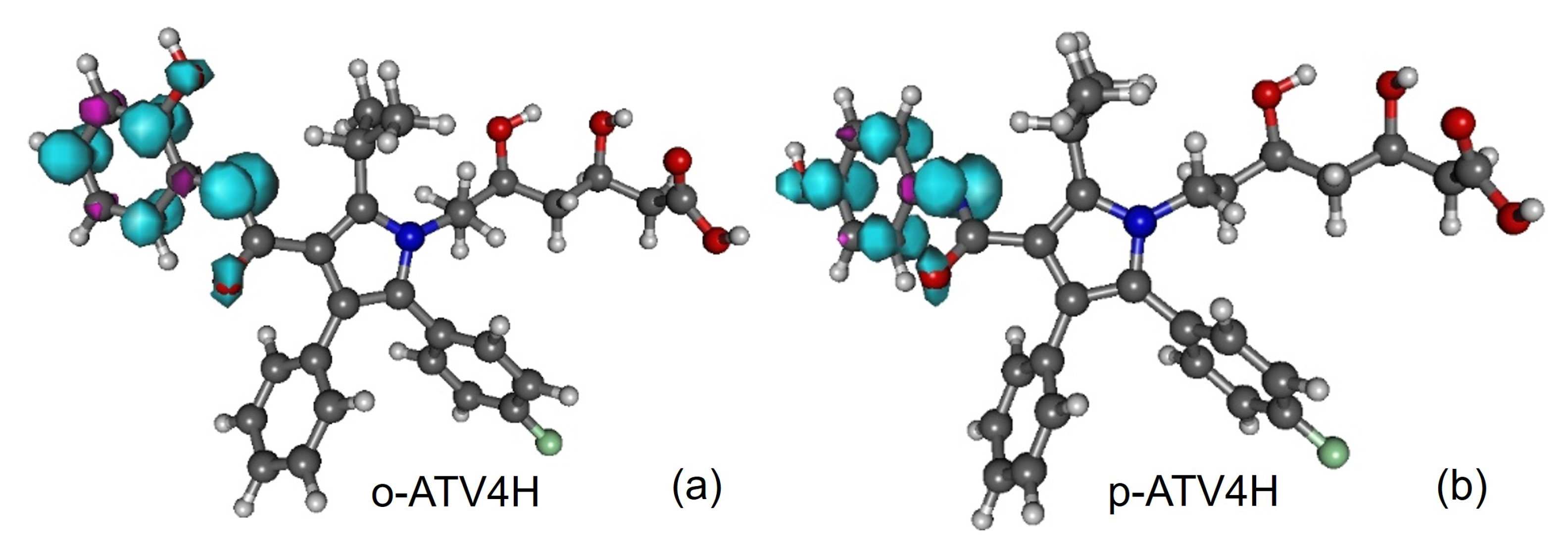
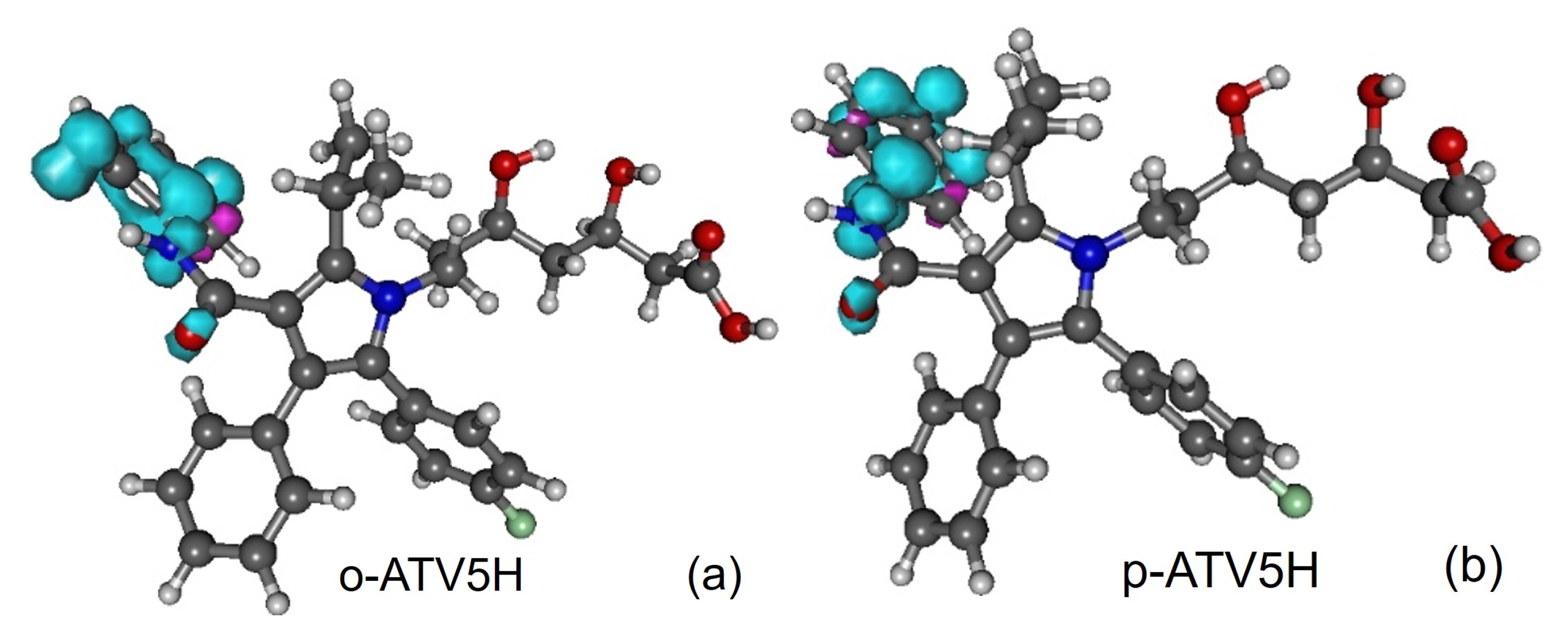
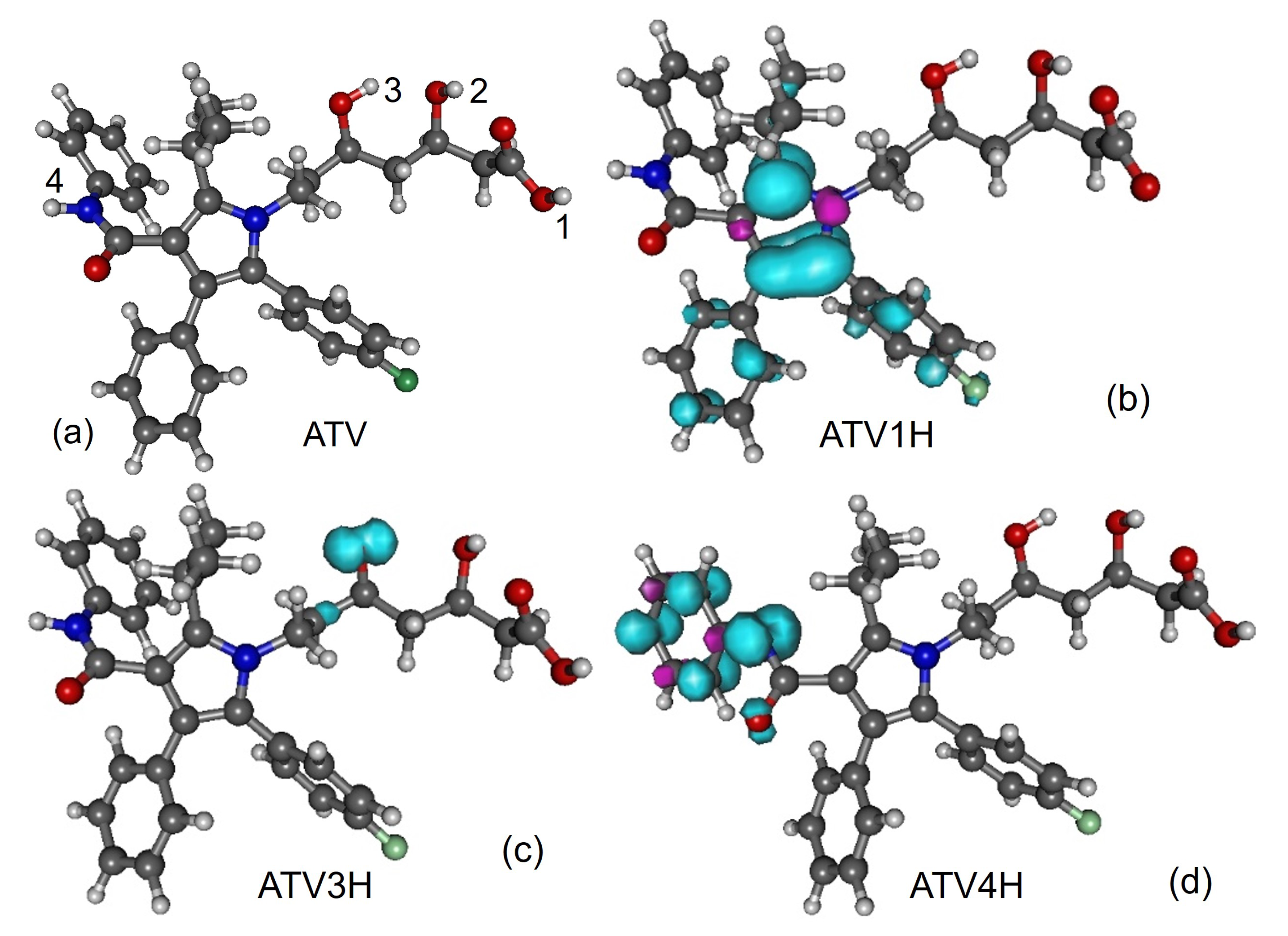
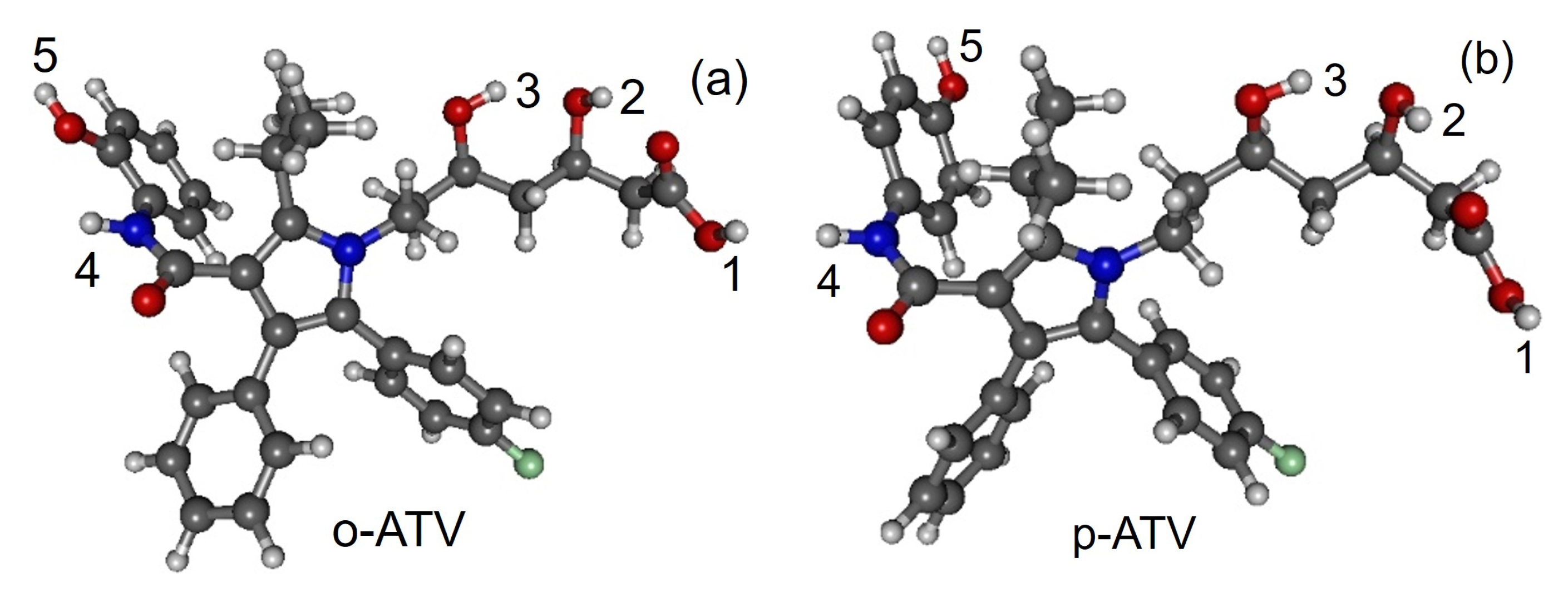
3.1. Molecular Geometries
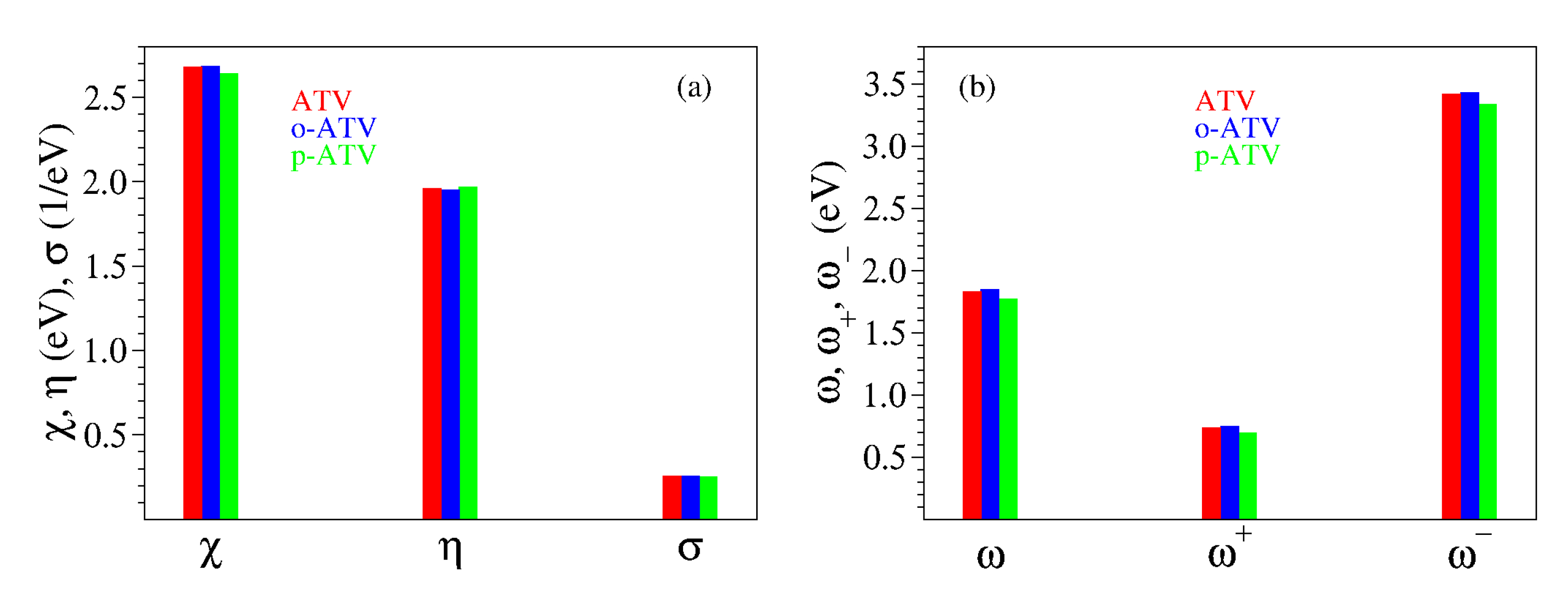
3.2. Chemical Reactivity Indices
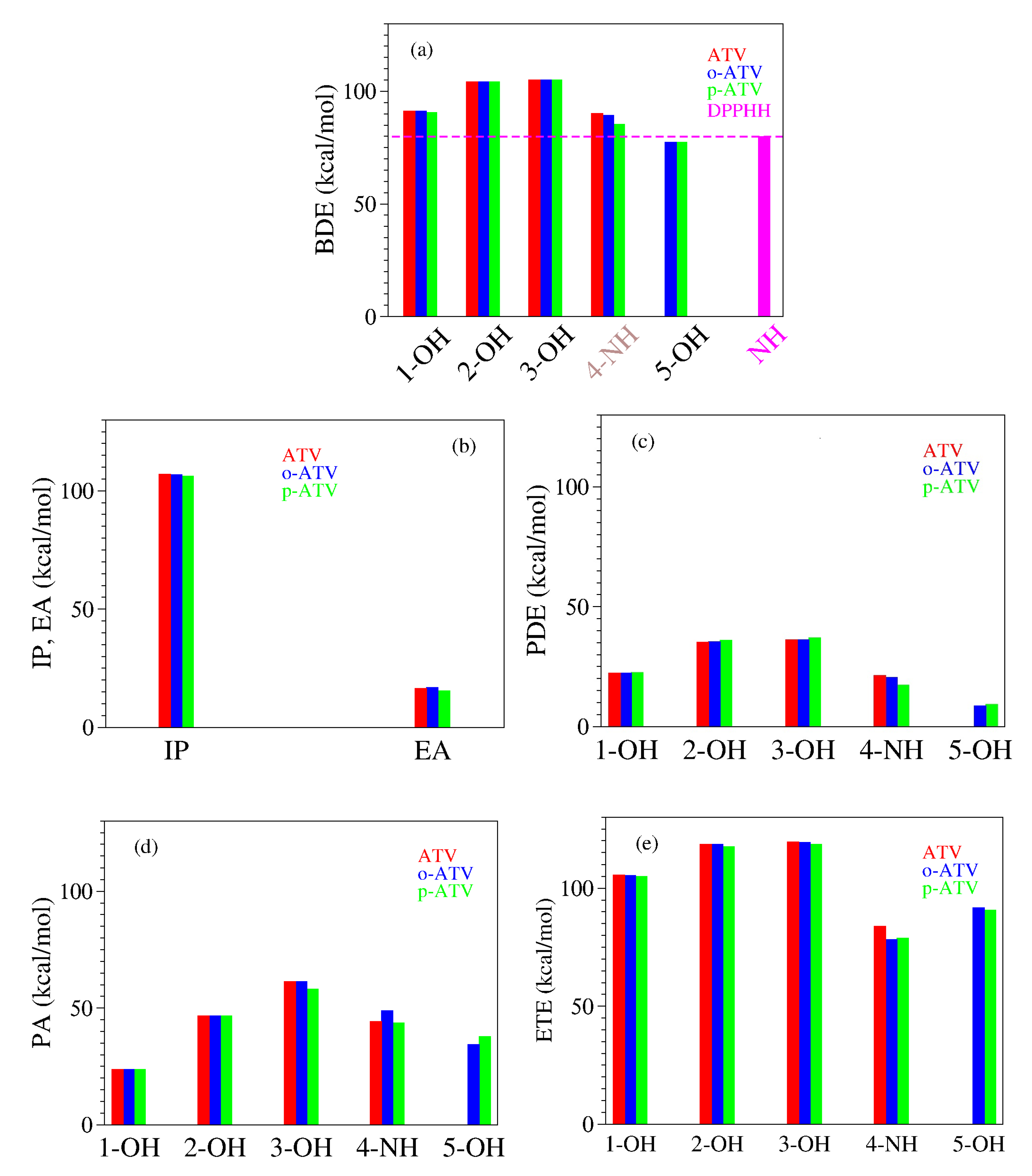
3.3. Antioxidant Mechanisms and Pertaining Enthalpies of Reaction
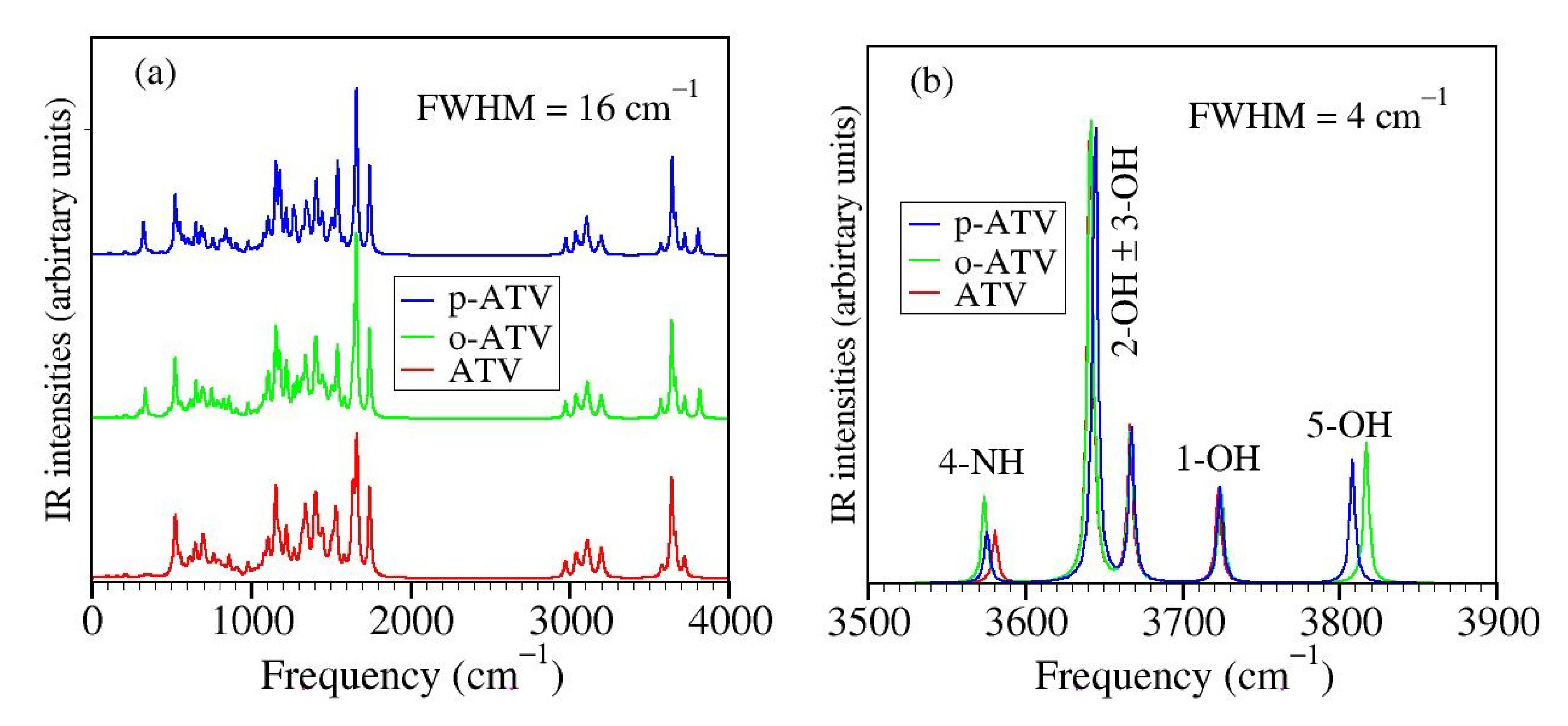
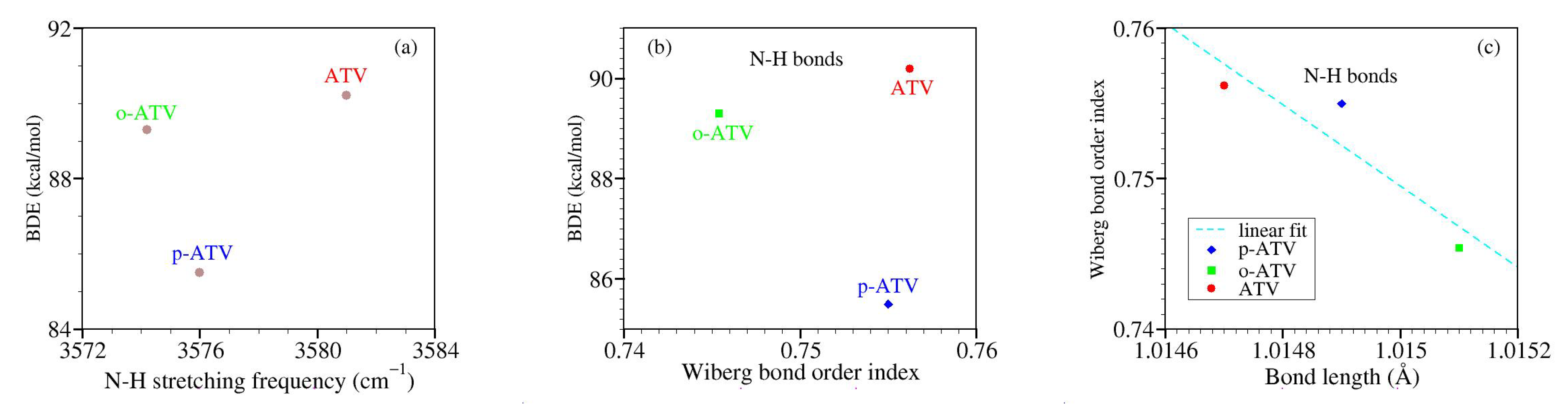
3.4. Alternative Approaches to the O-H and N-H Bond Strengths: Vibrational Frequencies and Bond Order Indices
- (i)
- although the BDE of ATV and its metabolites at position 1-OH is lower than at positions 2-OH and 3-OH, the streching mode at position 1-OH has a higher frequency than at positions 2-OH and 3-OH;
- (ii)
- although o-ATV and p-ATV have at position 5-OH a smaller BDE than for all OH-positions of the parent ATV, the 5-OH stretching mode of the metabolites is higher than those of all O-H streching mode of ATV;
- (iii)
- although o-ATV’s and ATV’s N-H BDE are equal, the frequency of the N-H of the former is smaller than that of the latter;
- (iv)
- although o-ATV’s BDE and p-ATV’s BDE are different, their N-H streching modes have the same frequency;
- (v)
- although o-ATV and p-ATV have equal BDE at position 5-OH, the o-ATV’s O-H streching frequency is higher than that of p-ATV.
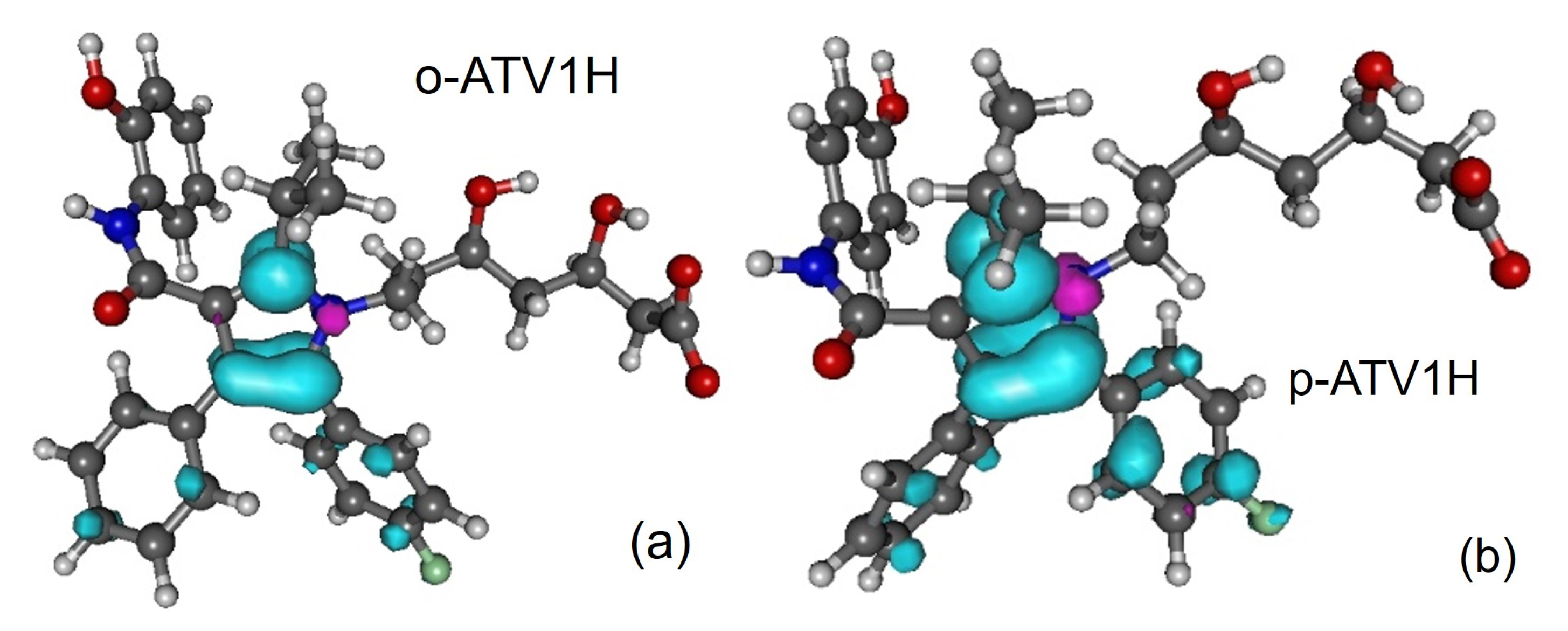
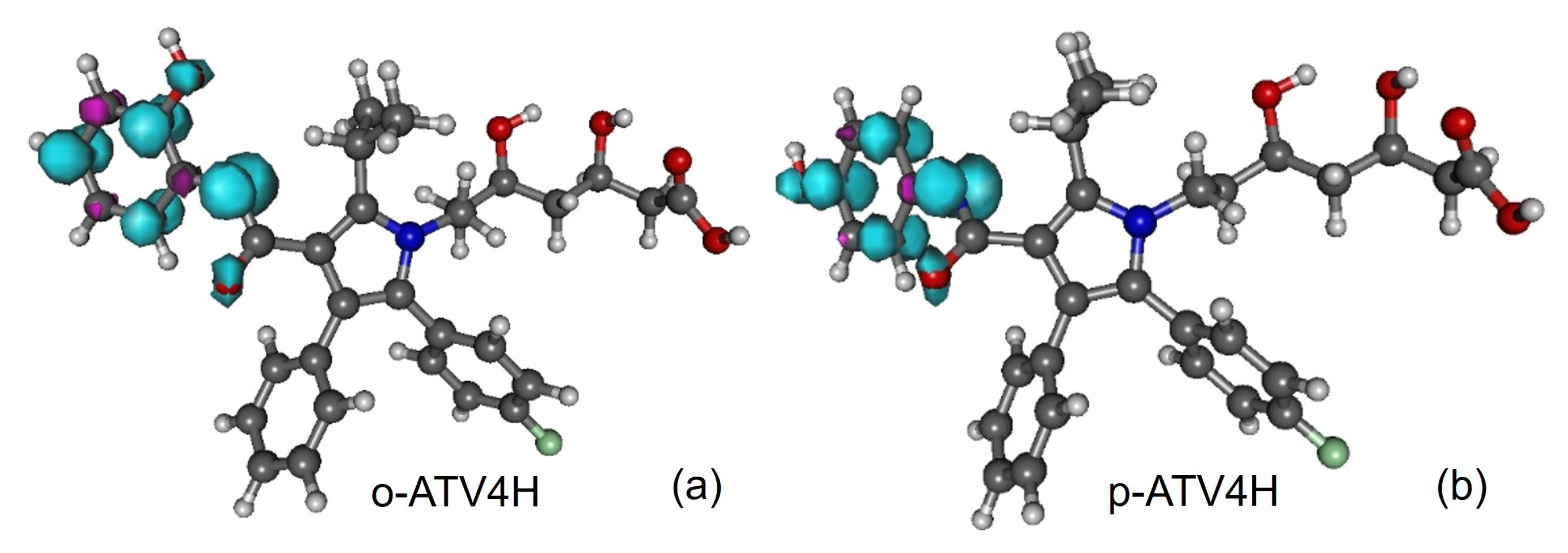
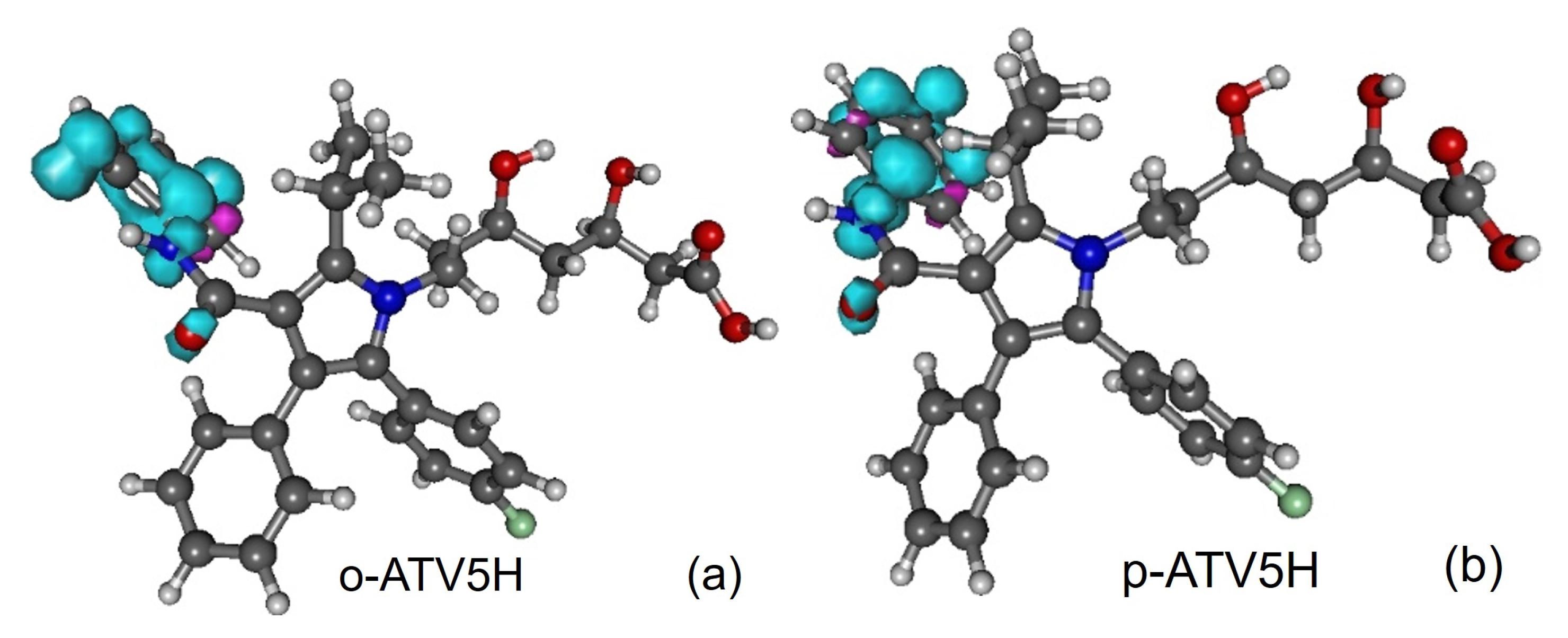
3.5. Assessing the Radical Scavenging Activity—A Specific Example
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | |||||||
|---|---|---|---|---|---|---|---|
| F | |||||||
| O | 1 | B1 | |||||
| O | 1 | B2 | 2 | A1 | |||
| N | 3 | B3 | 1 | A2 | 2 | D1 | 0 |
| C | 2 | B4 | 1 | A3 | 4 | D2 | 0 |
| C | 3 | B5 | 1 | A4 | 4 | D3 | 0 |
| C | 4 | B6 | 3 | A5 | 1 | D4 | 0 |
| C | 4 | B7 | 3 | A6 | 1 | D5 | 0 |
| C | 8 | B8 | 4 | A7 | 3 | D6 | 0 |
| C | 6 | B9 | 3 | A8 | 1 | D7 | 0 |
| C | 7 | B10 | 4 | A9 | 3 | D8 | 0 |
| C | 8 | B11 | 4 | A10 | 3 | D9 | 0 |
| C | 8 | B12 | 4 | A11 | 3 | D10 | 0 |
| C | 12 | B13 | 8 | A12 | 4 | D11 | 0 |
| C | 7 | B14 | 4 | A13 | 3 | D12 | 0 |
| H | 15 | B15 | 7 | A14 | 4 | D13 | 0 |
| H | 15 | B16 | 7 | A15 | 4 | D14 | 0 |
| H | 15 | B17 | 7 | A16 | 4 | D15 | 0 |
| C | 10 | B18 | 6 | A17 | 3 | D16 | 0 |
| H | 19 | B19 | 10 | A18 | 6 | D17 | 0 |
| C | 19 | B20 | 10 | A19 | 6 | D18 | 0 |
| H | 21 | B21 | 19 | A20 | 10 | D19 | 0 |
| C | 21 | B22 | 19 | A21 | 10 | D20 | 0 |
| H | 23 | B23 | 21 | A22 | 19 | D21 | 0 |
| C | 23 | B24 | 21 | A23 | 19 | D22 | 0 |
| H | 25 | B25 | 23 | A24 | 21 | D23 | 0 |
| C | 25 | B26 | 23 | A25 | 21 | D24 | 0 |
| H | 27 | B27 | 25 | A26 | 23 | D25 | 0 |
| C | 14 | B28 | 12 | A27 | 8 | D26 | 0 |
| H | 29 | B29 | 14 | A28 | 12 | D27 | 0 |
| C | 29 | B30 | 14 | A29 | 12 | D28 | 0 |
| H | 31 | B31 | 29 | A30 | 14 | D29 | 0 |
| C | 31 | B32 | 29 | A31 | 14 | D30 | 0 |
| H | 33 | B33 | 31 | A32 | 29 | D31 | 0 |
| C | 33 | B34 | 31 | A33 | 29 | D32 | 0 |
| H | 35 | B35 | 33 | A34 | 31 | D33 | 0 |
| C | 35 | B36 | 33 | A35 | 31 | D34 | 0 |
| H | 37 | B37 | 35 | A36 | 33 | D35 | 0 |
| C | 13 | B38 | 8 | A37 | 4 | D36 | 0 |
| H | 39 | B39 | 13 | A38 | 8 | D37 | 0 |
| C | 9 | B40 | 8 | A39 | 4 | D38 | 0 |
| H | 41 | B41 | 9 | A40 | 8 | D39 | 0 |
| C | 9 | B42 | 8 | A41 | 4 | D40 | 0 |
| H | 43 | B43 | 9 | A42 | 8 | D41 | 0 |
| C | 43 | B44 | 9 | A43 | 8 | D42 | 0 |
| H | 45 | B45 | 43 | A44 | 9 | D43 | 0 |
| C | 4 | B46 | 3 | A45 | 1 | D44 | 0 |
| H | 47 | B47 | 4 | A46 | 3 | D45 | 0 |
| H | 47 | B48 | 4 | A47 | 3 | D46 | 0 |
| C | 47 | B49 | 4 | A48 | 3 | D47 | 0 |
| H | 50 | B50 | 47 | A49 | 4 | D48 | 0 |
| H | 50 | B51 | 47 | A50 | 4 | D49 | 0 |
| C | 5 | B52 | 2 | A51 | 1 | D50 | 0 |
| H | 53 | B53 | 5 | A52 | 2 | D51 | 0 |
| H | 53 | B54 | 5 | A53 | 2 | D52 | 0 |
| O | 50 | B55 | 47 | A54 | 4 | D53 | 0 |
| H | 56 | B56 | 50 | A55 | 47 | D54 | 0 |
| O | 53 | B57 | 5 | A56 | 2 | D55 | 0 |
| H | 58 | B58 | 53 | A57 | 5 | D56 | 0 |
| O | 5 | B59 | 2 | A58 | 1 | D57 | 0 |
| H | 60 | B60 | 5 | A59 | 2 | D58 | 0 |
| N | 6 | B61 | 3 | A60 | 1 | D59 | 0 |
| H | 62 | B62 | 6 | A61 | 3 | D60 | 0 |
| C | 7 | B63 | 4 | A62 | 3 | D61 | 0 |
| H | 64 | B64 | 7 | A63 | 4 | D62 | 0 |
| C | 64 | B65 | 7 | A64 | 4 | D63 | 0 |
| H | 66 | B66 | 64 | A65 | 7 | D64 | 0 |
| H | 66 | B67 | 64 | A66 | 7 | D65 | 0 |
| H | 66 | B68 | 64 | A67 | 7 | D66 | 0 |
| C | 47 | B69 | 4 | A68 | 3 | D67 | 0 |
| H | 70 | B70 | 47 | A69 | 4 | D68 | 0 |
| H | 70 | B71 | 47 | A70 | 4 | D69 | 0 |
| C | 56 | B72 | 50 | A71 | 47 | D70 | 0 |
| H | 73 | B73 | 56 | A72 | 50 | D71 | 0 |
| C | 58 | B74 | 53 | A73 | 5 | D72 | 0 |
| H | 75 | B75 | 58 | A74 | 53 | D73 | 0 |
| Element | RB3LYP | RPBE0 |
|---|---|---|
| B1 | 7.63688039 | 7.57842858 |
| B2 | 9.51376438 | 9.49797976 |
| B3 | 4.46432846 | 4.45735736 |
| B4 | 1.22436588 | 1.22105960 |
| B5 | 1.24094609 | 1.23460721 |
| B6 | 1.38313941 | 1.37429179 |
| B7 | 1.39258446 | 1.38313248 |
| B8 | 4.26421886 | 4.25026562 |
| B9 | 2.55737607 | 2.53336720 |
| B10 | 1.39352326 | 1.38966003 |
| B11 | 1.38854352 | 1.38531627 |
| B12 | 1.48198028 | 1.47533742 |
| B13 | 1.47996148 | 1.47276739 |
| B14 | 2.54735210 | 2.53021178 |
| B15 | 1.09461863 | 1.09441258 |
| B16 | 1.09443324 | 1.09451231 |
| B17 | 1.09548377 | 1.09521443 |
| B18 | 1.40411033 | 1.39956240 |
| B19 | 1.08656542 | 1.08702976 |
| B20 | 1.39590982 | 1.39231014 |
| B21 | 1.08596263 | 1.08625533 |
| B22 | 1.39827379 | 1.39423877 |
| B23 | 1.08557774 | 1.08587383 |
| B24 | 1.39829551 | 1.39470047 |
| B25 | 1.08613361 | 1.08644255 |
| B26 | 1.39684075 | 1.39270470 |
| B27 | 1.08266424 | 1.08414373 |
| B28 | 1.40754480 | 1.40342299 |
| B29 | 1.08542085 | 1.08622867 |
| B30 | 1.39703298 | 1.39303810 |
| B31 | 1.08651573 | 1.08675442 |
| B32 | 1.39866698 | 1.39488556 |
| B33 | 1.08611015 | 1.08634947 |
| B34 | 1.39859436 | 1.39482202 |
| B35 | 1.08646176 | 1.08669302 |
| B36 | 1.39688653 | 1.39286539 |
| B37 | 1.08594257 | 1.08685598 |
| B38 | 1.40608245 | 1.40185966 |
| B39 | 1.08526008 | 1.08604603 |
| B40 | 1.38854404 | 1.38594990 |
| B41 | 1.08466653 | 1.08507152 |
| B42 | 1.38886026 | 1.38604194 |
| B43 | 1.08464963 | 1.08507327 |
| B44 | 1.39673177 | 1.39264953 |
| B45 | 1.08575599 | 1.08651262 |
| B46 | 1.47041402 | 1.45879308 |
| B47 | 1.09044919 | 1.09154067 |
| B48 | 1.08676249 | 1.08820032 |
| B49 | 3.14830814 | 3.11381339 |
| B50 | 1.09708441 | 1.09727967 |
| B51 | 1.09665251 | 1.09755904 |
| B52 | 1.51042629 | 1.50242427 |
| B53 | 1.09777914 | 1.09781828 |
| B54 | 1.09295806 | 1.09309313 |
| B55 | 2.46095421 | 2.44275249 |
| B56 | 0.97500252 | 0.97301949 |
| B57 | 2.45826509 | 2.43975864 |
| B58 | 0.97546854 | 0.97377704 |
| B59 | 1.34162105 | 1.33146502 |
| B60 | 0.97475723 | 0.97138697 |
| B61 | 1.37559401 | 1.36892180 |
| B62 | 1.01471448 | 1.01312538 |
| B63 | 1.51639092 | 1.50803841 |
| B64 | 1.09438086 | 1.09568883 |
| B65 | 1.54383200 | 1.53441635 |
| B66 | 1.09274556 | 1.09354200 |
| B67 | 1.09394603 | 1.09430081 |
| B68 | 1.09550804 | 1.09534859 |
| B69 | 1.53735984 | 1.52751574 |
| B70 | 1.09529399 | 1.09601347 |
| B71 | 1.09519081 | 1.09581652 |
| B72 | 1.43702671 | 1.42351134 |
| B73 | 1.10229127 | 1.10342439 |
| B74 | 1.44374367 | 1.43034604 |
| B75 | 1.09511016 | 1.09602489 |
| A1 | 80.07624165 | 79.42212905 |
| A2 | 36.81921669 | 36.41767374 |
| A3 | 44.89960818 | 44.80116201 |
| A4 | 69.40323071 | 68.51578435 |
| A5 | 38.40240012 | 38.36009230 |
| A6 | 74.54408034 | 74.60701401 |
| A7 | 124.00299971 | 123.75578641 |
| A8 | 142.06293646 | 143.13044343 |
| A9 | 107.05456306 | 107.03652983 |
| A10 | 108.31353197 | 108.33842834 |
| A11 | 122.99231254 | 122.86893832 |
| A12 | 126.63463731 | 126.55972259 |
| A13 | 106.44925702 | 106.31547335 |
| A14 | 89.06053861 | 89.10098490 |
| A15 | 97.89435637 | 97.80052634 |
| A16 | 142.49393445 | 142.62052327 |
| A17 | 137.34771594 | 137.15532746 |
| A18 | 119.50192439 | 119.48359463 |
| A19 | 120.38662171 | 120.33363957 |
| A20 | 119.38771650 | 119.39817314 |
| A21 | 120.33528025 | 120.33582946 |
| A22 | 120.40428227 | 120.38589644 |
| A23 | 119.22951772 | 119.27084737 |
| A24 | 120.07799529 | 120.08000208 |
| A25 | 120.85846463 | 120.80411660 |
| A26 | 119.79850536 | 119.90398307 |
| A27 | 121.00661427 | 120.96513207 |
| A28 | 119.36095172 | 119.32269019 |
| A29 | 121.04376065 | 120.97477881 |
| A30 | 119.57781238 | 119.59079132 |
| A31 | 120.32876554 | 120.32506969 |
| A32 | 120.35906970 | 120.34690449 |
| A33 | 119.30913374 | 119.33935667 |
| A34 | 120.13215959 | 120.12543035 |
| A35 | 120.27437657 | 120.26315929 |
| A36 | 119.21937981 | 119.25183302 |
| A37 | 120.20314915 | 120.00645252 |
| A38 | 119.42757095 | 119.34799906 |
| A39 | 61.14506295 | 61.02049583 |
| A40 | 120.29133823 | 120.18802069 |
| A41 | 61.56595278 | 61.54130326 |
| A42 | 120.28619011 | 120.18989970 |
| A43 | 118.21356561 | 118.29730405 |
| A44 | 118.97682184 | 119.03604297 |
| A45 | 160.61076752 | 161.02034138 |
| A46 | 107.85730043 | 108.01405941 |
| A47 | 108.36720304 | 108.53503060 |
| A48 | 155.00270587 | 155.09486109 |
| A49 | 93.02162927 | 93.36411343 |
| A50 | 65.83600761 | 65.46008045 |
| A51 | 124.63027507 | 124.38270955 |
| A52 | 106.33950994 | 106.35485078 |
| A53 | 109.65684048 | 109.75541245 |
| A54 | 62.50431090 | 62.44180079 |
| A55 | 79.14840553 | 78.61332450 |
| A56 | 94.38442272 | 93.89685773 |
| A57 | 81.94439843 | 81.33655214 |
| A58 | 122.30128587 | 122.38085758 |
| A59 | 108.69599817 | 108.46009440 |
| A60 | 118.51678659 | 118.96829710 |
| A61 | 112.14562665 | 112.66775833 |
| A62 | 125.70871686 | 125.55440414 |
| A63 | 104.10605874 | 104.05408726 |
| A64 | 115.66124171 | 115.41491837 |
| A65 | 112.85938504 | 112.90944315 |
| A66 | 111.09046713 | 111.04846793 |
| A67 | 109.12827188 | 109.12112805 |
| A68 | 112.50720874 | 112.20795577 |
| A69 | 108.93102542 | 108.90860024 |
| A70 | 109.76379473 | 109.82714850 |
| A71 | 35.55295345 | 35.64055684 |
| A72 | 108.78209604 | 109.15267108 |
| A73 | 35.76405946 | 35.85622083 |
| A74 | 104.88212372 | 105.29541039 |
| D1 | 23.44707166 | 22.86899643 |
| D2 | −143.01621213 | −145.69511584 |
| D3 | 38.42108435 | 38.12739399 |
| D4 | 167.03950592 | 166.71102018 |
| D5 | 11.19248174 | 10.26295325 |
| D6 | −166.27299883 | −167.50854474 |
| D7 | 154.43296848 | 154.87100444 |
| D8 | −24.72393257 | −23.90813959 |
| D9 | 14.87760390 | 14.33966315 |
| D10 | −166.33850331 | −167.39527774 |
| D11 | −178.68215577 | −179.05559597 |
| D12 | 116.12498790 | 117.52135961 |
| D13 | −96.95997467 | −97.21214362 |
| D14 | 10.99862661 | 10.70745397 |
| D15 | 143.71999664 | 143.27578873 |
| D16 | 53.45687997 | 56.95553385 |
| D17 | −13.05739505 | −15.48602055 |
| D18 | 166.77990428 | 164.20618170 |
| D19 | 179.92648481 | 179.96617851 |
| D20 | −0.52972049 | −0.55875352 |
| D21 | −179.66796941 | −179.58911616 |
| D22 | 0.73133137 | 0.86390555 |
| D23 | 179.16217965 | 179.06507149 |
| D24 | 0.01070889 | −0.03915718 |
| D25 | 179.03888636 | 179.05702057 |
| D26 | 48.07112323 | 45.04719980 |
| D27 | 0.74469490 | 0.91641539 |
| D28 | −179.96524676 | −179.74189717 |
| D29 | −179.85477901 | −179.85236519 |
| D30 | −0.39506461 | −0.41589868 |
| D31 | −179.79735693 | −179.79071017 |
| D32 | 0.01649873 | 0.03030576 |
| D33 | 179.92823773 | 179.93269511 |
| D34 | 0.28491317 | 0.27631314 |
| D35 | 179.18887066 | 179.19810092 |
| D36 | −116.75958591 | −117.79096246 |
| D37 | 1.86219749 | 1.56294201 |
| D38 | −115.86020598 | −117.02116664 |
| D39 | −179.58717365 | −179.74490671 |
| D40 | 63.79739074 | 62.83877093 |
| D41 | 178.95201872 | 179.06783274 |
| D42 | −0.28698798 | −0.13593353 |
| D43 | 178.22649816 | 178.07298377 |
| D44 | −148.93368537 | −152.08506467 |
| D45 | 111.42783975 | 113.18142815 |
| D46 | −4.05565899 | −2.40606604 |
| D47 | −179.15665881 | −177.51084506 |
| D48 | −12.57964549 | −12.42277636 |
| D49 | −120.44409694 | −120.19086848 |
| D50 | 98.09202903 | 96.64992442 |
| D51 | 91.15604637 | 91.57000780 |
| D52 | −153.66577605 | −153.20632535 |
| D53 | 135.69337038 | 135.76291529 |
| D54 | 154.19232954 | 153.37488771 |
| D55 | −1.14110681 | −0.79096165 |
| D56 | 4.84906073 | 4.80879280 |
| D57 | −83.86223058 | −85.19930682 |
| D58 | −1.48047919 | −1.48833544 |
| D59 | 161.82475625 | 161.81654714 |
| D60 | 8.46484636 | 8.87965902 |
| D61 | 147.77400083 | 149.20907105 |
| D62 | −178.10763280 | −178.26760363 |
| D63 | 66.16427504 | 65.99502020 |
| D64 | −71.70727202 | −71.00674962 |
| D65 | 50.12782255 | 50.90167682 |
| D66 | 168.95273147 | 169.66688376 |
| D67 | −126.11657682 | −124.36123085 |
| D68 | 54.49143257 | 54.30008219 |
| D69 | −62.36290369 | −62.63339700 |
| D70 | −66.30729976 | −66.82133358 |
| D71 | −118.50510691 | −118.64472010 |
| D72 | −128.64978340 | −128.59930105 |
| D73 | −115.36964313 | −115.74853701 |
| Atom | |||||||
|---|---|---|---|---|---|---|---|
| F | |||||||
| O | 1 | B1 | |||||
| O | 1 | B2 | 2 | A1 | |||
| N | 3 | B3 | 1 | A2 | 2 | D1 | 0 |
| C | 2 | B4 | 1 | A3 | 4 | D2 | 0 |
| C | 3 | B5 | 1 | A4 | 4 | D3 | 0 |
| C | 4 | B6 | 3 | A5 | 1 | D4 | 0 |
| C | 4 | B7 | 3 | A6 | 1 | D5 | 0 |
| C | 8 | B8 | 4 | A7 | 3 | D6 | 0 |
| C | 6 | B9 | 3 | A8 | 1 | D7 | 0 |
| C | 7 | B10 | 4 | A9 | 3 | D8 | 0 |
| C | 8 | B11 | 4 | A10 | 3 | D9 | 0 |
| C | 8 | B12 | 4 | A11 | 3 | D10 | 0 |
| C | 12 | B13 | 8 | A12 | 4 | D11 | 0 |
| C | 7 | B14 | 4 | A13 | 3 | D12 | 0 |
| H | 15 | B15 | 7 | A14 | 4 | D13 | 0 |
| H | 15 | B16 | 7 | A15 | 4 | D14 | 0 |
| H | 15 | B17 | 7 | A16 | 4 | D15 | 0 |
| C | 10 | B18 | 6 | A17 | 3 | D16 | 0 |
| H | 19 | B19 | 10 | A18 | 6 | D17 | 0 |
| C | 19 | B20 | 10 | A19 | 6 | D18 | 0 |
| H | 21 | B21 | 19 | A20 | 10 | D19 | 0 |
| C | 21 | B22 | 19 | A21 | 10 | D20 | 0 |
| H | 23 | B23 | 21 | A22 | 19 | D21 | 0 |
| C | 23 | B24 | 21 | A23 | 19 | D22 | 0 |
| H | 25 | B25 | 23 | A24 | 21 | D23 | 0 |
| C | 25 | B26 | 23 | A25 | 21 | D24 | 0 |
| H | 27 | B27 | 25 | A26 | 23 | D25 | 0 |
| C | 14 | B28 | 12 | A27 | 8 | D26 | 0 |
| H | 29 | B29 | 14 | A28 | 12 | D27 | 0 |
| C | 29 | B30 | 14 | A29 | 12 | D28 | 0 |
| H | 31 | B31 | 29 | A30 | 14 | D29 | 0 |
| C | 31 | B32 | 29 | A31 | 14 | D30 | 0 |
| H | 33 | B33 | 31 | A32 | 29 | D31 | 0 |
| C | 33 | B34 | 31 | A33 | 29 | D32 | 0 |
| H | 35 | B35 | 33 | A34 | 31 | D33 | 0 |
| C | 35 | B36 | 33 | A35 | 31 | D34 | 0 |
| H | 37 | B37 | 35 | A36 | 33 | D35 | 0 |
| C | 13 | B38 | 8 | A37 | 4 | D36 | 0 |
| H | 39 | B39 | 13 | A38 | 8 | D37 | 0 |
| C | 9 | B40 | 8 | A39 | 4 | D38 | 0 |
| H | 41 | B41 | 9 | A40 | 8 | D39 | 0 |
| C | 9 | B42 | 8 | A41 | 4 | D40 | 0 |
| H | 43 | B43 | 9 | A42 | 8 | D41 | 0 |
| C | 43 | B44 | 9 | A43 | 8 | D42 | 0 |
| H | 45 | B45 | 43 | A44 | 9 | D43 | 0 |
| C | 4 | B46 | 3 | A45 | 1 | D44 | 0 |
| H | 47 | B47 | 4 | A46 | 3 | D45 | 0 |
| H | 47 | B48 | 4 | A47 | 3 | D46 | 0 |
| C | 5 | B49 | 2 | A48 | 1 | D47 | 0 |
| H | 50 | B50 | 5 | A49 | 2 | D48 | 0 |
| H | 50 | B51 | 5 | A50 | 2 | D49 | 0 |
| C | 5 | B52 | 2 | A51 | 1 | D50 | 0 |
| H | 53 | B53 | 5 | A52 | 2 | D51 | 0 |
| H | 53 | B54 | 5 | A53 | 2 | D52 | 0 |
| O | 50 | B55 | 5 | A54 | 2 | D53 | 0 |
| H | 56 | B56 | 50 | A55 | 5 | D54 | 0 |
| O | 53 | B57 | 5 | A56 | 2 | D55 | 0 |
| H | 58 | B58 | 53 | A57 | 5 | D56 | 0 |
| O | 5 | B59 | 2 | A58 | 1 | D57 | 0 |
| N | 6 | B60 | 3 | A59 | 1 | D58 | 0 |
| H | 61 | B61 | 6 | A60 | 3 | D59 | 0 |
| C | 7 | B62 | 4 | A61 | 3 | D60 | 0 |
| H | 63 | B63 | 7 | A62 | 4 | D61 | 0 |
| C | 63 | B64 | 7 | A63 | 4 | D62 | 0 |
| H | 65 | B65 | 63 | A64 | 7 | D63 | 0 |
| H | 65 | B66 | 63 | A65 | 7 | D64 | 0 |
| H | 65 | B67 | 63 | A66 | 7 | D65 | 0 |
| C | 47 | B68 | 4 | A67 | 3 | D66 | 0 |
| H | 69 | B69 | 47 | A68 | 4 | D67 | 0 |
| H | 69 | B70 | 47 | A69 | 4 | D68 | 0 |
| C | 56 | B71 | 50 | A70 | 5 | D69 | 0 |
| H | 72 | B72 | 56 | A71 | 50 | D70 | 0 |
| C | 58 | B73 | 53 | A72 | 5 | D71 | 0 |
| H | 74 | B74 | 58 | A73 | 53 | D72 | 0 |
| Element | UB3LYP | ROB3LYP | UPBE0 |
|---|---|---|---|
| B1 | 8.44235812 | 8.44847690 | 8.35899747 |
| B2 | 9.48151258 | 9.48004054 | 9.44104389 |
| B3 | 4.45293368 | 4.45205992 | 4.45170279 |
| B4 | 1.28075664 | 1.28075532 | 1.27612637 |
| B5 | 1.23516216 | 1.23520893 | 1.22884511 |
| B6 | 1.39121019 | 1.39036552 | 1.38136414 |
| B7 | 1.36865245 | 1.36927061 | 1.36158073 |
| B8 | 4.22903226 | 4.22867569 | 4.21410980 |
| B9 | 2.54297346 | 2.54296359 | 2.51827392 |
| B10 | 1.41411564 | 1.41273105 | 1.41276955 |
| B11 | 1.39814133 | 1.39906265 | 1.39123927 |
| B12 | 1.45764082 | 1.45731631 | 1.45172243 |
| B13 | 1.46214878 | 1.46225268 | 1.45692318 |
| B14 | 2.52804795 | 2.52736311 | 2.50801737 |
| B15 | 1.09361662 | 1.09358464 | 1.09350906 |
| B16 | 1.09280828 | 1.09278512 | 1.09298184 |
| B17 | 1.09400195 | 1.09401889 | 1.09384406 |
| B18 | 1.40294782 | 1.40295766 | 1.39868090 |
| B19 | 1.08608989 | 1.08608772 | 1.08659742 |
| B20 | 1.39528664 | 1.39528456 | 1.39148253 |
| B21 | 1.08563236 | 1.08563462 | 1.08591759 |
| B22 | 1.39885297 | 1.39887769 | 1.39500026 |
| B23 | 1.08539151 | 1.08539203 | 1.08568270 |
| B24 | 1.39725830 | 1.39725954 | 1.39357881 |
| B25 | 1.08569301 | 1.08569202 | 1.08600487 |
| B26 | 1.39754666 | 1.39755038 | 1.39365084 |
| B27 | 1.08290419 | 1.08290428 | 1.08428078 |
| B28 | 1.41359961 | 1.41373605 | 1.40819345 |
| B29 | 1.08383502 | 1.08382196 | 1.08499596 |
| B30 | 1.39307164 | 1.39303645 | 1.38966728 |
| B31 | 1.08532894 | 1.08532521 | 1.08564174 |
| B32 | 1.39877845 | 1.39870695 | 1.39488367 |
| B33 | 1.08554363 | 1.08554068 | 1.08589662 |
| B34 | 1.40107536 | 1.40113592 | 1.39694939 |
| B35 | 1.08531301 | 1.08531095 | 1.08561477 |
| B36 | 1.39114277 | 1.39108971 | 1.38781466 |
| B37 | 1.08454080 | 1.08454217 | 1.08578418 |
| B38 | 1.41428406 | 1.41456146 | 1.40940924 |
| B39 | 1.08378663 | 1.08375477 | 1.08486309 |
| B40 | 1.39026744 | 1.39013657 | 1.38630505 |
| B41 | 1.08392114 | 1.08391505 | 1.08433610 |
| B42 | 1.39295578 | 1.39307149 | 1.39002859 |
| B43 | 1.08393715 | 1.08393322 | 1.08438619 |
| B44 | 1.38976598 | 1.38960925 | 1.38585905 |
| B45 | 1.08386009 | 1.08382658 | 1.08487947 |
| B46 | 1.48288423 | 1.48290265 | 1.46968125 |
| B47 | 1.08655848 | 1.08656906 | 1.08771365 |
| B48 | 1.08526299 | 1.08526229 | 1.08700526 |
| B49 | 3.13186572 | 3.13214456 | 3.09809470 |
| B50 | 1.09749147 | 1.09749242 | 1.09754851 |
| B51 | 1.09723187 | 1.09723227 | 1.09817660 |
| B52 | 1.54597249 | 1.54596976 | 1.53696411 |
| B53 | 1.09826990 | 1.09827142 | 1.09817381 |
| B54 | 1.09366646 | 1.09366220 | 1.09361307 |
| B55 | 2.45496563 | 2.45493465 | 2.43617051 |
| B56 | 0.98121613 | 0.98120462 | 0.98045950 |
| B57 | 2.44581966 | 2.44580722 | 2.42582935 |
| B58 | 1.00162018 | 1.00160987 | 1.00474416 |
| B59 | 1.25555505 | 1.25555586 | 1.24897137 |
| B60 | 1.36463937 | 1.36473756 | 1.35864347 |
| B61 | 1.01542708 | 1.01542689 | 1.01391007 |
| B62 | 1.50493077 | 1.50429573 | 1.49670728 |
| B63 | 1.09337036 | 1.09333772 | 1.09467810 |
| B64 | 1.54747843 | 1.54768005 | 1.53733187 |
| B65 | 1.09151271 | 1.09148630 | 1.09246030 |
| B66 | 1.09323237 | 1.09319168 | 1.09361961 |
| B67 | 1.09407784 | 1.09408957 | 1.09401377 |
| B68 | 1.53535621 | 1.53533608 | 1.52584909 |
| B69 | 1.09518094 | 1.09518217 | 1.09596063 |
| B70 | 1.09496161 | 1.09497326 | 1.09558744 |
| B71 | 1.43537977 | 1.43540753 | 1.42193713 |
| B72 | 1.10242221 | 1.10243533 | 1.10351859 |
| B73 | 1.44492447 | 1.44492195 | 1.43098159 |
| B74 | 1.09747052 | 1.09747351 | 1.09843948 |
| A1 | 76.58822830 | 76.56600564 | 76.18592634 |
| A2 | 36.60887483 | 36.62337281 | 36.65315300 |
| A3 | 40.02401754 | 40.02725714 | 40.37741291 |
| A4 | 69.22460650 | 69.21215273 | 69.11650364 |
| A5 | 37.06294802 | 37.05454543 | 37.33294209 |
| A6 | 75.97461715 | 75.97019252 | 75.54658203 |
| A7 | 125.60763591 | 125.62294801 | 125.53862522 |
| A8 | 145.46690797 | 145.43385778 | 146.47703392 |
| A9 | 107.94505539 | 107.97228749 | 107.96898197 |
| A10 | 108.38689995 | 108.36146722 | 108.27547546 |
| A11 | 124.74777225 | 124.75087571 | 124.69207445 |
| A12 | 127.97840469 | 127.96666547 | 128.15656185 |
| A13 | 105.59510463 | 105.58982134 | 105.42968231 |
| A14 | 89.06762423 | 89.07379124 | 89.13648017 |
| A15 | 98.38552433 | 98.38081510 | 98.24507700 |
| A16 | 141.42365452 | 141.39940160 | 141.62200086 |
| A17 | 135.11423751 | 135.13907569 | 134.99049648 |
| A18 | 119.61460474 | 119.61458063 | 119.55946866 |
| A19 | 120.09632412 | 120.09653690 | 120.05115014 |
| A20 | 119.42313169 | 119.42315027 | 119.43302709 |
| A21 | 120.32365928 | 120.32410586 | 120.32294735 |
| A22 | 120.29030763 | 120.28966185 | 120.26610493 |
| A23 | 119.45834121 | 119.45848930 | 119.50076102 |
| A24 | 120.18704632 | 120.18699066 | 120.19049653 |
| A25 | 120.64768821 | 120.64769441 | 120.59629785 |
| A26 | 119.56030742 | 119.56407910 | 119.66008477 |
| A27 | 120.97200577 | 120.97164632 | 120.89552896 |
| A28 | 119.95204121 | 119.93947727 | 119.96571886 |
| A29 | 120.50494184 | 120.51489097 | 120.36553094 |
| A30 | 119.59499068 | 119.60198784 | 119.61841115 |
| A31 | 120.22405430 | 120.21574498 | 120.19645130 |
| A32 | 120.10222516 | 120.09856981 | 120.06034754 |
| A33 | 119.87268934 | 119.88010650 | 119.94634847 |
| A34 | 120.16892282 | 120.16545793 | 120.17885990 |
| A35 | 120.16243769 | 120.16144382 | 120.12026330 |
| A36 | 119.28472624 | 119.28635927 | 119.38889210 |
| A37 | 119.13990753 | 119.14065136 | 118.93302300 |
| A38 | 119.93397869 | 119.91880819 | 119.92762403 |
| A39 | 120.71429714 | 120.72333881 | 120.60124236 |
| A40 | 121.46593615 | 121.47547577 | 121.48976173 |
| A41 | 61.90781157 | 61.91592578 | 61.89487363 |
| A42 | 120.24240110 | 120.24137519 | 120.17005220 |
| A43 | 118.35259175 | 118.34576673 | 118.40770516 |
| A44 | 118.86184831 | 118.86297431 | 118.93351992 |
| A45 | 158.71586453 | 158.72856275 | 159.46262187 |
| A46 | 107.97807868 | 107.98166144 | 108.16165610 |
| A47 | 107.59850751 | 107.60908685 | 107.84756046 |
| A48 | 157.42614280 | 157.44924436 | 157.60290007 |
| A49 | 94.85343455 | 94.84153771 | 95.55489296 |
| A50 | 64.95144615 | 64.95648523 | 64.45763585 |
| A51 | 116.19161990 | 116.19053946 | 115.94779395 |
| A52 | 106.86069836 | 106.86047108 | 106.91121971 |
| A53 | 109.53392738 | 109.53458463 | 109.64753221 |
| A54 | 62.07557911 | 62.08044425 | 61.96848271 |
| A55 | 76.49524293 | 76.49674331 | 75.79460665 |
| A56 | 92.76536137 | 92.76506052 | 92.30427679 |
| A57 | 73.57144767 | 73.57345073 | 72.89199367 |
| A58 | 125.45137451 | 125.45215395 | 125.49766270 |
| A59 | 120.73260155 | 120.70386918 | 121.19251908 |
| A60 | 112.50449394 | 112.51148946 | 113.02222681 |
| A61 | 125.77099330 | 125.75901888 | 125.58026516 |
| A62 | 104.23702836 | 104.25410004 | 104.23745904 |
| A63 | 115.40821162 | 115.42354499 | 115.23936294 |
| A64 | 113.24698603 | 113.25125527 | 113.34985866 |
| A65 | 111.19362011 | 111.18145366 | 111.20358677 |
| A66 | 107.92265897 | 107.90449558 | 107.87759561 |
| A67 | 112.10461860 | 112.12909104 | 111.84315602 |
| A68 | 109.13041373 | 109.12442468 | 109.15330530 |
| A69 | 110.22123305 | 110.22217326 | 110.32638994 |
| A70 | 35.66580947 | 35.66718568 | 35.77797455 |
| A71 | 108.97338188 | 108.96922402 | 109.34397229 |
| A72 | 36.23210769 | 36.23253122 | 36.38166941 |
| A73 | 106.11400594 | 106.11371323 | 106.59889713 |
| D1 | 20.32357302 | 20.28511825 | 19.87637559 |
| D2 | −162.17511087 | −162.33220490 | −162.80694109 |
| D3 | 43.63016177 | 43.60181174 | 42.29185870 |
| D4 | 164.26479778 | 164.21734263 | 164.45615232 |
| D5 | 10.77189784 | 10.75274670 | 10.05902947 |
| D6 | −166.43746347 | −166.45509545 | −167.30190503 |
| D7 | 158.44374263 | 158.23289402 | 158.80659445 |
| D8 | −28.08220344 | −28.08496724 | −26.82514391 |
| D9 | 1.91266449 | 1.90212780 | 1.62776737 |
| D10 | −165.86995934 | −165.89347603 | −166.61940143 |
| D11 | 175.37358525 | 175.35516221 | 176.13954461 |
| D12 | 113.64232131 | 113.65660017 | 114.91433970 |
| D13 | −95.76535683 | −95.82640318 | −96.19390095 |
| D14 | 12.62820769 | 12.57590279 | 12.17768961 |
| D15 | 145.45934742 | 145.38955744 | 144.82678344 |
| D16 | 54.19138739 | 54.30206745 | 57.11046377 |
| D17 | −17.85450383 | −17.82296579 | −19.82581123 |
| D18 | 161.85312253 | 161.88684573 | 159.76981057 |
| D19 | 179.94673200 | 179.94762681 | 179.95512591 |
| D20 | −0.60528557 | −0.60218788 | −0.68612039 |
| D21 | −179.59203284 | −179.59336859 | −179.50090877 |
| D22 | 0.90546409 | 0.90366406 | 1.03940801 |
| D23 | 178.93404376 | 178.93542059 | 178.83527738 |
| D24 | 0.05603713 | 0.05403935 | 0.03326550 |
| D25 | 177.82860566 | 177.83470660 | 177.66605428 |
| D26 | −137.16331958 | −137.09552508 | −138.22471461 |
| D27 | 0.43265772 | 0.51931825 | 0.61777653 |
| D28 | 178.15552655 | 178.21539172 | 178.43299456 |
| D29 | −178.95651780 | −178.95670070 | −178.99276897 |
| D30 | 0.79493565 | 0.77240094 | 0.77488924 |
| D31 | 179.51072096 | 179.53026575 | 179.50412377 |
| D32 | −0.68679275 | −0.67915786 | −0.68858323 |
| D33 | 179.67314075 | 179.67322011 | 179.71709289 |
| D34 | −0.20109695 | −0.20087651 | −0.18063234 |
| D35 | 179.30402096 | 179.29000187 | 179.32243696 |
| D36 | −130.28427078 | −130.51938776 | −130.58633908 |
| D37 | 1.84741835 | 1.91470640 | 1.67453246 |
| D38 | 179.74200519 | 179.74365748 | 179.60779307 |
| D39 | −178.45418197 | −178.42099743 | −178.44652258 |
| D40 | 51.73180726 | 51.51509192 | 51.55090486 |
| D41 | 178.78350784 | 178.76463222 | 178.80834185 |
| D42 | −0.73877044 | −0.75662840 | −0.65322109 |
| D43 | 177.62465046 | 177.60834940 | 177.40162657 |
| D44 | −156.11290080 | −156.22713857 | −158.14021510 |
| D45 | 116.06952548 | 116.07911206 | 117.63013987 |
| D46 | 0.99099890 | 1.01912297 | 2.42551191 |
| D47 | −171.42587327 | −171.42390819 | −169.95676548 |
| D48 | −15.20357679 | −15.21183395 | −14.84818756 |
| D49 | −122.34742832 | −122.36008992 | −121.73630690 |
| D50 | 88.06897802 | 87.95008988 | 88.85753223 |
| D51 | 83.56556644 | 83.56187427 | 83.14352949 |
| D52 | −160.76875154 | −160.77109993 | −161.02867903 |
| D53 | 132.32183428 | 132.32174256 | 132.43569018 |
| D54 | 149.97524265 | 149.98218167 | 148.79908686 |
| D55 | −9.90827522 | −9.91152885 | −10.13040806 |
| D56 | 9.74285779 | 9.73770241 | 9.81321453 |
| D57 | −93.97703409 | −94.06732013 | −93.02985254 |
| D58 | 160.87867630 | 160.75346463 | 161.09930653 |
| D59 | 6.66281917 | 6.73900407 | 7.18339300 |
| D60 | 145.35128108 | 145.39894308 | 146.79423835 |
| D61 | −177.00884009 | −177.06601025 | −177.27755993 |
| D62 | 67.06790752 | 66.97955721 | 66.63767044 |
| D63 | −71.82425328 | −71.79410371 | −71.15947356 |
| D64 | 50.84104068 | 50.87713734 | 51.65171208 |
| D65 | 169.00058929 | 169.03088651 | 169.71030546 |
| D66 | −120.64476837 | −120.63362997 | −119.11115232 |
| D67 | 51.89044308 | 51.87027494 | 51.66320139 |
| D68 | −65.58155270 | −65.59104262 | −65.93846062 |
| D69 | −66.67428542 | −66.66835138 | −67.19000316 |
| D70 | −118.76410502 | −118.75497303 | −118.87135263 |
| D71 | −132.76879862 | −132.76775335 | −132.41059726 |
| D72 | −117.45364759 | −117.45138885 | −117.81963777 |
References
- Roth, B.D. The Discovery and Development of Atorvastatin, A Potent Novel Hypolipidemic Agent. Prog. Med. Chem. 2002, 40, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mikulic, M. Worldwide revenue of Pfizer’s Lipitor from 2003 to 2019. 2021. Available online: https://www.statista.com/statistics/254341/pfizers-worldwide-viagra-revenues-since-2003/ (accessed on 11 July 2022).
- Alnajjar, R.; Mohamed, N.; Kawafi, N. Bicyclo[1.1.1]Pentane as Phenyl Substituent in Atorvastatin Drug to improve Physicochemical Properties: Drug-likeness, DFT, Pharmacokinetics, Docking, and Molecular Dynamic Simulation. J. Mol. Struct. 2021, 1230, 129628. [Google Scholar] [CrossRef]
- Hoffmann, M.; Nowosielski, M. DFT study on hydroxy acid-lactone interconversion of statins: The case of atorvastatin. Org. Biomol. Chem. 2008, 6, 3527–3531. [Google Scholar] [CrossRef]
- Duque, L.; Guerrero, G.; Colorado, J.H.; Restrepo, J.A.; Velez, E. Theoretical Insight into mechanism of antioxidant capacity of atorvastatin and its o-hydroxy and p-hydroxy metabolites, using DFT methods. Comput. Theor. Chem. 2022, 1214, 113758. [Google Scholar] [CrossRef]
- Bâldea, I. Critical analysis of radical scavenging properties of atorvastatin in methanol recently estimated via density functional theory. arXiv 2022, arXiv:2206.13990. [Google Scholar] [CrossRef]
- Portes, E.; Gardrat, C.; Castellan, A. A comparative study on the antioxidant properties of tetrahydrocurcuminoids and curcuminoids. Tetrahedron 2007, 63, 9092–9099. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis 1998, 138, 271–280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- bwHPC. bwHPC 2.0 (JUSTUS cluster_2.0, bwUniCluster_2.0, MLS&WISO_2.0) Supported by the State of Baden-Württemberg Supported by the State of Baden-Württemberg and the German Research Foundation (DFG) through Grant no INST 40/575-1 FUGG. Available online: https://wiki.bwhpc.de/e/Category:BwForCluster_JUSTUS_2 (accessed on 12 June 2022).
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry. I. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A Complete Basis Set Model Chemistry. II. Open-Shell Systems and the Total Energies of the First-Row Atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, J.F.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States. J. Phys. Chem. A 2006, 110, 13126–13130. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Bâldea, I. Comprehensive Quantum Chemical Characterization of the Astrochemically Relevant HCnH Chain Family. An Attempt to Aid Astronomical Observations. Adv. Theor. Simul. 2022, 2200244. [Google Scholar] [CrossRef]
- Kaiser, R.I.; Sun, B.J.; Lin, H.M.; Chang, A.H.H.; Mebel, A.M.; Kostko, O.; Ahmed, M. An Experimental and Theoretical Study on the Ionization Energies of Polyynes H–(C≡C)n–H; n = 1–9). Astrophys. J. 2010, 719, 1884. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. A Universal Approach to Solvation Modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Perspective on Foundations of Solvation Modeling: The Electrostatic Contribution to the Free Energy of Solvation. J. Chem. Theory Comput. 2008, 4, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.R. Gabedit: A Graphical User Interface For Computational Chemistry Softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Glendening, E.; Badenhoop, J.; Reed, A.; Carpenter, J.; Bohmann, J.; Morales, C.; Weinhold, F. NBO Code Version 6.0. 2012. Available online: https://nbo6.chem.wisc.edu/ (accessed on 7 April 2022).
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Mayer, I. Bond Order and Valence Indices: A Personal Account. J. Comput. Chem. 2007, 28, 204–221. [Google Scholar] [CrossRef]
- Bâldea, I. Chemical bonding in representative astrophysically relevant neutral, cation, and anion HCnH chains. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Clarendon, UK, 1989; p. 149. [Google Scholar]
- Gázquez, J.L.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Rajan, V.K.; Hasna, C.K.; Muraleedharan, K. The natural food colorant Peonidin from cranberries as a potential radical scavenger—A DFT based mechanistic analysis. Food Chem. 2018, 262, 184–190. [Google Scholar] [CrossRef]
- Bâldea, I. Impact of Molecular Conformation on Transport and Transport-Related Properties at the Nanoscale. Appl. Surf. Sci. 2019, 487, 593–600. [Google Scholar] [CrossRef]
- Bâldea, I. Alternation of Singlet and Triplet States in Carbon-Based Chain Molecules and Its Astrochemical Implications: Results of an Extensive Theoretical Study. Adv. Theory Simul. 2019, 2, 1900084. [Google Scholar] [CrossRef]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef] [PubMed]
- Bâldea, I. A Quantum Chemical Study from a Molecular Transport Perspective: Ionization and Electron Attachment Energies for Species Often Used to Fabricate Single-Molecule Junctions. Faraday Discuss. 2014, 174, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Chem. Phys. 1996, 100, 12974–12980, Notice that after eq. (19), the authors of this reference state: “The individual eigenfunctions and eigenvalues, φj and εj, of the Kohn-Sham equations have no strict physical significance…”. [Google Scholar] [CrossRef] [Green Version]
- Godby, R.W.; Schlüter, M.; Sham, L.J. Self-energy operators and exchange-correlation potentials in semiconductors. Phys. Rev. B 1988, 37, 10159–10175. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, V.; Baldereschi, A. Dielectric Scaling of the Self-Energy Scissor Operator in Semiconductors and Insulators. Phys. Rev. B 1995, 51, 17196–17198. [Google Scholar] [CrossRef]
- Bâldea, I. Demonstrating Why DFT-Calculations For Molecular Transport in Solvents Need Scissor Corrections. Electrochem. Commun. 2013, 36, 19–21. [Google Scholar] [CrossRef]
- Bâldea, I. Profiling C4N Radicals of Astrophysical Interest. Mon. Not. R. Astron. Soc. 2020, 493, 2506–2510. [Google Scholar] [CrossRef]
- Bâldea, I. Profiling Astrophysically Relevant MgC4H Chains. An Attempt to Aid Astronomical Observations. Mon. Not. R. Astron. Soc. 2020, 498, 4316–4326. [Google Scholar] [CrossRef]
- Zhou, Z.; Parr, R.G. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar]
- Gázquez, J.L. Perspectives on the Density Functional Theory of Chemical Reactivity. J. Mex. Chem. Soc. 2008, 52, 3–10. [Google Scholar]
- Domingo, L.R.; Aurell, M.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Burton, G.W.; Doba, T.; Gabe, E.; Hughes, L.; Lee, F.L.; Prasad, L.; Ingold, K.U. Autoxidation of biological molecules. 4. Maximizing the antioxidant activity of phenols. J. Am. Chem. Soc. 1985, 107, 7053–7065. [Google Scholar] [CrossRef]
- de Heer, M.I.; Mulder, P.; Korth, H.G.; Ingold, K.U.; Lusztyk, J. Hydrogen Atom Abstraction Kinetics from Intramolecularly Hydrogen Bonded Ubiquinol-0 and Other (Poly)methoxy Phenols. J. Am. Chem. Soc. 2000, 122, 2355–2360. [Google Scholar] [CrossRef]
- Mayer, I.; Salvador, P. Overlap populations, bond orders and valences for “fuzzy” atoms. Chem. Phys. Lett. 2004, 383, 368–375. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M.G. Flavonoids as Antioxidants. J. Am. Chem. Soc. 1994, 116, 4846–4851. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Steenken, S.; Hara, Y.; Simic, M.G. Reduction potentials of flavonoid and model phenoxyl radicals. Which ring in flavonoids is responsible for antioxidant activity? J. Chem. Soc. Perkin Trans. 1996, 2, 2497–2504. [Google Scholar] [CrossRef]
- Litwinienko, G.; Ingold, K.U. Abnormal Solvent Effects on Hydrogen Atom Abstractions. 1. The Reactions of Phenols with 2,2-Diphenyl-1-picrylhydrazyl (DPPH•) in Alcohols. J. Org. Chem. 2003, 68, 3433–3438. [Google Scholar] [CrossRef] [Green Version]
- Litwinienko, G.; Ingold, K.U. Abnormal Solvent Effects on Hydrogen Atom Abstraction. 2. Resolution of the Curcumin Antioxidant Controversy. The Role of Sequential Proton Loss Electron Transfer. J. Org. Chem. 2004, 69, 5888–5896. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The Density Functional Formalism, Its Applications and Prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Bâldea, I. Extending the Newns-Anderson Model to Allow Nanotransport Studies Through Molecules with Floppy Degrees of Freedom. Europhys. Lett. 2012, 99, 47002. [Google Scholar] [CrossRef] [Green Version]
- Fifen, J.J. Thermodynamics of the Electron Revisited and Generalized. J. Chem. Theory Comput. 2013, 9, 3165–3169. [Google Scholar] [CrossRef] [PubMed]
- Fifen, J.J.; Dhaouadi, Z.; Nsangou, M. Revision of the Thermodynamics of the Proton in Gas Phase. J. Phys. Chem. A 2014, 118, 11090–11097. [Google Scholar] [CrossRef] [PubMed]
- Markovic, Z.; Tosovic, J.; Milenkovic, D.; Markovic, S. Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Rimarcik, J.; Lukes, V.; Klein, E.; Ilcin, M. Study of the solvent effect on the enthalpies of homolytic and heterolytic N-H bond cleavage in p-phenylenediamine and tetracyano-p-phenylenediamine. J. Mol. Struct. THEOCHEM 2010, 952, 25–30. [Google Scholar] [CrossRef]
- Xu, B.; Tao, N.J. Measurement of Single-Molecule Resistance by Repeated Formation of Molecular Junctions. Science 2003, 301, 1221–1223. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Xiao, X.; Tao, N.J. Measurements of Single-Molecule Electromechanical Properties. J. Am. Chem. Soc. 2003, 125, 16164–16165. [Google Scholar] [CrossRef]
- Bruot, C.; Hihath, J.; Tao, N. Mechanically controlled molecular orbital alignment in single molecule junctions. Nat. Nano 2011, 7, 35–40. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Bâldea, I. Long Carbon-Based Chains of Interstellar Medium Can Have a Triplet Ground State. Why Is This Important for Astrochemistry? ACS Earth Space Chem. 2019, 3, 863–872. [Google Scholar] [CrossRef]
- Luo, Y.R. (Ed.) Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, USA, 2003; p. 239. [Google Scholar] [CrossRef]
- Bâldea, I. Extensive Quantum Chemistry Study of Neutral and Charged C4N Chains: An Attempt to Aid Astronomical Observations. ACS Earth Space Chem. 2020, 4, 434–448. [Google Scholar] [CrossRef]
- Li, Y.; Haworth, N.L.; Xiang, L.; Ciampi, S.; Coote, M.L.; Tao, N. Mechanical Stretching-Induced Electron-Transfer Reactions and Conductance Switching in Single Molecules. J. Am. Chem. Soc. 2017, 139, 14699–14706. [Google Scholar] [CrossRef] [PubMed]
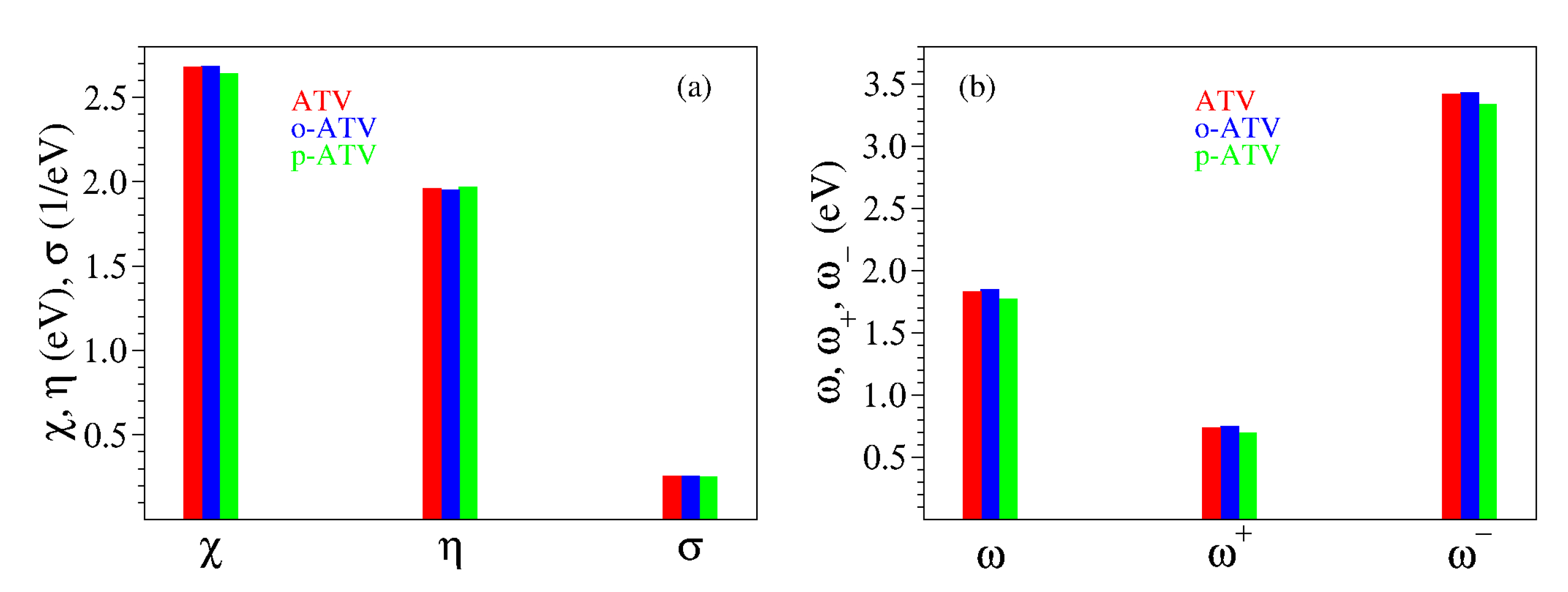

| Molecule | IP | EA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ATV | 4.64 | 0.72 | 3.92 | 1.96 | −2.68 | 0.26 | 1.83 | 0.74 | 3.42 |
| o-ATV | 4.64 | 0.73 | 3.90 | 1.95 | −2.68 | 0.26 | 1.85 | 0.75 | 3.43 |
| p-ATV | 4.60 | 0.67 | 3.93 | 1.97 | −2.64 | 0.25 | 1.77 | 0.70 | 3.34 |
| Phenol | 5.43 | 0.31 | 5.12 | 2.56 | −2.87 | 0.20 | 1.61 | 0.49 | 3.36 |
| Trolox | 4.51 | 0.76 | 3.76 | 1.88 | −2.63 | 0.27 | 1.85 | 0.77 | 3.40 |
| Molecule | Method | IP | EA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ATV | UB3LYP/IEFPCM | 4.64 | 0.72 | 3.92 | 1.96 | −2.68 | 0.26 | 1.83 | 0.74 | 3.42 |
| B3LYP/SMD | 4.39 | 0.63 | 3.76 | 1.88 | −2.51 | 0.27 | 1.67 | 0.65 | 3.16 | |
| ROB3LYP/IEFPCM | 4.69 | 0.70 | 3.98 | 1.99 | −2.69 | 0.25 | 1.82 | 0.72 | 3.42 | |
| UPBE0/IEFPCM | 4.67 | 0.71 | 3.96 | 1.98 | −2.69 | 0.25 | 1.82 | 0.73 | 3.41 | |
| UM062x/IEFPCM | 4.95 | 0.73 | 4.22 | 2.11 | −2.84 | 0.24 | 1.91 | 0.75 | 3.59 | |
| o-ATV | UB3LYP/IEFPCM | 4.64 | 0.73 | 3.90 | 1.95 | −2.68 | 0.26 | 1.85 | 0.75 | 3.43 |
| UB3LYP/SMD | 4.38 | 0.63 | 3.75 | 1.87 | −2.51 | 0.27 | 1.68 | 0.66 | 3.16 | |
| p-ATV | UB3LYP/IEFPCM | 4.60 | 0.67 | 3.93 | 1.97 | −2.64 | 0.25 | 1.77 | 0.70 | 3.34 |
| UB3LYP/SMD | 4.37 | 0.61 | 3.77 | 1.88 | −2.49 | 0.27 | 1.65 | 0.64 | 3.13 |
| Species | ||
|---|---|---|
| Electron | +0.001194 a | −0.030204 c |
| Proton | +0.002339 b | −0.405508 c |
| H-atom | −0.497912 | +0.001904 d |
| Molecule | Position | BDE | IP | PDE | PA | ETE |
|---|---|---|---|---|---|---|
| ATV | 1-OH | 91.4 | 107.0 | 22.4 | 23.8 | 105.7 |
| 2-OH | 104.2 | 35.3 | 46.7 | 118.5 | ||
| 3-OH | 105.2 | 36.3 | 61.5 | 119.5 | ||
| 4-NH | 90.2 | 21.3 | 44.4 | 83.9 | ||
| o-ATV | 1-OH | 91.2 | 106.9 | 22.4 | 23.8 | 105.5 |
| 2-OH | 104.2 | 35.4 | 46.8 | 118.5 | ||
| 3-OH | 105.1 | 36.3 | 61.5 | 119.4 | ||
| 4-NH | 89.3 | 20.5 | 49.0 | 78.4 | ||
| 5-OH | 77.5 | 8.7 | 34.4 | 91.8 | ||
| p-ATV | 1-OH | 90.7 | 106.2 | 22.6 | 23.8 | 105.0 |
| 2-OH | 104.2 | 36.0 | 46.8 | 117.6 | ||
| 3-OH | 105.1 | 37.0 | 58.2 | 118.5 | ||
| 4-NH | 85.5 | 17.4 | 43.8 | 79.0 | ||
| 5-OH | 77.4 | 9.2 | 37.9 | 90.8 |
| Molecule | Method | Position | BDE | IP | PDE | PA | ETE |
|---|---|---|---|---|---|---|---|
| ATV | UPBE0/IEFPCM | 1-OH | 93.5 | 107.7 | 23.2 | 24.5 | 106.4 |
| UPBE0/IEFPCM | 4-OH | 109.8 | 39.4 | 45.4 | 101.7 | ||
| ATV | UB3LYP/IEFPCM | 1-OH | 91.4 | 107.0 | 22.4 | 23.8 | 105.7 |
| ATV | UB3LYP/IEFPCM | 4-NH | 90.2 | 21.3 | 44.4 | 83.9 | |
| ATV | ROB3LYP/IEFPCM | 1-OH | 92.4 | 108.0 | 21.4 | (23.8) | 106.7 |
| ATV | ROB3LYP/IEFPCM | 4-NH | 92.2 | 22.2 | (44.4) | 85.9 | |
| ATV | UB3LYP/SMD | 1-OH | 85.9 | 101.2 | 22.7 | 24.0 | 100.0 |
| UB3LYP/SMD | 4-NH | 90.7 | 27.6 | 44.0 | 84.8 | ||
| o-ATV | UB3LYP/IEFPCM | 5-OH | 77.5 | 106.9 | 8.7 | 34.4 | 91.8 |
| UB3LYP/SMD | 5-OH | 79.6 | 101.0 | 16.8 | 34.6 | 83.1 | |
| p-ATV | UB3LYP/IEFPCM | 5-OH | 77.4 | 106.2 | 9.2 | 37.9 | 90.8 |
| UB3LYP/SMD | 5-OH | 78.0 | 100.9 | 15.2 | 36.5 | 79.5 |
| Molecule | Position | Wiberg | Length | BDE | |
|---|---|---|---|---|---|
| ATV | 1-OH | 0.6789 | 0.9748 | 91.4 | 3723.2 |
| 2-OH | 0.6732 | 0.9755 | 104.2 | 3667.0 | |
| 3-OH | 0.6721 | 0.9750 | 105.2 | 3641.6 | |
| 4-NH | 0.7562 | 1.0147 | 90.2 | 3581.0 | |
| o-ATV | 1-OH | 0.6789 | 0.9747 | 91.2 | 3724.8 |
| 2-OH | 0.6733 | 0.9754 | 104.2 | 3641.7 | |
| 3-OH | 0.6720 | 0.9750 | 105.1 | 3667.5 | |
| 4-NH | 0.7454 | 1.0151 | 89.3 | 3574.2 | |
| 5-OH | 0.6946 | 0.9676 | 77.5 | 3817.1 | |
| p-ATV | 1-OH | 0.6789 | 0.9748 | 90.7 | 3724.2 |
| 2-OH | 0.6732 | 0.9755 | 104.2 | 3667.9 | |
| 3-OH | 0.6730 | 0.9748 | 105.1 | 3644.7 | |
| 4-NH | 0.7550 | 1.0149 | 85.5 | 3576.0 | |
| 5-OH | 0.7029 | 0.9679 | 77.4 | 3808.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bâldea, I. Why Ortho- and Para-Hydroxy Metabolites Can Scavenge Free Radicals That the Parent Atorvastatin Cannot? Important Pharmacologic Insight from Quantum Chemistry. Molecules 2022, 27, 5036. https://doi.org/10.3390/molecules27155036
Bâldea I. Why Ortho- and Para-Hydroxy Metabolites Can Scavenge Free Radicals That the Parent Atorvastatin Cannot? Important Pharmacologic Insight from Quantum Chemistry. Molecules. 2022; 27(15):5036. https://doi.org/10.3390/molecules27155036
Chicago/Turabian StyleBâldea, Ioan. 2022. "Why Ortho- and Para-Hydroxy Metabolites Can Scavenge Free Radicals That the Parent Atorvastatin Cannot? Important Pharmacologic Insight from Quantum Chemistry" Molecules 27, no. 15: 5036. https://doi.org/10.3390/molecules27155036
APA StyleBâldea, I. (2022). Why Ortho- and Para-Hydroxy Metabolites Can Scavenge Free Radicals That the Parent Atorvastatin Cannot? Important Pharmacologic Insight from Quantum Chemistry. Molecules, 27(15), 5036. https://doi.org/10.3390/molecules27155036