2.1. Equilibrium Binding of ALKBH2 to Metal Ions, Cosubstrate, and Methylated DNA
To assess the affinity of ALKBH2 for its cofactor, to a cosubstrate, and to methylated and nonmethylated DNA (
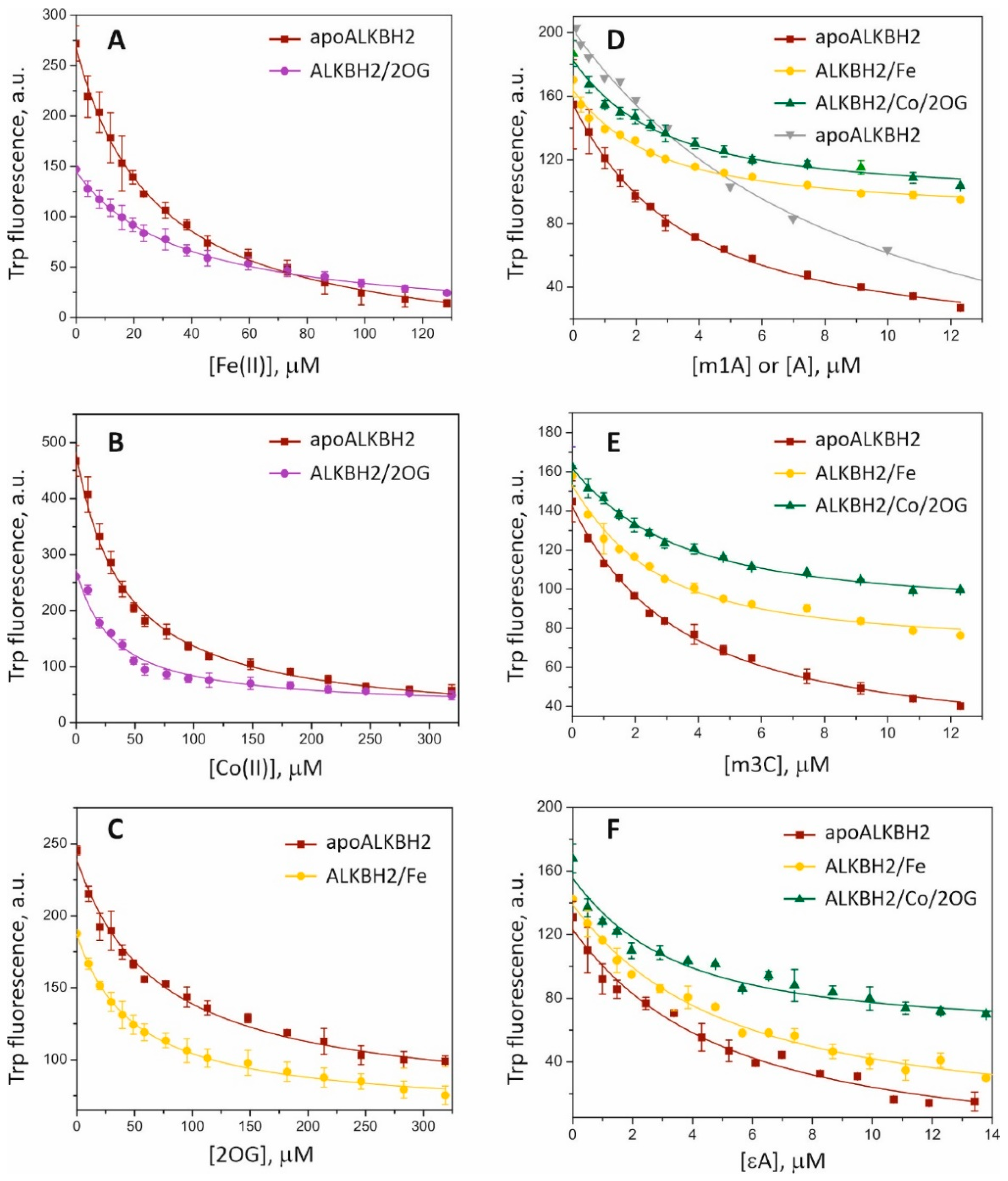
Table 1), Trp fluorescence of the protein was monitored upon titration with the ligands (
Figure 2). It was found that equilibrium dissociation constant (
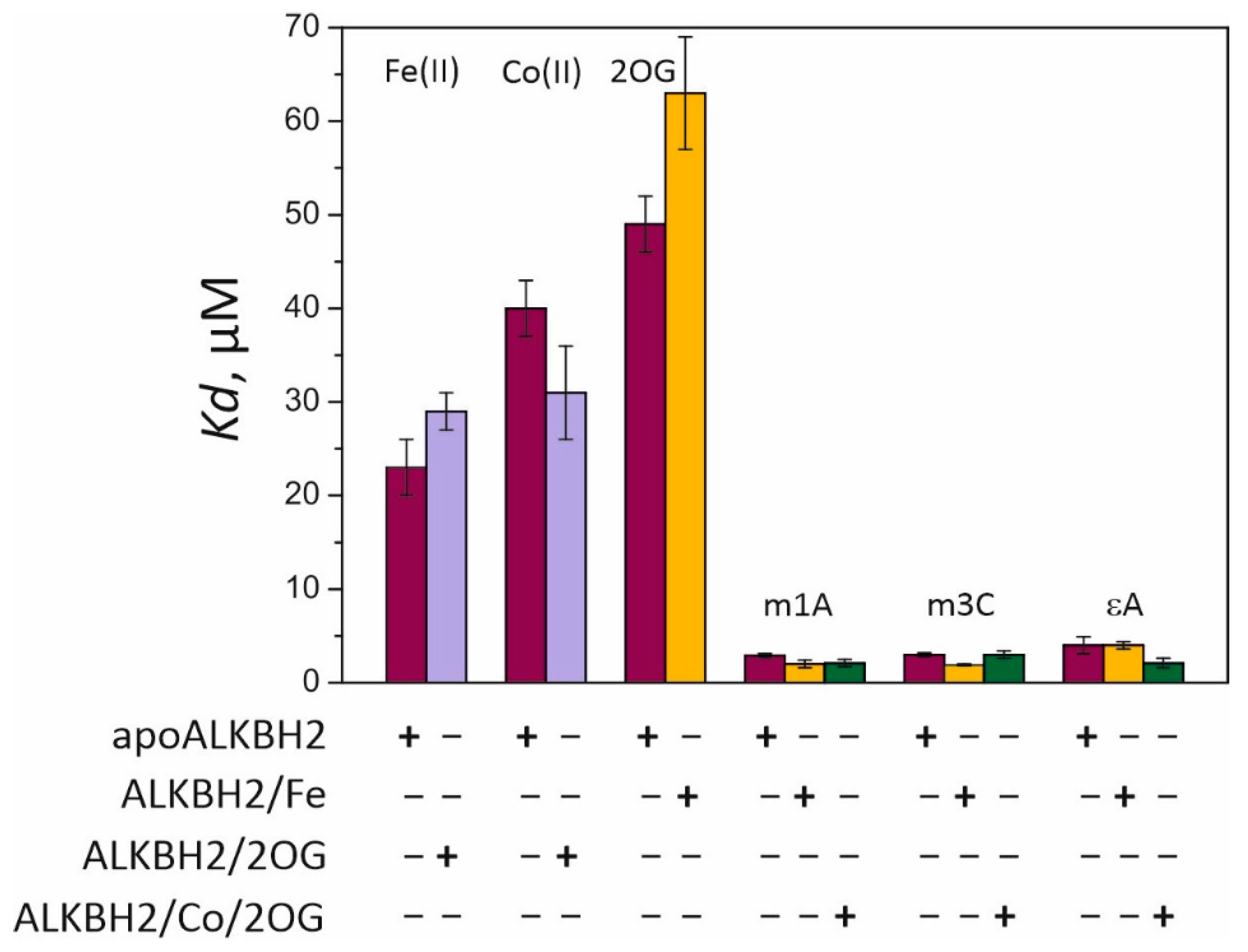
Kd) values for complexes of the free enzyme (apoALKBH2) with Fe(II) and Co(II) are 27 and 40 μM, respectively (
Figure 3). Thus, Co(II) can compete with Fe(II) for the iron-binding site only at concentrations twofold lower than those observed for Ni(II) [
27]. Incubation of ALKBH2 with 2OG prior to the titration with metal ions slightly reduced binding affinity for Fe(II) and increased affinity for Co(II). The lowest stability was observed for the complex of ALKBH2 with its cosubstrate 2OG, while preincubation of the protein with Fe(II) did not improve but rather reduced the binding efficiency of 2OG. In our previous work [
28], we studied an ability of the EcAlkB protein to bind different metal cofactors, a cosubstrate, and methylated DNA. Those results indicated that EcAlkB has 2–4 times higher affinity for Fe(II) and 2OG than for Co(II), and the formation of the complex between the dioxygenase and 2OG is not affected by preincubation with a metal ion.
It is known that transition metals other than Fe(II) inhibit the catalytic activity of AlkB-like proteins while maintaining their ability to bind DNA substrates [
17,
27,
29,
30]. Here, we determined the binding affinity of ALKBH2 toward alkylated DNA in the presence of Co(II) ions. ApoALKBH2 and preformed complexes ALKBH2/Fe, ALKBH2/Co/2OG (1 μM) were titrated with 15 nt dsODN containing N1-methyladenine (m1A), N3-methylcytosine (m3C), or εA.
Kd values for methylated-DNA binding were calculated by means of Equation (2) (see
Section 4.3 for details). They are in the range of 1.9–4.0 μM, which is one order of magnitude lower than that of metal ions and 2OG, indicating higher affinity of ALKBH2 for the DNA substrate than for its cofactor and cosubstrate. Similar affinity patterns have been observed during fluorescent titration of the EcAlkB protein [
28,
29]. Incubation of ALKBH2 with the Fe(II) ion prior to the titration resulted in a 1.5-fold increase in the affinity for the m1A- and m3C-containing substrates but did not affect the efficiency of binding to the εA-containing substrate. In a comparison of the efficiency of DNA binding to preformed complex ALKBH2/Co/2OG vs. apoALKBH2, the most pronounced effect was observed in the case of the εA-containing substrate accompanied by a 2-fold reduction in
Kd. As a control, we also measured Trp fluorescence quenching upon titration of ALKBH2 with an intact 15 nt dsODN (
Figure 2D, gray triangles). The resultant
Kd reached 7.8 ± 1.2 μM, which meant that ALKBH2 binds to alkylated DNA in a more specific manner. Overall, the fluorescence spectroscopy data indicated that the binding of damaged DNA by the ALKBH2 protein is preferable over the binding to the metal cofactor and cosubstrate.
2.2. ALKBH2 Repair Activity toward DNA Lesions m1A, m3C, and εA
Activity of the recombinant human ALKBH2 protein toward damaged dsDNA substrates was measured by a restriction enzyme-coupled assay followed by gel electrophoresis digestion. The reaction mixture contained equimolar amounts of the enzyme and its substrate preincubated with Fe(II) and 2OG. After termination of the reaction at each time point, samples were treated with a specific restriction endonuclease to cleave the intact DNA product (see
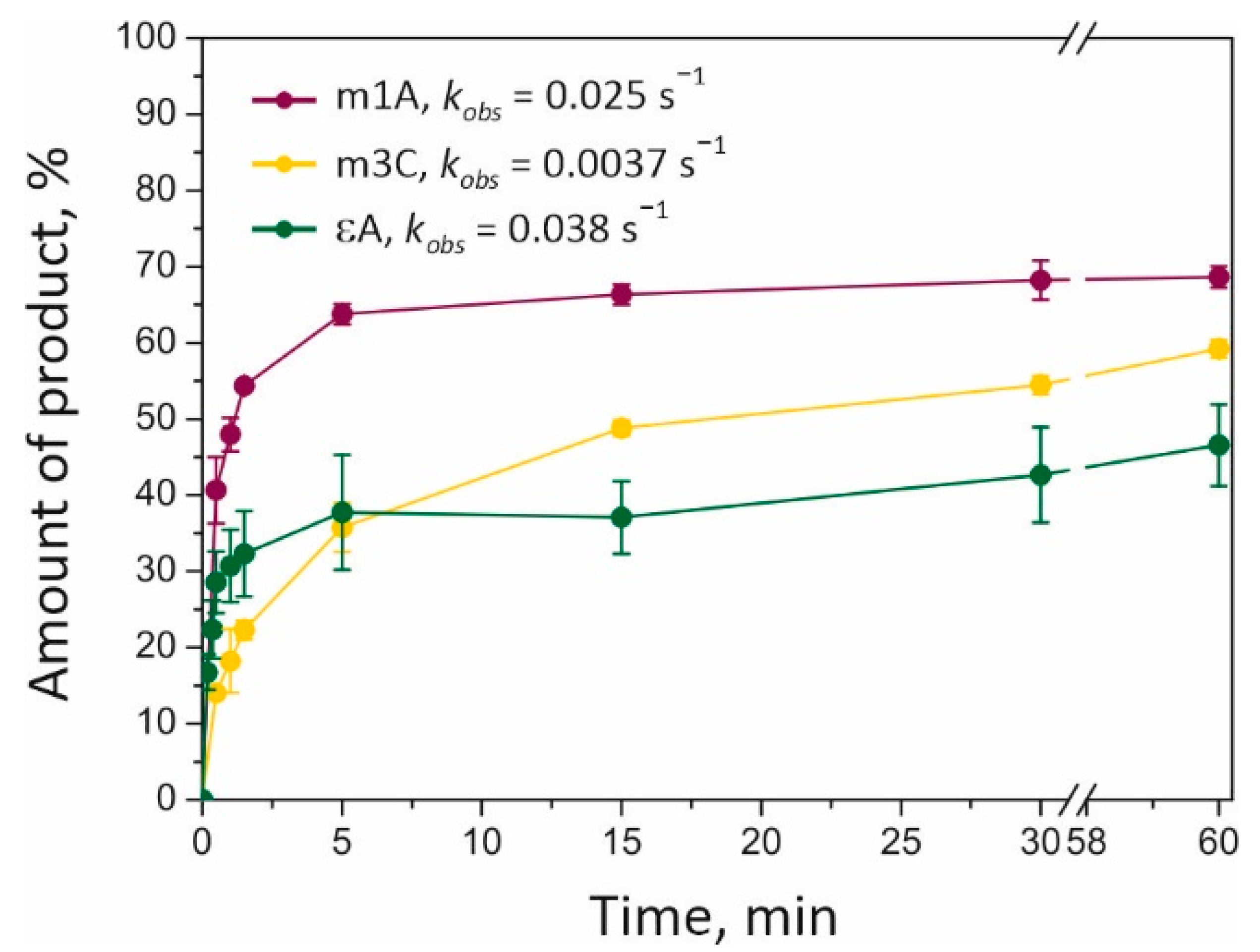
Section 4.4 for details). A shortened reaction product was next visualized on a denaturing polyacrylamide gel, revealing time courses of the substrate dealkylation (
Figure 4). The amount of dealkylated DNA varied from 40 to 70% depending on the lesion type (m1A > m3C > εA). Repair of the m1A base is carried out somewhat more slowly but more efficiently than the repair of the εA base is. The lowest rate of repair (with a medium amount of the product: ~58% after 2 h) was detected in the case of the m3C-containing substrate (m3C). Our data indicated that m1A is the most effective substrate for ALKBH2 among those analyzed.
The observed rate constant (
kobs; see
Section 4.4 for details) of dealkylation reactions depended on the lesion as follows: εA (0.038 s
−1) > m1A (0.025 s
−1) > m3C (0.0037 s
−1). Our data suggested that an initial reaction rate does not correlate with the maximum amount of dealkylated product formed for each lesion. The highest reaction rate was documented for the repair of the εA-containing substrate, which also gave the lowest amount of the product. The presence of fast and slow phases of product accumulation in the case of εA base oxidation may be attributed to a tight binding of the reaction product within the ALKBH2 active site; this event impairs dissociation of the enzyme–product complex. This hypothesis can find support in previous findings about EcAlkB and ALKBH2. As shown by NMR spectroscopy, the EcAlkB/Fe/Suc complex has higher flexibility as compared to the 2OG-bound complex, and this property is believed to be important for an effective product release [
31]. On the other hand, recent data obtained by the QM/MM method indicate that the ALKBH2/dsDNA complex is more compact and rigid (than the EcAlkB/dsDNA complex), and these properties may result in tighter binding of the product [
21]. Thus, slow dissociation of the enzyme–product complex can inhibit the activity on the next enzymatic cycle.
So far, no data have been published that describe in vitro kinetic parameters for both methylated and etheno lesions under the same conditions (reagent amounts, temperature, and a nucleotide context). Two research groups have investigated Michaelis–Menten kinetics of ALKBH2 toward m1A- and m3C-containing DNA and reported markedly distinct values of
kcat and
KM [
8,
32]. For example,
kcat for lesion m3C within a 24 nt dsODN was found to be 8.8 s
−1 [
8], whereas
kcat for the same lesion within a 16 nt dsODN was 0.043 s
−1 [
32]. As for lesion εA, no steady-state kinetics of its hydroxylation by ALKBH2 have been reported at all. Single-turnover kinetics measured by Ringvoll et al. [
10] revealed the efficiency of εA removal of 0.094 min
−1 in terms of the turnover number, and the largest amount of the dealkylated product did not exceed 15%. Another study suggests that the specific repair activity of ALKBH2 against εA is 69.4 fmol min
−1 pmol enzyme
−1, with 70% of the repaired dsODN detected after 10 min of incubation [
9]. The existence of such fragmentary or controversial data may be due to variation of the proportion of an active fraction in enzyme samples among studies. We detected a similar problem in the course of the protein purification; therefore, all experiments were conducted on the same ALKBH2 sample that manifested the highest activity level.
2.3. SF Fluorescence Measurements of Conformational Changes in the Complex of ALKBH2 with a Monoalkyl Lesion (m1A or m3C)
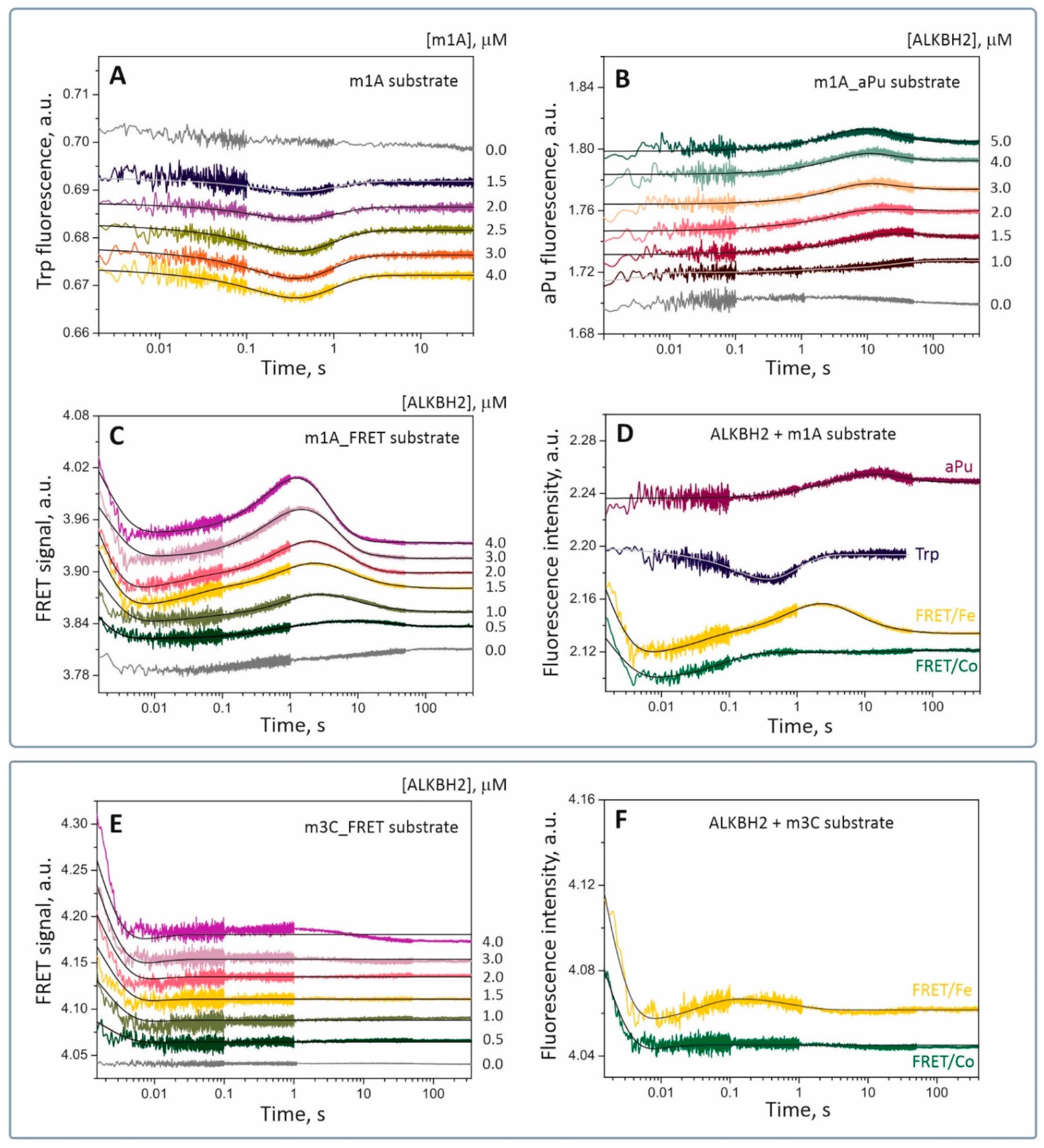
To investigate in detail the changes in the conformation of the enzyme–substrate complex in the course of DNA dealkylation, we measured a fluorescent signal from various fluorescent probes sensitive to changes in their local environment. Equimolar amounts of ALKBH2 and a model DNA substrate containing a lesion (m1A or m3C) were mixed in an SF spectrometer cell in the presence of Fe(II) and 2OG. Time-dependent conformational changes of the protein were recorded as changes in the fluorescence of its Trp residues. Detection of the conformational transitions in the DNA substrate structure was carried out using two fluorophores. The first one was a fluorescent analog of adenine aPu introduced into the damaged strand 3′ to the m1A base, and the second was a dye-quencher pair FAM-BHQ1 located at the 5′ end and 3′ end of the damaged strand, respectively, for measuring fluorescence resonance energy transfer (FRET; see
Table 1). According to preliminary experiments, interaction of ALKBH2 with the substrate containing the m1A lesion caused significant changes in the local environment of each fluorescent probe, whereas binding to an m3C-containing substrate had very little effect on fluorescence intensity of the labels. Further SF analysis was performed with fluorophores sensitive to changes in the local environment. A series of SF traces was obtained for each substrate under approximately single-turnover conditions for a fixed concentration of the fluorescent component (1.5 or 2.0 μM) and various concentrations of its nonfluorescent counterpart (0.5 to 5.0 μM;
Figure 5). In the case of substrate m3C, we were able to detect a concentration-dependent change in the FRET signal. In each case, a control curve was obtained by mixing the fluorescent component with reaction buffer to avoid detection of nonspecific changes in the signal (
Figure 5, gray curves).
It was found that conformational dynamics of the ALKBH2 protein during interactions with substrate m1A are biphasic (
Figure 5A). In the first phase, Trp fluorescence is quenched (up to time point 300 ms), and then it gradually grows in the interval 0.3 s to 5 s. By analyzing available X-ray structural data [
17,
18], we attempted to estimate the possible contribution of each of the four Trp residues of ALKBH2 (see
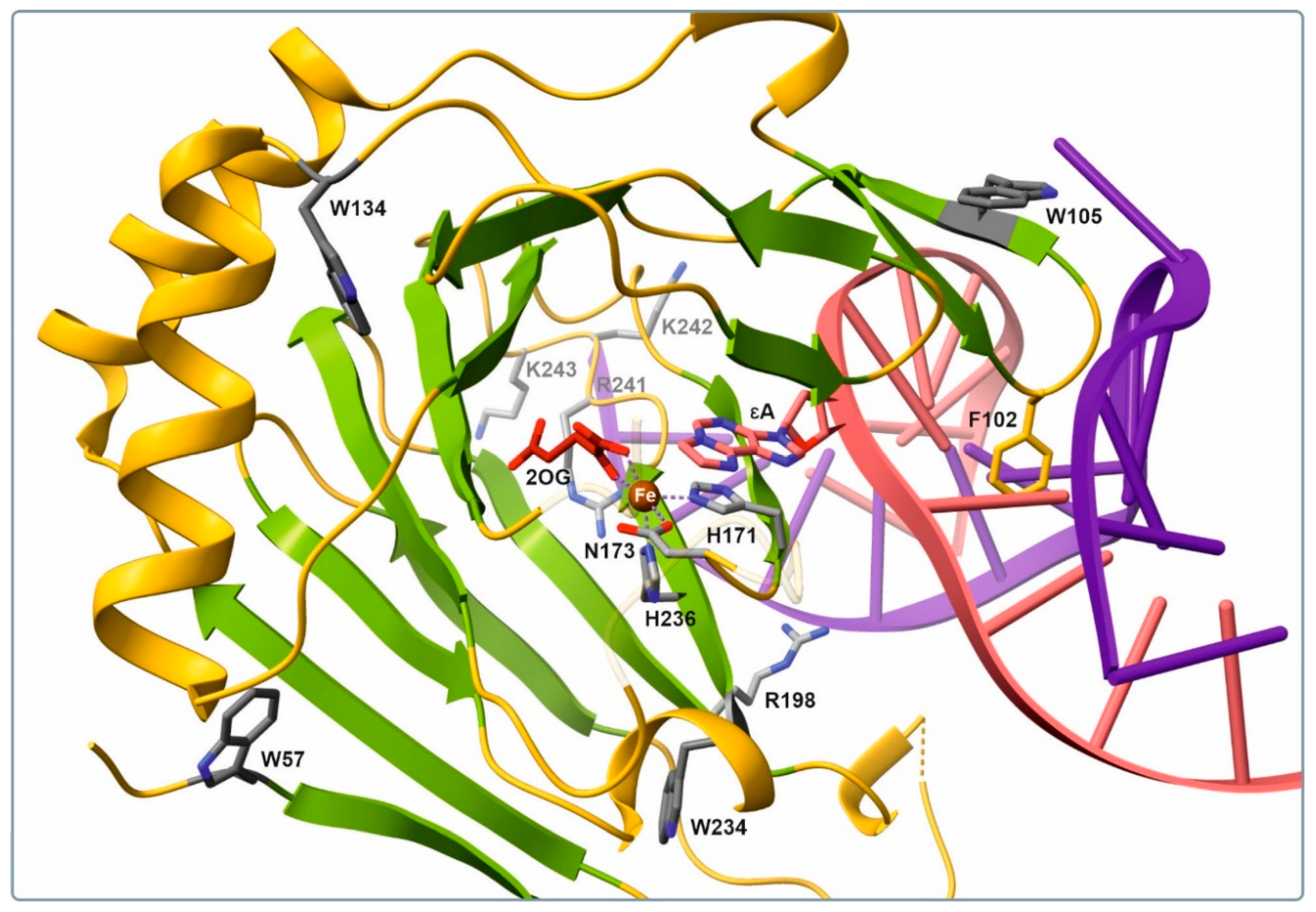
Figure 1) to the overall signal detected by the SF analysis. A side chain of Trp-105 proved to be the most important contributor because it belongs to the aromatic β3–β4 hairpin, which interacts closely with the damaged strand of the substrate duplex. This finger motif participated in capture of the substrate via a side chain of Phe-102, which occupies the vacant space created by methylated-base flipping. Other residues (Trp-57, -134, and -234) are likely to make only a minor contribution to the changes in fluorescence intensity because they are located on the protein surface far from the active site. Therefore, conformational changes of the enzyme observed by the SF method may represent fluctuations in the Trp-105 microenvironment in the course of embedding of the specific loop into the DNA substrate groove.
Behavior of the aPu fluorescent probe located adjacently to the m1A base showed no specific changes in the local environment of the fluorophore immediately after mixing with ALKBH2 (
Figure 5B). Nonetheless, starting from 400 ms, the aPu fluorescent signal underwent noticeable growth and reached its maximum in the range 10 s to 30 s, depending on the enzyme concentration. Next, there was a moderate decrease in fluorescence intensity up to 100 s. The fluorescent properties of aPu are described in the literature [
33,
34]. In particular, the fluorescence of aPu is quenched in solution by a hydrophobic environment and is enhanced after the transition to a more hydrophilic medium. Thus, we can assume that the interaction of enzyme ALKBH2 with substrate m1A in the middle and late stages of the catalytic cycle is accompanied by a bending of the DNA duplex in the region of the damaged nucleotide, as evidenced by an increase in solvent polarity around the aPu.
Analyses of protein–DNA interactions on the basis of the FRET phenomenon are widely used in biochemistry [
35]. Interactions between the enzyme and FRET-labeled damaged DNA cause a bending of the substrate duplex, resulting in fluctuation of the distance between the donor and acceptor and changes in FRET efficiency. Therefore, by measuring the intensity of the fluorescence transfer from the donor dye, we can detect protein-induced conformational changes in the DNA substrate with high accuracy. Here, we measured the FRET signal from the 5′-FAM/3′-BHQ1 pair during interactions of ALKBH2 with substrate m1A or m3C. As follows from
Figure 5C,E, the interaction of ALKBH2 with the FRET-labeled substrate caused multiple changes in the signal. The initial parts of the FRET curves for both substrates contain a sharp decrease in the signal from 1 ms to 5 ms, pointing to a spatial convergence of the fluorophore and quencher, and may mean the bending of the DNA helix when it binds to the dioxygenase. This conformational transition in the substrate molecule most likely accompanies the formation of a nonspecific collision complex. The next stage of substrate m1A processing was followed by a pronounced two-phase growth of the FRET signal with a peak at 80 ms and 1.5–2.0 s, respectively. These transitions denote spatial separation of FAM and BHQ1 and likely correspond to fine tuning of the nucleoprotein complex toward a catalytically active state. As for substrate m3C, low-amplitude alterations of the signal were detected after 10 ms on FRET curves corresponding to high ALKBH2 concentrations. A possible interpretation of this type of signal change is weak or slow conformational rearrangements in catalytic complex ALKBH2/m3C.
Some structural studies on AlkB family proteins have shown that bacterial and human 2OG dioxygenases can bind ions of transition metals other than Fe(II) (e.g., Co(II), Ni(II), Mn(II) or Cu(II)) with high affinity while retaining the proper tertiary structure [
17,
36,
37,
38]. At the same time, replacing Fe(II) with other metal ions leads to complete inhibition of the dealkylating activity [
27,
30]. This phenomenon can be exploited for the detection of substrate recognition and binding processes separately from hydroxylation by the SF method.
In the present study, we implemented this strategy by replacing Fe(II) with the Co(II) ion at the ALKBH2 active site. As we demonstrated by fluorescent titration (
Section 2.1,
Figure 2B and
Figure 3), cobalt binds to the ALKBH2 active site with the same efficiency as iron does in the presence of 2OG. To elucidate conformational dynamics of the enzyme–substrate complex at steps preceding the catalysis, 1.5 μM ALKBH2 was mixed with 1.5 μM substrate m1A or m3C in the presence of 100 μM CoSO
4.
Figure 5D,F depict time courses of the FRET signal (green curves) registered by the SF method under conditions similar to the Fe-containing system. In a comparison of FRET curves obtained in the presence of Fe(II) (yellow curves) and Co(II) ions, one can see that shapes of the curves matched in the interval 1–400 ms for substrate m1A and 1–30 ms for substrate m3C. After this period, no specific changes in fluorescence were noted on the curves in the presence of Co(II). Subsequent changes in the signal detected in the presence of Fe(II) most probably correspond to specific transitions that require the presence of a natural cofactor. For the ALKBH2/Co(II)/2OG/m3C complex, the FRET curve contained a single conformational transition (in the interval 1–20 ms) similar to that of the ALKBH2/Fe(II)/2OG/m3C complex (
Figure 5F). Consequently, replacement of the natural metal-cofactor with Co(II) ion does not affect the initial binding but complicates subsequent transformation of the enzyme–substrate complex.
2.4. Assessment of ALKBH2 Conformational Dynamics and Kinetic Parameters for the m1A- and m3C-Containing dsODNs
To quantitatively describe and interpret the observed conformational transitions in the light of specific stages of the molecular-kinetic mechanism, data obtained under similar conditions by different methods were analyzed. In particular, the fluorescent curves determined by the SF method were compared with time courses of reaction product accumulation revealed by polyacrylamide gel electrophoresis (PAGE). Half-conversion time (τ
1/2) of substrate m1A (
Figure 4), as determined by means of ratio ln2/
kobs, is approximately 30 s. This time matches the phase of the decrease in aPu fluorescence intensity between time points 20 and 80 s (
Figure 5B) and overlaps with the phase of a FRET signal reduction between 2 s and 50 s (
Figure 5C). Accordingly, these two conformational transitions of substrate m1A may accompany the steps of its hydroxylation and dissociation of the enzyme–product complex. As follows from PAGE analysis of ALKBH2 repair activity (
Figure 4), hydroxylation of substrate m3C takes much more time (τ
1/2 ≈ 180 s) as compared to substrate m1A. Therefore, the changes in substrate m3C′s conformation as recorded by FRET (
Figure 5E) may reflect the binding process at the ALKBH2 active site and DNA duplex adjustment on the way to the catalytically active complex but do not provide information about the catalytic step.
To compare the conformational transitions observed using different fluorophores, one curve with clearly distinguishable conformational transitions was selected from each kinetic series, and individual phases were compared qualitatively (
Figure 5D,F). This approach revealed that the initial binding of ALKBH2 to substrate m1A (up to time point ~8–10 ms) does not cause significant alterations of protein conformation in the region of intercalating loop β3–β4 (no change in Trp fluorescence). The DNA duplex undergoes some bending, which brings its ends closer together, but does not affect solvent polarity near the damaged nucleotide (no change in aPu fluorescence). Once the enzyme has detected the presence of a methylated base in the DNA substrate, its conformation changes, and a specific interaction with the substrate begins, as evidenced by synchronous quenching of Trp fluorescence (embedding of the intercalating loop into the DNA groove) and an increase in the FRET signal (eversion of the damaged nucleotide into the active site pocket) (up to time point ~100 ms). As a result, the aPu residue adjacent to m1A becomes more accessible to the solvent, and its fluorescence begins to increase gradually (up to ~10 s). At the same time, conformational transitions in the catalytic complex continue, accompanied by a more pronounced increase and a subsequent decrease in the FRET signal as well as a return of Trp fluorescence to the initial level. By comparing all of the data, we gained important knowledge that allowed us to correlate the observed conformational transitions with individual phases of the catalytic cycle. For example, the first two phases of FRET signal changes appeared to be associated with the formation of a specific complex between ALKBH2 and substrate m1A.
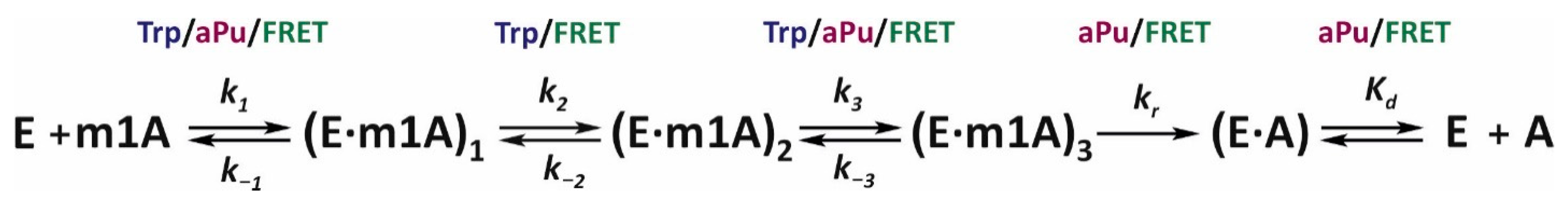
To choose an appropriate kinetic scheme and to determine the kinetic parameters, the DynaFit 4 software was employed [
39]. During the selection of the kinetic scheme, its sequential complication was carried out, and deviations of the theoretical curves from the experimental ones were evaluated. The rate constants for each elementary step were obtained via optimization of the parameters during numerical integration of the system of differential equations [
25,
40]. As a result, it was determined that the lowest number of steps needed to describe the experimental curves for the demethylation of substrate m1A is five. These are three reversible steps described by rate constants
ki and
k−i (
i = 1, 2, and 3) and one irreversible step (
kr), followed by the decay of the enzyme–product complex (
Kd) (
Scheme 1). Thus, the sequence of changes in the conformation of the enzyme–substrate complex can be described as follows. The initial binding of ALKBH2 to a DNA duplex leads to the formation of a nonspecific complex (E∙m1A)
1, which isomerizes successively into specific (E∙m1A)
2 and catalytically competent (E∙m1A)
3 complexes. This event is followed by a step of irreversible hydroxylation of the substrate to form the enzyme–product complex (E∙A) and its dissociation. It is important to note that not every stage of the mechanism could be detected by each of the three fluorophores. Particularly, Trp fluorescence did not provide information about the dynamics of the ALKBH2 conformation at the substrate hydroxylation step, and aPu fluorescence failed to sense the second stage of isomerization of the initial complex.
In
Table 2, the rate constants corresponding to each reaction of
Scheme 1 are summarized. The
k1 constants derived by means of different fluorophores suggest that the formation of complex (E∙m1A)
1FRET (detected by FRET) precedes the formation of the complex (E∙m1A)
1Trp and (E∙m1A)
1aPu (detected via fluorescence intensities of Trp and aPu), respectively. This means that recognition and binding of DNA by ALKBH2 first leads to an alteration in the conformation of the substrate and only then causes changes in the local environment of the protein Trp residues. If one assumes that the main contribution to the fluorescence of the enzyme is made by residue Trp-105, then the observed changes in protein conformation should be associated with the dynamics of the β3–β4 hairpin, which interacts with the damaged DNA strand to retain the substrate. Consequently, we can theorize that damage recognition within the DNA duplex begins with its bending by the enzyme. The second conformational transition in the structure of the enzyme–substrate complex is characterized by forward rate constant
k2 values 7.2 and 10 s
−1 for the Trp fluorescence and FRET signal, respectively. Given that this difference is statistically insignificant, these constants may be describing the same step of the mechanism. On the other hand, this conformational transition should proceed more efficiently in the protein (
k−2Trp = 3.2 s
−1) than in the substrate (
k−2FRET = 21 s
−1). The third reversible step in the kinetic scheme is described by the
k3 values in the range of 0.67–2.2 s
−1, with the lowest value found for aPu, which shows the long increase in the signal. Taking into account fluorescent properties of aPu, we supposed that this base becomes more accessible to the hydrophilic environment at this step, most likely owing to eversion of the m1A base into the active site of the enzyme. Summarizing the findings, we can conclude that the conformational transitions at steps 2 and 3 reflect the process of damage eversion and incorporation of the intercalating finger Phe-102 into the resulting gap. Nevertheless, it is still difficult to say exactly to which step each event belongs. The fourth and fifth steps of the mechanism represent irreversible hydroxylation of the substrate and disintegration of the substrate–protein complex because the fluorescence of aPu diminishes, pointing to restoration of stacking interactions within the duplex. The values of the substrate hydroxylation step constant (
kr) are consistent with the observed rate constant obtained by PAGE: within the same order of magnitude.
In this work, conformational dynamics in the ALKBH2/Fe(II)/2OG/m3C complex did not cause any specific shifts in the Trp and aPu fluorescence. The only label sensitive to alterations in the conformation of the enzyme–substrate complex was the FAM/BHQ1 pair, although FRET was not able to detect the step of irreversible hydroxylation of the substrate (
Figure 5E). The kinetic curves contained several clearly visible phases of signal change, which lasted for 10 s from the moment of mixing at low enzyme concentrations and up to 50 s at high concentrations (3.0 and 4.0 μM). The results of PAGE analysis (
Figure 4) mean that demethylation of the m3C base engaging ALKBH2 requires more time as compared to other tested lesions. Because the estimated half-conversion time for the m3C-containing substrate is 190 s, it can be stated with high confidence that the conformational transitions observed by the SF method correspond to the DNA-binding steps and isomerization of the initial complex but not the step of substrate demethylation. X-ray structures of complexes of ALKBH2 with DNA substrates [
17,
18] suggest that this enzyme is able to bind various lesions in the active-site pocket. In this context, conversion efficiency of a given substrate depends on how close to the activated metal the oxidized alkyl group is located. It is likely that alkylated analogs of adenine, owing to their larger size as compared to cytosine, can quickly occupy an optimal position deep inside the DNA-binding pocket. The similar shapes of initial parts of the FRET curves obtained for substrates m1A and m3C support this hypothesis. After data fitting, a minimal scheme was determined, which comprises two reversible steps of binding and isomerization of the initial complex (
Scheme 2,
Table 2).
2.5. Conformational Dynamics of εA-Containing DNA during Interactions with ALKBH2
1,N6-Ethenoadenine can simultaneously serve as a DNA lesion and as a fluorescent probe, providing an opportunity for detection of changes in the local environment of the damaged base in the course of interactions with ALKBH2. On the other hand, detection of Trp fluorescence can be impeded by the presence of εA owing to an overlap of their emission and excitation spectra. This is exactly what we observed in preliminary experiments (data not shown). Therefore, in the present study, conformational dynamics of the ALKBH2/Fe(II)/2OG/εA complex were measured by means of the fluorescence of εA and of the FAM/BHQ1 FRET pair.
On the SF fluorescence curves obtained at a fixed concentration of εA, several phases of successive changes in fluorescence intensity were observed (
Figure 6A). A pronounced increase in the signal, whose amplitude depended on the ALKBH2 concentration, began at 20 to 30 ms and reached a maximum at ~2 s, followed by quenching of the εA fluorescence in the interval between 2 s and 70 s. It is known that the fluorescence of εA and its derivatives is quenched by hydrophobic and stacking interactions and strengthens in a polar environment [
41]. In addition, fluorescence intensity of εA in single- and double-stranded oligonucleotides is affected by the nature of the neighboring bases. In particular, purine nucleotides, and especially guanine, that are in the same strand as εA, significantly reduce the fluorescence of the latter owing to strong electrostatic interactions [
42]. The model DNA duplexes tested in the present work contained etheno-adenine in the context GεAT, which can strongly quench fluorescence of the substrate. The interaction of substrate εA with ALKBH2 turned out to be accompanied by the eversion of the damaged base from the DNA duplex into the more polar environment of the active site. Accordingly, the increase in fluorescence observed on kinetic curves probably corresponds to the step of εA eversion and its fixation in the active site of the enzyme (
Figure 6A). The subsequent signal decline, not detected at low protein concentrations, is most likely driven by the εA-to-A transformation, which is accompanied by the destruction of the fluorophore group. This supposition is supported by the finding that the τ
1/2 determined by PAGE analysis for substrate εA is 19 s (see
Section 2.2), which corresponds to the phase of εA fluorescence quenching on the SF curves.
Changes in the FRET signal describing the conformational dynamics of substrate εA when mixed with ALKBH2 are displayed in
Figure 6B. The kinetic curves contain two or three consecutive phases of signal growth (periods 3–70 ms, 0.1–1.0 s, and 10–50 s), whose magnitude depends on the concentration of the enzyme. These transitions may be explained by an alteration in the nature of the interaction between FAM and BHQ1 as a consequence of the DNA substrate helix loosening as well as its bending. In the case of εA, there was no initial drop in the FRET signal on the curves, as documented for the m1A- and m3C-containing substrates above. This finding may indicate differences in structural and dynamic behavior of the system during the initial stages of interaction with damaged bases of different size and structure.
To assess the conformational dynamics of ALKBH2 in the course of binding to substrate εA in the presence of Co(II) ions, the SF kinetic curves were recorded in a buffer containing cobalt instead of iron (
Figure 6C, green curve). The change in the FRET signal of the ALKBH2/Co(II)/2OG/εA complex contained one maximum, instead of the two observed for catalytically active complex ALKBH2/Fe(II)/2OG/εA (
Figure 6C, yellow curve). Then, the curve for the Co(II)-containing system featured a weak and prolonged decrease in the fluorescent signal, apparently associated with a nonproductive enzyme–substrate interaction. The initial phase of the increase between 2 ms and 30 ms corresponds to the first phase of the increase detected in the presence of Fe(II). Therefore, the growth of the FRET signal in the above period describes the step of initial binding of the enzyme to the DNA.
After the quantitative analysis of the kinetic traces obtained for both types of fluorescent probes (εA and FRET) by the nonlinear-regression method, a four-step molecular-kinetic mechanism was established (
Scheme 3). There was good general agreement between the values of kinetic constants obtained using the two fluorophores (
Table 2). Thus, the change in the polarity of the εA environment occurred simultaneously with the change in the distance between labels FAM and BHQ1 at each step of interactions with the enzyme.
The initial binding is characterized by almost identical values of rate constants
k1 and
k−1, derived from εA and FRET fluorescence (see
Figure 6 and
Table 2). After comparing the values of forward and reverse rate constants of the first step among the three lesions (m1A, m3C, and εA), we can conclude that these constants are lower for εA than for the other lesions. Nevertheless, equilibrium association constant
Ka =
k1/
k−1, which describes the formation of the bimolecular complex (E∙εA), is 8.6 × 10
5 M
−1, which is about twice as high as these values for substrate m1A (3.6 × 10
6 M
−1) and substrate m3C (2.3 × 10
5 M
−1). Subsequent isomerization of (E∙εA) into a more specific complex, (E∙εA)
1, also proceeds with high efficiency in terms of the
k2/
k−2 ratio. This process enhances overall affinity of the ALKBH2 active site for εA, as determined by Equation (1):
This value (KaSF= 1.9 × 106 M−1) is 4–5 times higher than this protein’s affinity for m1A (6.0 × 105 M−1) and m3C (4.6 × 105 M−1). In other words, despite the lower rate of formation of the initial complex between ALKBH2 and εA-containing DNA, the efficiency of this interaction is very high. According to the εA fluorescence data and FRET analysis, kr (the rate of the irreversible hydroxylation step) is 0.067 and 0.034 s−1, respectively, and these values are close to the hydroxylation rate constant obtained for substrate m1A (0.065 s−1). The nature of the applied estimation method does not allow one to determine individual rate constants of forward and reverse reactions of the decay of the enzyme’s complex with the reaction product (E∙A), but make it possible to determine their ratio in terms of equilibrium dissociation constant Kd. The Kd values measured by means of εA fluorescence and FRET are 2.2 and 0.4 μM, respectively. This is much lower than this parameter for the ALKBH2 complex with intact DNA containing adenine at the site of damage (7.8 μM). Consequently, we propose that the post-catalytic complex is quite stable and does not decompose immediately.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
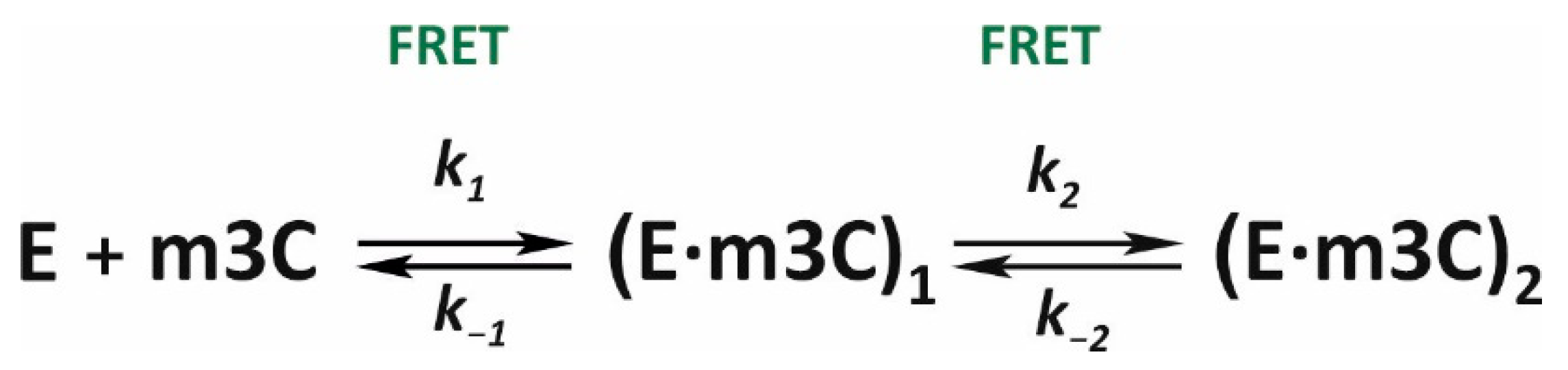{kind=link}
{kind=link}