
Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Preparation of New Compounds
2.2. Instrumentation and Methods
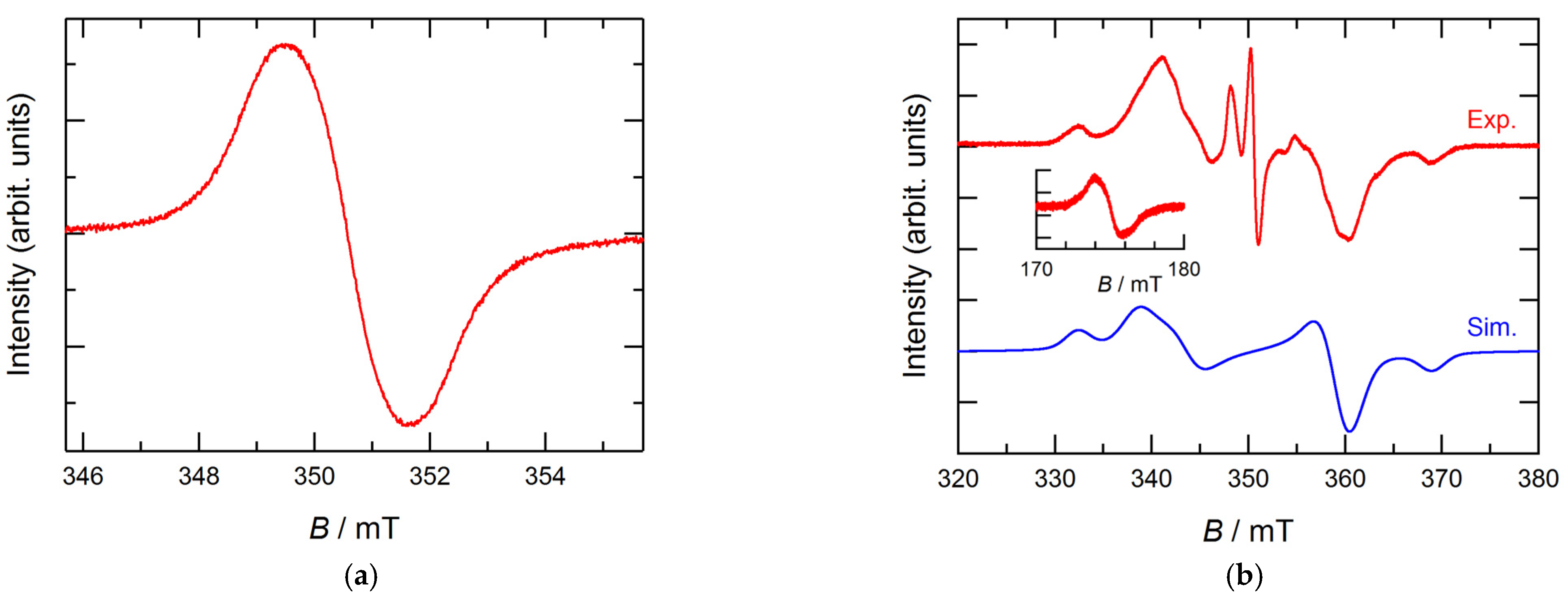
3. Results and Discussion
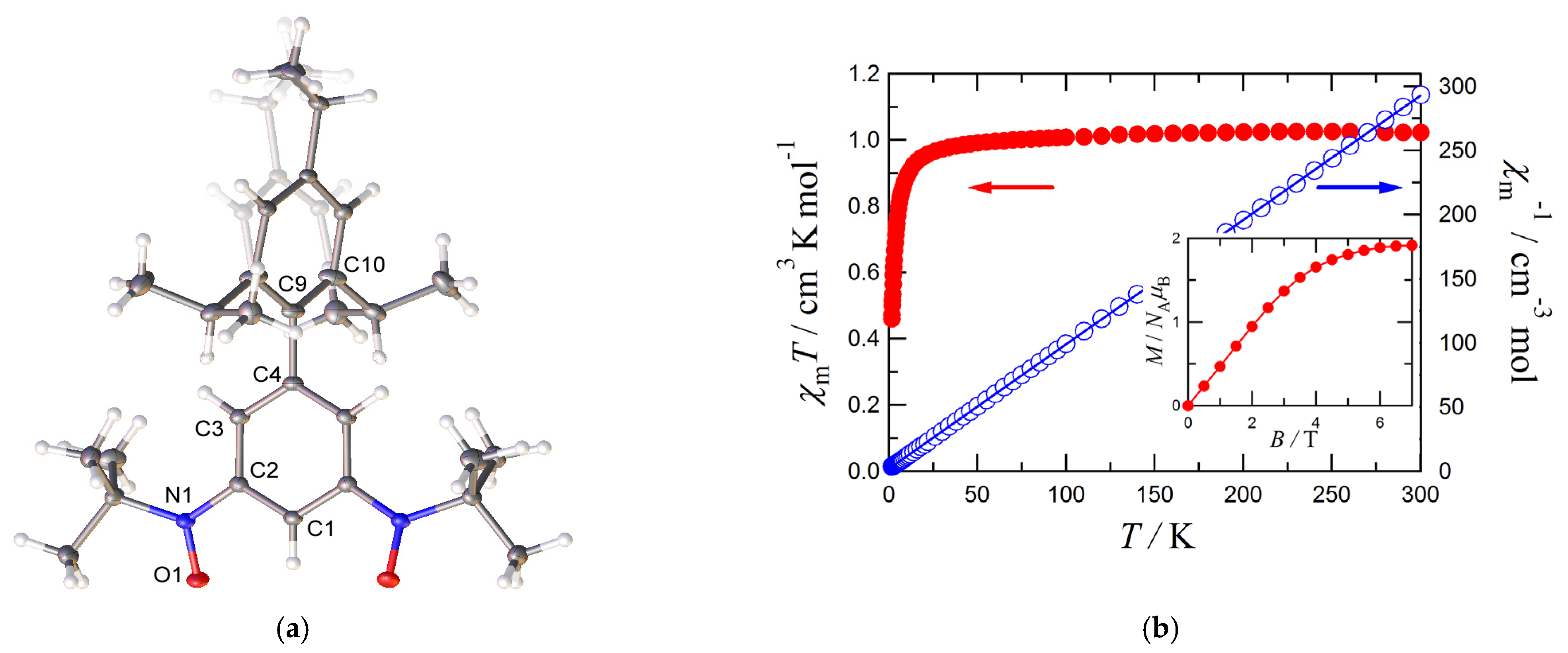
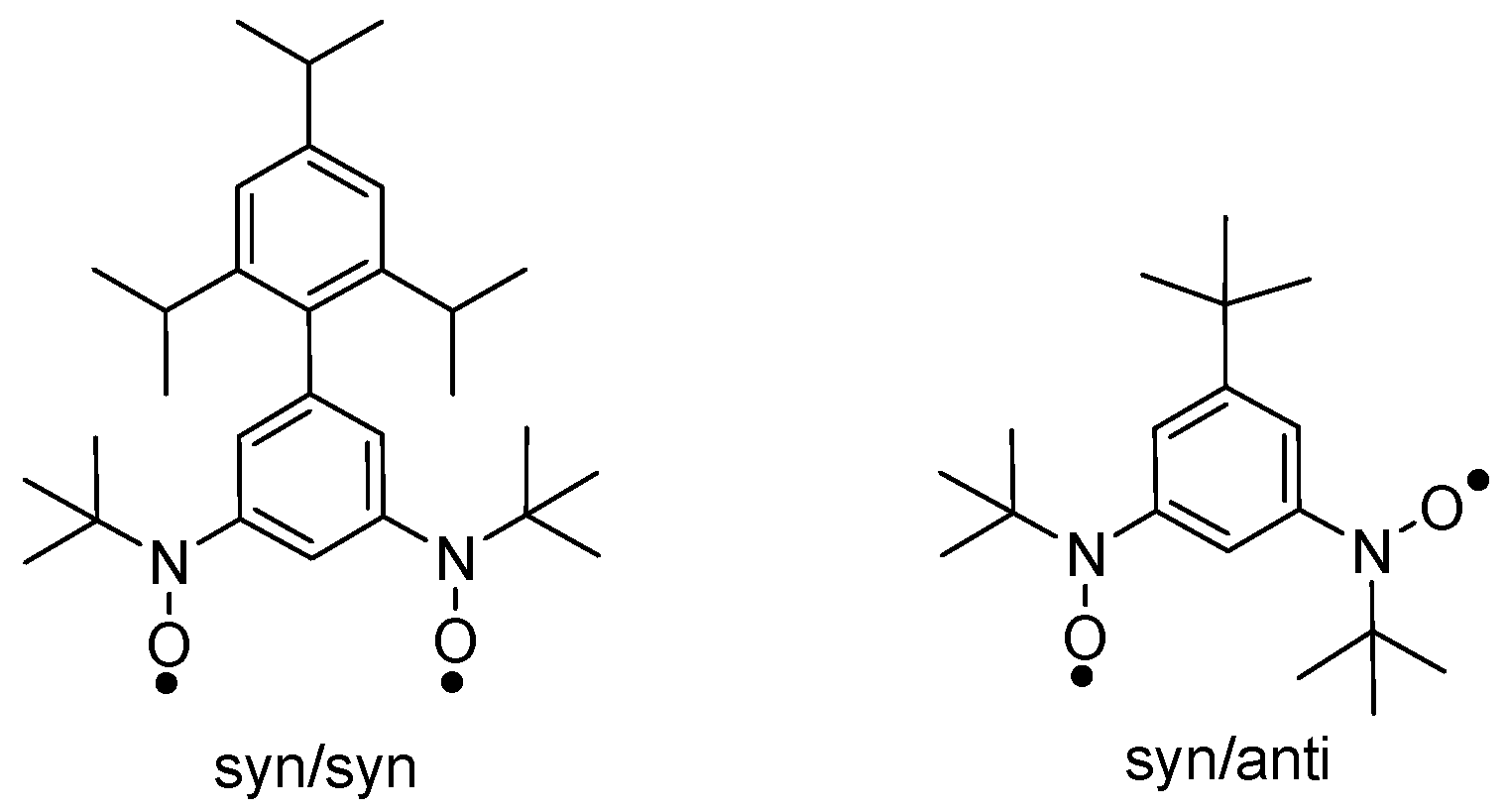
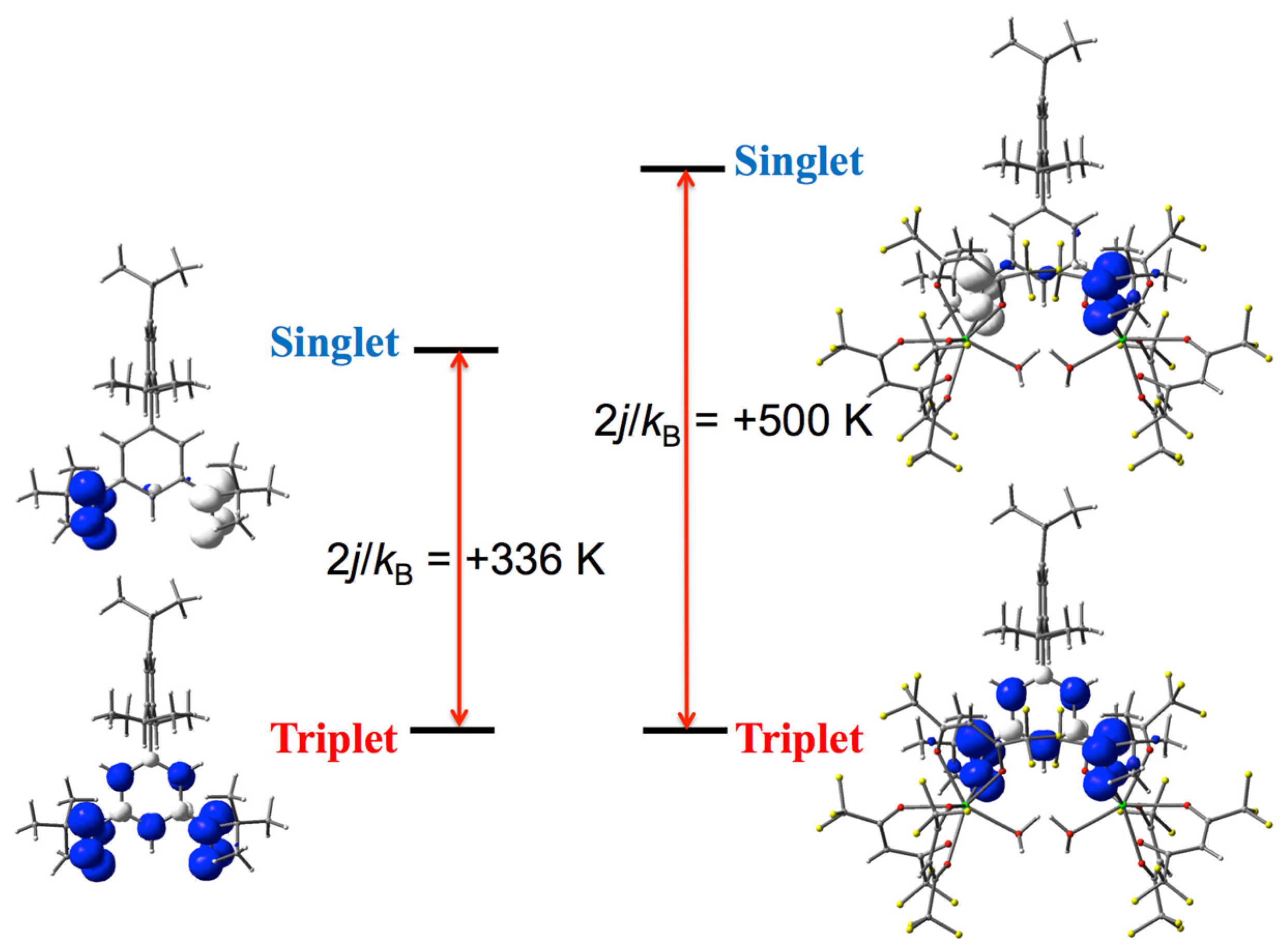
3.1. Structure and Magnetic Properties of iPr3BPBN
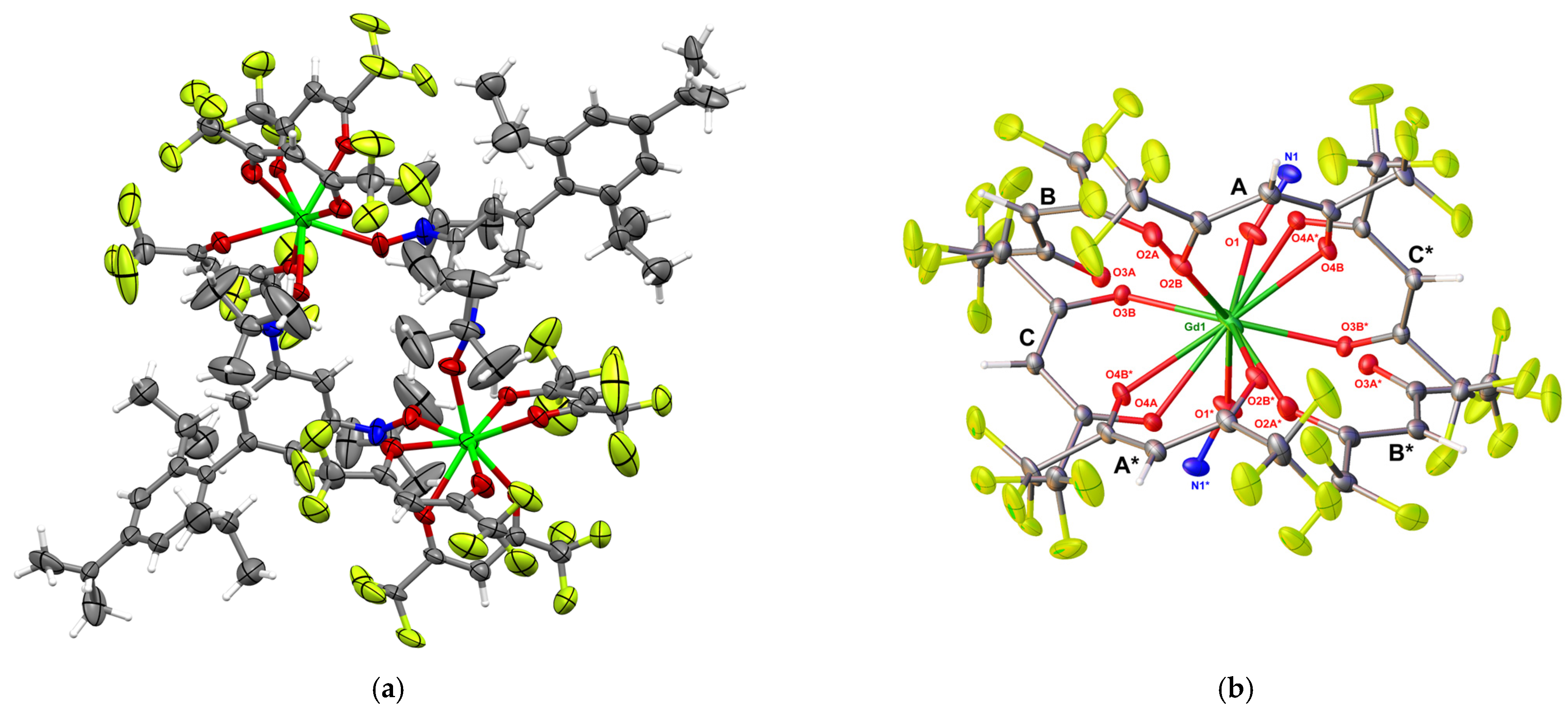
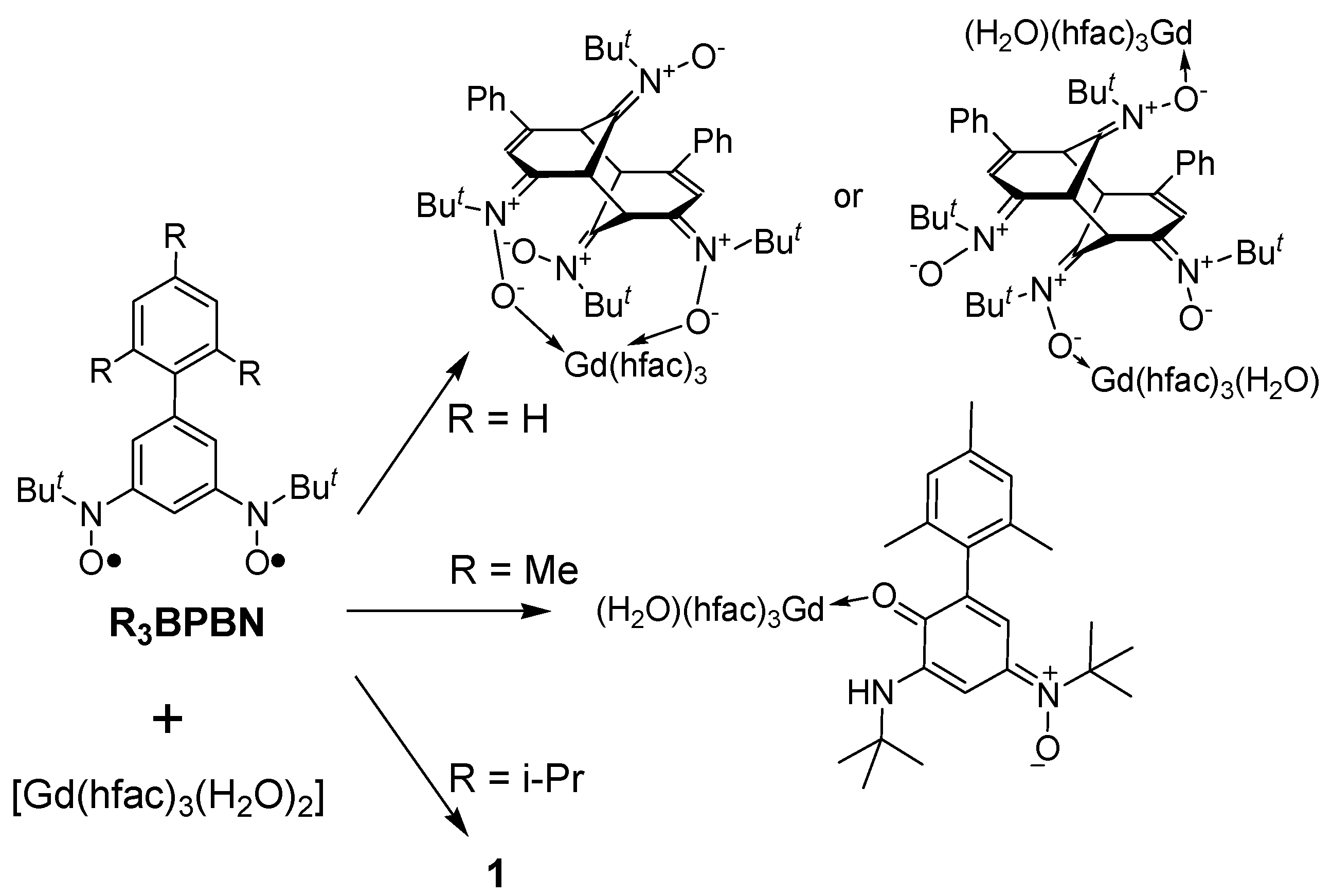
3.2. Synthesis and Crystal Structure of 1
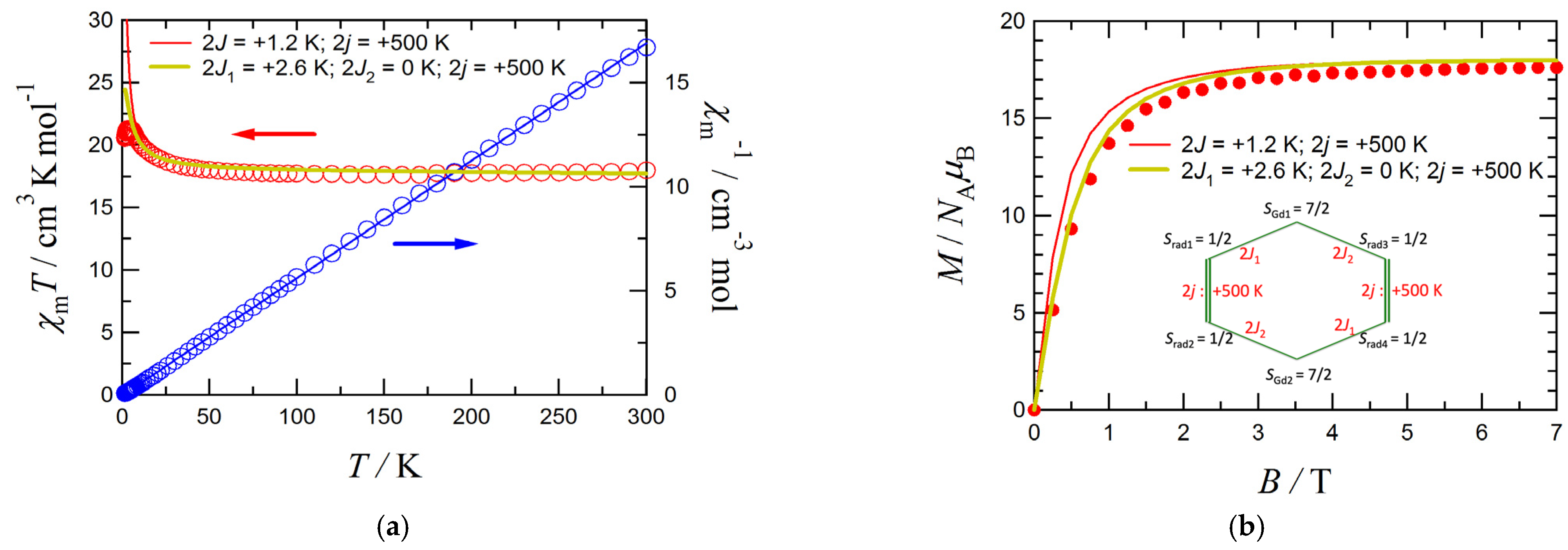
3.3. Magnetic Properties of 1
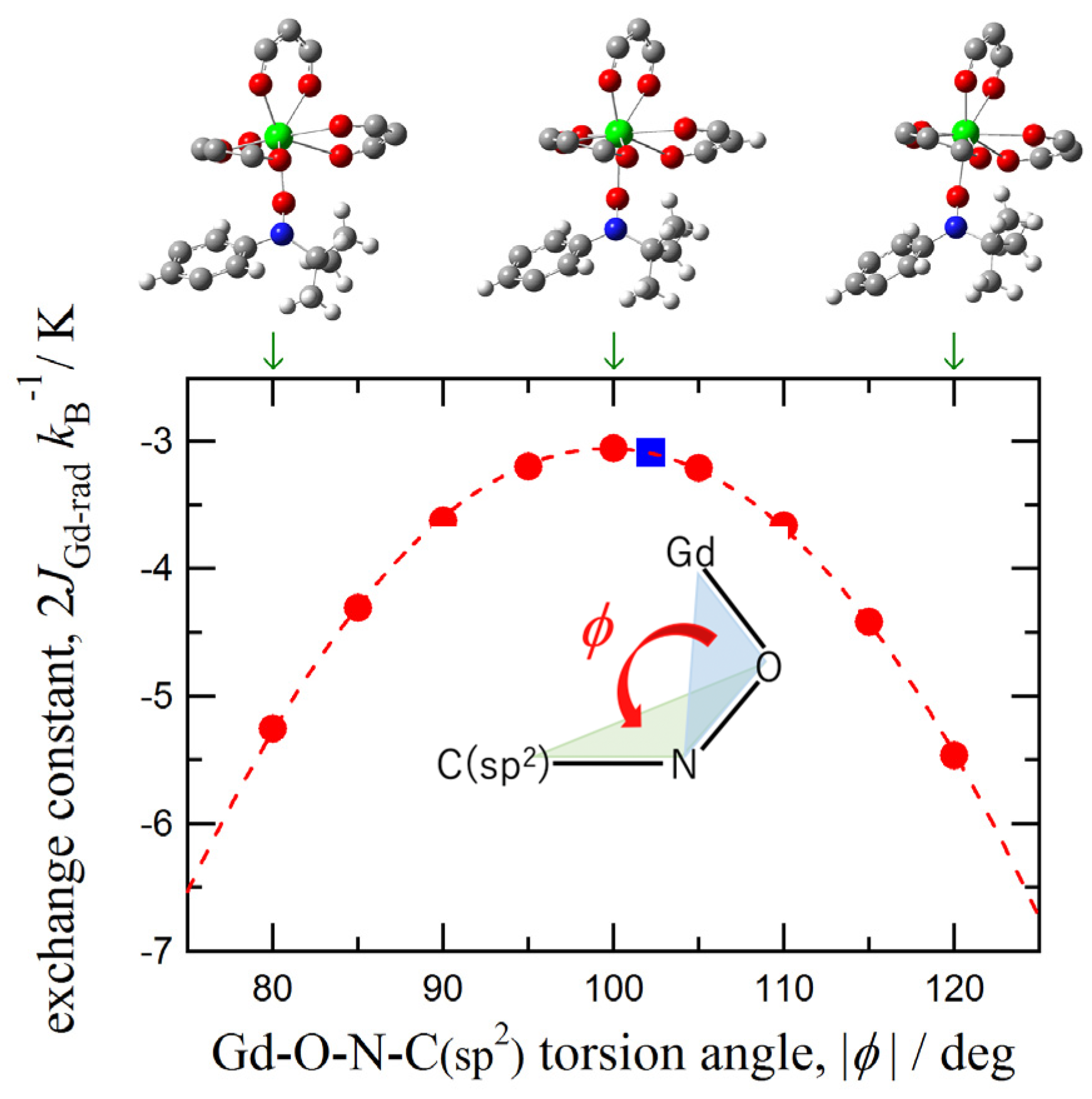
3.4. DFT Calculation on the Gd3+-iPr3BPNB System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Marin, R.; Brunet, G.; Murugesu, M. Shining New Light on Multifunctional Lanthanide Single-Molecule Magnets. Angew. Chem. Int. Ed. 2021, 60, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T.; van Slageren, J. Improving f-element single molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, M.G.; Andruh, M. Molecule-based magnetic materials constructed from paramagnetic organic ligands and two different metal ions. Coord. Chem. Rev. 2021, 427, 213611. [Google Scholar] [CrossRef]
- Demir, S.; Jeon, I.R.; Long, J.R.; Harris, T.D. Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 2015, 289, 149–176. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Shi, W.; Cheng, P. Magnetism in one-dimensional metal–nitronyl nitroxide radical system. Coord. Chem. Rev. 2019, 378, 134–150. [Google Scholar] [CrossRef]
- Likhtenshtein, G.I. (Ed.) Nitroxides: Brief History, Fundamentals, and Recent Developments; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Rey, P. Toward molecular magnets: The metal-radical approach. Acc. Chem. Res. 1989, 22, 392–398. [Google Scholar] [CrossRef]
- Train, C.; Norel, L.; Baumgarten, M. Organic radicals, a promising route towards original molecule-based magnetic materials. Coord. Chem. Rev. 2009, 253, 2342–2351. [Google Scholar] [CrossRef]
- Ferrando-Soria, J.; Vallejo, J.; Castellano, M.; Martinez-Lillo, J.; Pardo, E.; Cano, J.; Castro, I.; Lloret, F.; Ruiz-Garcia, R.; Julve, M. Molecular magnetism, quo vadis? A historical perspective from a coordination chemist viewpoint. Coord. Chem. Rev. 2017, 339, 17–103. [Google Scholar] [CrossRef]
- Ishida, T.; Ito, S.; Homma, Y.; Kyoden, Y. Molecular S = 2 High-Spin, S = 0 Low-Spin and S = 0 ⇄ 2 Spin-Transition/-Crossover Nickel(II)-Bis(nitroxide) Coordination Compounds. Inorganics 2021, 9, 10. [Google Scholar] [CrossRef]
- Nakamura, T.; Kanetomo, T.; Ishida, T. Strong Antiferromagnetic Interaction in a Gadolinium(III) Complex with Methoxy-TEMPO Radical: A Relation between the Coupling and the Gd–O–N Angle. Inorg. Chem. 2021, 60, 535–539. [Google Scholar] [CrossRef]
- Kanetomo, T.; Naoi, Y.; Enomoto, M. Gadolinium-Triradical Complex with Ground S = 10 State: Synthesis, Structural Characterization and Magnetic Studies. Eur. J. Inorg. Chem. 2021, 2021, 1130–1136. [Google Scholar] [CrossRef]
- Kanetomo, T.; Yoshitake, T.; Ishida, T. Strongest Ferromagnetic Coupling in Designed Gadolinium(III)–Nitroxide Coordination Compounds. Inorg. Chem. 2016, 55, 8140–8146. [Google Scholar] [CrossRef]
- Kanetomo, T.; Kihara, T.; Miyake, A.; Matsuo, A.; Tokunaga, M.; Kindo, K.; Nojiri, H.; Ishida, T. Giant Exchange Coupling Evidenced with a Magnetization Jump at 52 T for a Gadolinium-Nitroxide Chelate. Inorg. Chem. 2017, 56, 3310–3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanetomo, T.; Ishida, T. Strongest exchange coupling in gadolinium(III) and nitroxide coordination compounds. Inorg. Chem. 2014, 53, 10794–10796. [Google Scholar] [CrossRef] [PubMed]
- Wernsdorfer, W.; Aliaga-Alcalde, N.; Hendrickson, D.N.; Christou, G. Exchange-biased quantum tunnelling in a supramolecular dimer of single-molecule magnets. Nature 2002, 416, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Gonzalez, M.I.; Darago, L.E.; Evans, W.J.; Long, J.R. Giant coercivity and high magnetic blocking temperatures for N23− radical-bridged dilanthanide complexes upon ligand dissociation. Nat. Commun. 2017, 8, 2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamura, H. What role has organic chemistry played in the development of molecule-based magnets? Polyhedron 2013, 66, 3–14. [Google Scholar] [CrossRef]
- Baumgarten, M. High spin molecules directed towards molecular magnets. In EPR of Free Radicals in Solids II; Springer: Dordrecht, The Netherlands, 2012; pp. 205–244. [Google Scholar]
- Gallagher, N.M.; Olankitwanit, A.; Rajca, A. High-spin organic molecules. J. Org. Chem. 2015, 80, 1291–1298. [Google Scholar] [CrossRef]
- Chapyshev, S.V.; Mendez-Vega, E.; Sander, W. Molecular Magnets: The Synthesis and Characterization of High-Spin Nitrenes. Chem. Eur. J. 2021, 27, 1258–1269. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Ovcharenko, V.I.; Terent’ev, A.O.; Krylov, I.B.; Magdesieva, T.V.; Mazhukin, D.G.; Gritsan, N.P. Conjugated nitroxides. Russ. Chem. Rev. 2022, 91, RCR5025. [Google Scholar] [CrossRef]
- Yoshitake, T.; Ishida, T. Ferromagnetic chains of ground triplet 5-methoxy-1,3-phenylene bis(tert-butyl nitroxide). Chem. Lett. 2016, 45, 391–393. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, G.; Ishida, T.; Nogami, T. Remarkably strong intermolecular antiferromagnetic couplings in the crystal of biphenyl-3,5-diyl bis(tert-butyl nitroxide). Chem. Phys. Lett. 2004, 392, 74–79. [Google Scholar] [CrossRef]
- Ikegaya, N.; Kanetomo, T.; Murakami, R.; Ishida, T. Triply Radical-coordinated Gadolinium(III) Complex as a High-spin S = 5 Assembly. Chem. Lett. 2012, 41, 82–83. [Google Scholar] [CrossRef]
- Ishida, T.; Murakami, R.; Kanetomo, T.; Nojiri, H. Magnetic study on radical-gadolinium(III) complexes. Relationship between the exchange coupling and coordination structure. Polyhedron 2013, 66, 183–187. [Google Scholar] [CrossRef]
- Kawakami, H.; Tonegawa, A.; Ishida, T. A designed room-temperature triplet ligand from pyridine-2,6-diyl bis(tert-butyl nitroxide). Dalton Trans. 2016, 45, 1306–1309. [Google Scholar] [CrossRef]
- Kanetomo, T.; Yoshii, S.; Nojiri, H.; Ishida, T. Single-molecule magnet involving strong exchange coupling in terbium(III) complex with 2, 2′-bipyridin-6-yl tert-butyl nitroxide. Inorg. Chem. Front. 2015, 2, 860–866. [Google Scholar] [CrossRef]
- Kanetomo, T.; Yasui, M.; Ishida, T. Exchange-coupled terbium-radical complex Tb-phNO showing slow reversal of magnetization. Polyhedron 2017, 136, 30–34. [Google Scholar] [CrossRef]
- Kanetomo, T.; Ishida, T. Preparation and characterization of [Gd(hfac)3(DTBN)(H2O)](DTBN = di-t-butyl nitroxide). Ferromagnetic Gd3+–Gd3+ super–superexchange. Chem. Commun. 2014, 50, 2529–2531. [Google Scholar] [CrossRef]
- Yoshitake, T.; Kudo, H.; Ishida, T. Thermally Activated Paramagnets from Diamagnetic Polymers of Biphenyl-3,5-diyl Bis(tert-butyl Nitroxides) with Methyl and Fluoro Groups at the 2’- and 5’-Positions. Crystals 2016, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Kudo, H.; Ishida, T. Intermediate-paramagnetic phases with a half and a quarter spin entities in fluorinated biphenyl-3, 5-diyl bis(tert-butyl nitroxides). J. Mater. Chem. C 2015, 3, 7813–7818. [Google Scholar] [CrossRef]
- Sekine, H.; Ishida, T. Unexpected complexes from meta-phenylene bis(tert-butyl nitroxides) and gadolinium(III) 1,1,1,5,5,5-hexafluoropentane-2,4-dionate. AIP Conf. Proc. 2018, 1929, 020022. [Google Scholar]
- Richardson, M.F.; Wagner, W.F.; Sands, D.E. Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 1968, 30, 1275–1289. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C: Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Wever, R.T. WIN-EPR SimFonia, version 1.2; Bruker Instruments: Billerica, MA, USA, 1995. [Google Scholar]
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken symmetry approach to calculation of exchange coupling constants for homobinuclear and heterobinuclear transition metal complexes. J. Comput. Chem. 1999, 20, 1391–1400. [Google Scholar] [CrossRef]
- Tsipis, A.C. DFT flavor of coordination chemistry. Coord. Chem. Rev. 2014, 272, 1–29. [Google Scholar] [CrossRef]
- Maron, L.; Eisenstein, O. Do f Electrons Play a Role in the Lanthanide−Ligand Bonds? A DFT Study of Ln(NR2)3; R = H, SiH3. J. Phys. Chem. A 2000, 104, 7140–7143. [Google Scholar] [CrossRef]
- Demireva, M.; Kim, J.; Armentrout, P.B. Gadolinium (Gd) oxide, carbide, and carbonyl cation bond energies and evaluation of the Gd+ O → GdO+ + e− chemi-ionization reaction enthalpy. J. Phys. Chem. A 2016, 120, 8550–8563. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Kawakami, T.; Takano, Y.; Kitagawa, Y.; Yamashita, Y.; Fujita, H. Analytical and ab initio studies of effective exchange interactions, polyradical character, unpaired electron density, and information entropy in radical clusters (R)N: Allyl radical cluster (N = 2−10) and hydrogen radical cluster (N = 50). Int. J. Quantum Chem. 2002, 90, 370–385. [Google Scholar] [CrossRef]
- Okazawa, A.; Nagaichi, Y.; Nogami, T.; Ishida, T. Magneto-structure relationship in copper(II) and nickel(II) complexes chelated with stable tert-butyl 5-phenyl-2-pyridyl nitroxide and related radicals. Inorg. Chem. 2008, 47, 8859–8868. [Google Scholar] [CrossRef] [PubMed]
- Nishimaki, H.; Mashiyama, S.; Yasui, M.; Nogami, T.; Ishida, T. Bistable Polymorphs Showing Diamagnetic and Paramagnetic States of an Organic Crystalline Biradical Biphenyl-3,5-diyl Bis(tert-butylnitroxide). Chem. Mater. 2006, 18, 3602–3604. [Google Scholar] [CrossRef]
- Nishimaki, H.; Ishida, T. Organic two-step spin-transition-like behavior in a linear S = 1 array: 3’-methylbiphenyl-3,5-diyl bis(tert-butylnitroxide) and related compounds. J. Am. Chem. Soc. 2010, 132, 9598–9599. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Nagai, H.; Ishizu, K. The Proof of a Triplet Ground State in the N,N’-Di-t-butyl-m- phenylenebinitroxide Biradical. Bull. Chem. Soc. Jpn. 1975, 48, 2381–2382. [Google Scholar] [CrossRef] [Green Version]
- Calder, A.; Forrester, A.R.; James, P.G.; Luckhurst, G.R. Nitroxide radicals. V. N,N’-Di-tert-butyl-m-phenylenebinitroxide, a stable triplet. J. Am. Chem. Soc. 1969, 91, 3724–3727. [Google Scholar] [CrossRef]
- Scafuri, A.; Vivani, R.; Carniato, F.; Tei, L.; Botta, M.; Taddei, M.; Costantino, F. A structural and 1H NMR relaxometric study on novel layered carboxyalkylaminophosphonate nanocrystals with Gd(III) ions located in the framework. Dalton Trans. 2015, 44, 19072–19075. [Google Scholar] [CrossRef]
- Lluncll, M.; Casanova, D.; Circra, J.; Bofill, J.M.; Alcmany, P.; Alvarez, S.; Pinsky, M.; Avnir, D. SHAPE, version 2.1; University of Barcelona: Barcelona, Spain; Hebrew University of Jerusalem: Jerusalem, Israel, 2005. [Google Scholar]
- Sekine, H.; Ishida, T. Unexpected Complexes of a [3+3] Cycloadduct from Biphenyl-3,5-diyl Bis(tert-butyl nitroxide) with Gadolinium(III) 1,1,1,5,5,5-Hexafluoropentane-2,4-dionate. Chem. Lett. 2018, 47, 74–77. [Google Scholar] [CrossRef]
- Ito, S.; Takano, R.; Hatanaka, S.-I.; Ishida, T. Rare-Earth (RE = Y, Gd, Tb, Dy, Ho, and Er) Chains Bridged with a Triplet Biradical and Magnetic Hysteresis Recorded for RE = Tb. Inorg. Chem. 2022, 61, 10619–10623. [Google Scholar] [CrossRef]
- Borrás-Almenar, J.J.; Clemente-Juan, J.M.; Coronado, E.; Tsukerblat, B.S. MAGPACK A package to calculate the energy levels, bulk magnetic properties, and inelastic neutron scattering spectra of high nuclearity spin clusters. J. Comput. Chem. 2001, 22, 985–991. [Google Scholar] [CrossRef]
- Görlitz, G.; Hayamizu, T.; Itoh, T.; Matsuda, K.; Iwamura, H. Synthesis, Structure, and Magnetic Properties of a Cyclic Dimer of Bis(hexafluoroacetylacetonato){1,3-bis(N-tert-butyl-N-oxylamino)-5- tert-butylbenzene}manganese(II). Inorg. Chem. 1998, 37, 2083–2085. [Google Scholar] [CrossRef]
- Rabu, P.; Drillon, M.; Iwamura, H.; Görlitz, G.; Itoh, T.; Matsuda, K.; Koga, N.; Inoue, K. Exchange Coupling Parameters and Energy Levels for Cyclic Metal-Radical Complexes of Bis(hexafluoroacetylacetonato)manganese(II) with 5-tert-Butyl-1,3-phenylenebis(N-tert-butylaminoxyl) and (4-N-tert-Butyl-N-oxyamino)pyridine. Eur. J. Inorg. Chem. 2000, 2000, 211–216. [Google Scholar] [CrossRef]
- Numata, Y.; Inoue, K. Inoue, Synthesis, crystal structure and magnetic properties of a cyclic dimer of [{Co(hfac)2}·BNOt-Bu]2. J. Mag. Mag. Mater. 2007, 310, 1847–1848. [Google Scholar] [CrossRef]
- Ito, S.; Ishida, T. Practically Diamagnetic Macrocycle Consisting of Nickel-Biradical Heterospins with the Largest Out-of-Plane Torsion at Coordination Bonds. Chem. Lett. 2020, 49, 1062–1065. [Google Scholar] [CrossRef]
- Awaga, K.; Sugano, T.; Kinoshita, M. Ferromagnetic intermolecular interaction in the galvinoxyl radical: Cooperation of spin polarization and charge-transfer interaction. Chem. Phys. Lett. 1987, 141, 540–544. [Google Scholar] [CrossRef]
- Andruh, M.; Ramade, I.; Codjovi, E.; Guillou, O.; Kahn, O.; Trombe, J.C. Crystal structure and magnetic properties of [Ln2Cu4] hexanuclear clusters (where Ln = trivalent lanthanide). Mechanism of the gadolinium(III)-copper(II) magnetic interaction. J. Am. Chem. Soc. 1993, 115, 1822–1829. [Google Scholar] [CrossRef]
- Ungur, L.; Chibotaru, L.F. Computational Modelling of the Magnetic Properties of Lanthanide Compounds. In Lanthanides and Actinides in Molecular Magnetism; Layfield, R.A., Murugesu, M., Eds.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Kahn, O. Molecular Magnetism; VCH: New York, NY, USA, 1993; Chapter 8. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | iPr3BPBN | 1•(CH2Cl2)2 |
|---|---|---|
| Formula | C29H44N2O2 | C45H49Cl2F18GdN2O8 |
| Crystal system | monoclinic | tetragonal |
| Space group | C2/c | I41/acd |
| a/Å | 10.6857 (5) | 31.4028 (11) |
| b/Å | 18.0214 (8) | 31.4028 (11) |
| c/Å | 13.8637 (6) | 22.417 (3) |
| β/˚ | 96.604 (4) | 90 |
| V/Å3 | 2652.0 (2) | 22106 (3) |
| Z | 4 | 16 |
| dcalcd/g·cm−3 | 1.134 | 1.582 |
| μ (MoKα)/mm−1 | 0.070 | 1.406 |
| Rint | 0.0254 | 0.0450 |
| R(F) a, Rw(F2) b | 0.0397, 0.1113 | 0.1287, 0.2991 |
| GOF parameter | 1.049 | 1.130 |
| Δρmax, Δρmin/e Å−3 | 0.437, −0.233 | 1.876, −2.512 |
| T/K | 94 (2) | 94 (1) |
| CCDC Reference | 2192099 | 2077255 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, S.; Yoshitake, T.; Ishida, T. Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions. Molecules 2022, 27, 4930. https://doi.org/10.3390/molecules27154930
Ito S, Yoshitake T, Ishida T. Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions. Molecules. 2022; 27(15):4930. https://doi.org/10.3390/molecules27154930
Chicago/Turabian StyleIto, Saki, Toru Yoshitake, and Takayuki Ishida. 2022. "Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions" Molecules 27, no. 15: 4930. https://doi.org/10.3390/molecules27154930
APA StyleIto, S., Yoshitake, T., & Ishida, T. (2022). Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions. Molecules, 27(15), 4930. https://doi.org/10.3390/molecules27154930