
The Valence and Spin State Tuning of Iron(II/III) Porphyrazines with Bulky Pyrrolyl Periphery in Solution and Solid State

 , ,
, ,  , ,
, ,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
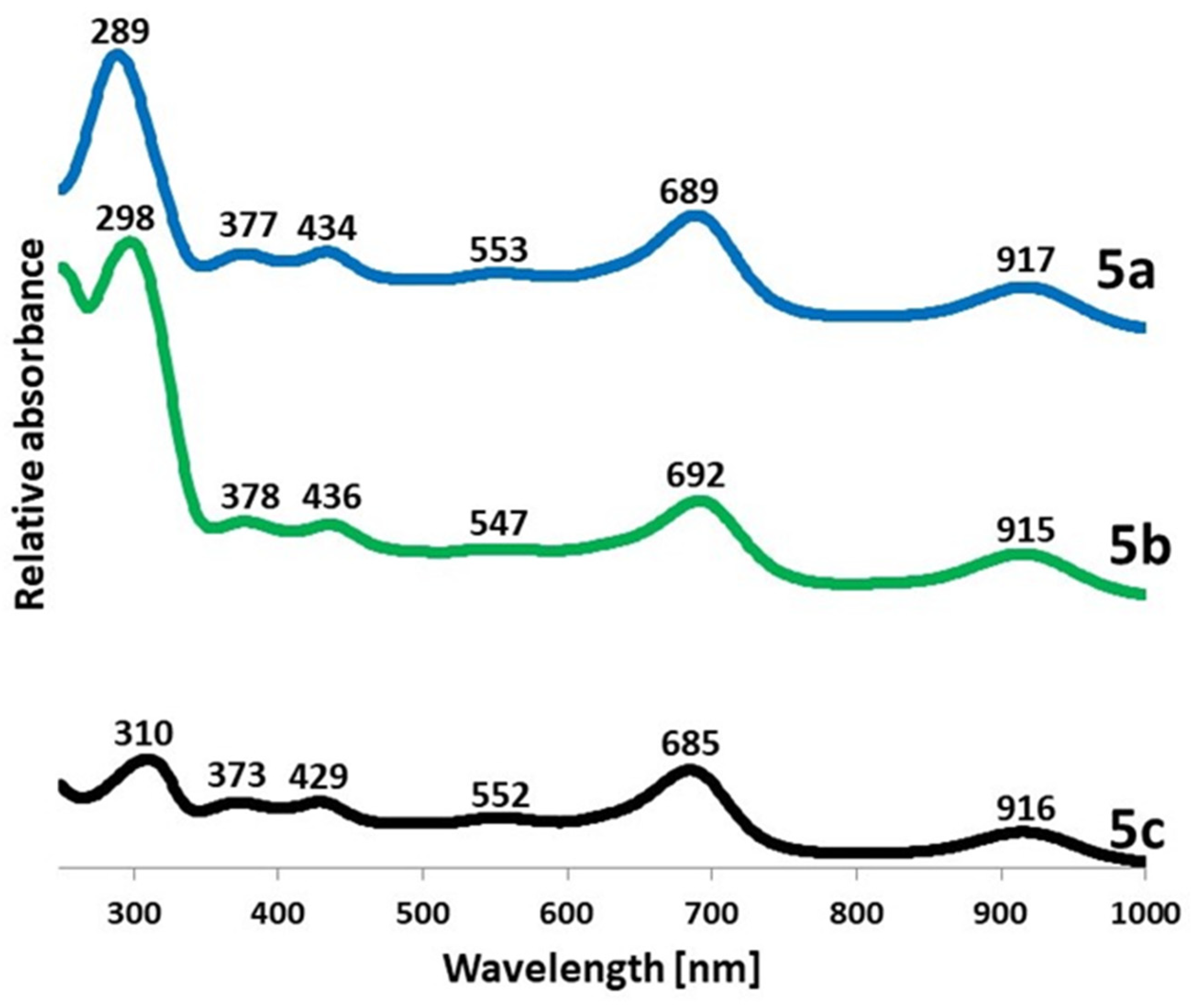
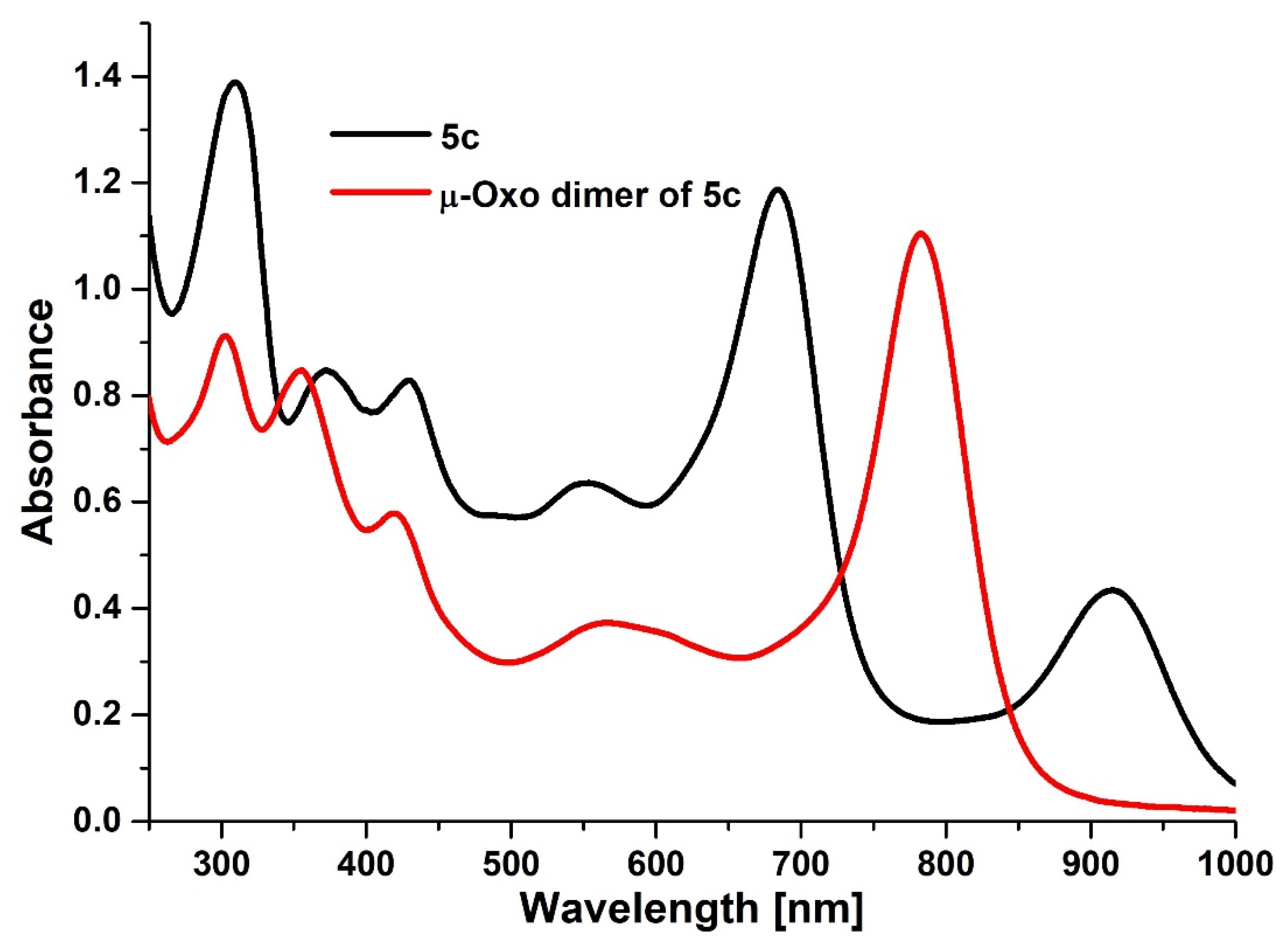
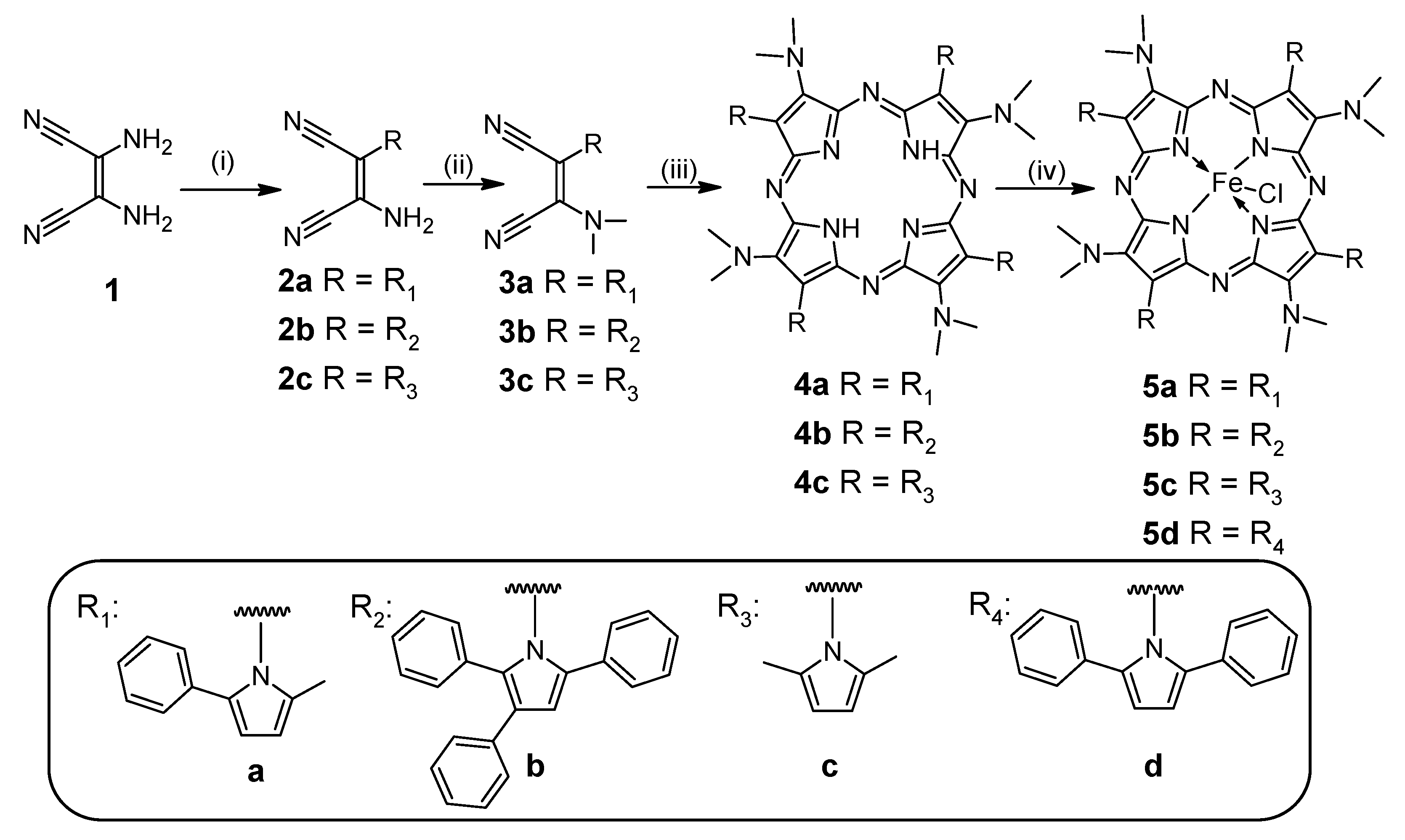
2.1. Synthesis and Physicochemical Characterization
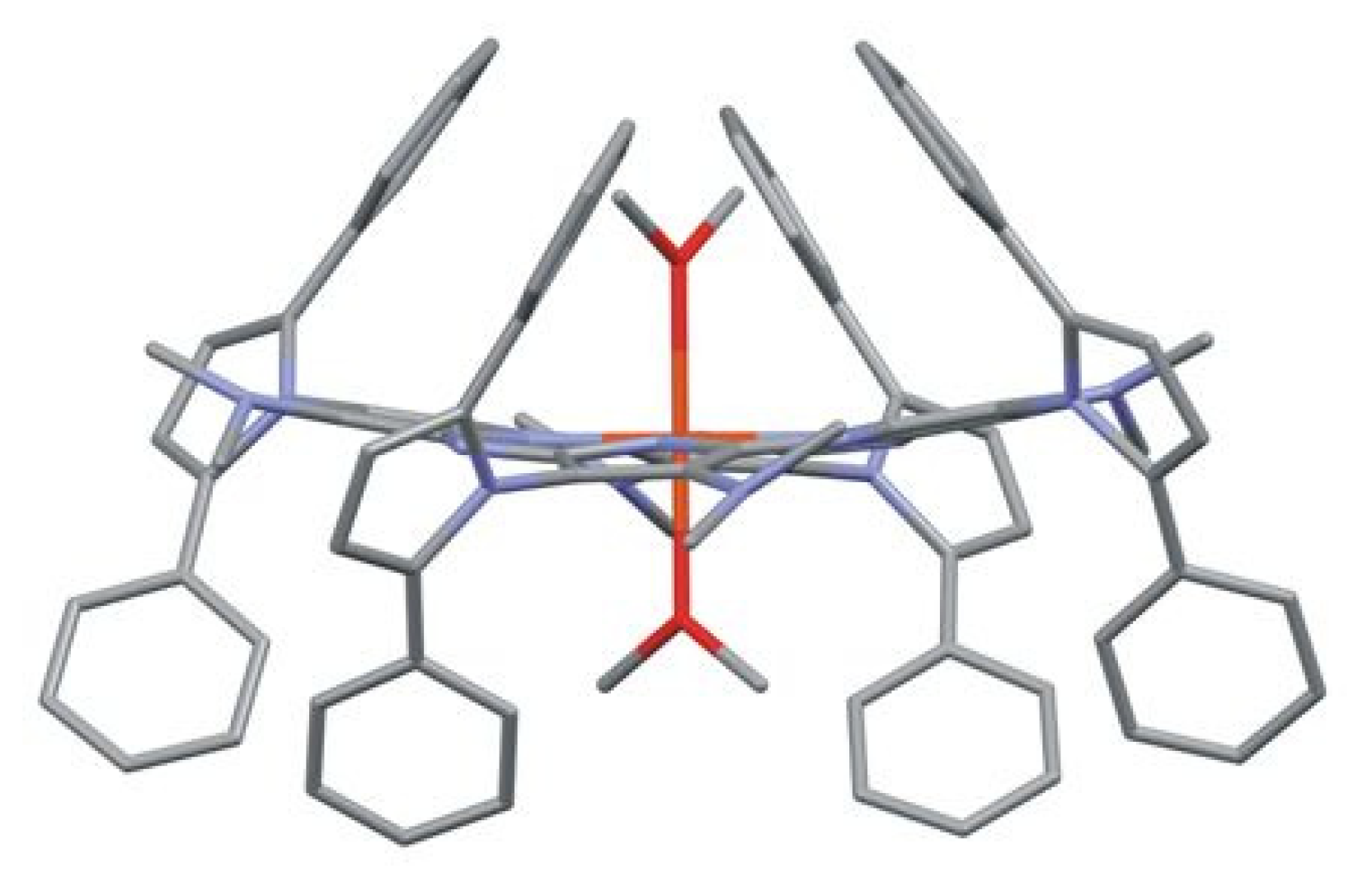
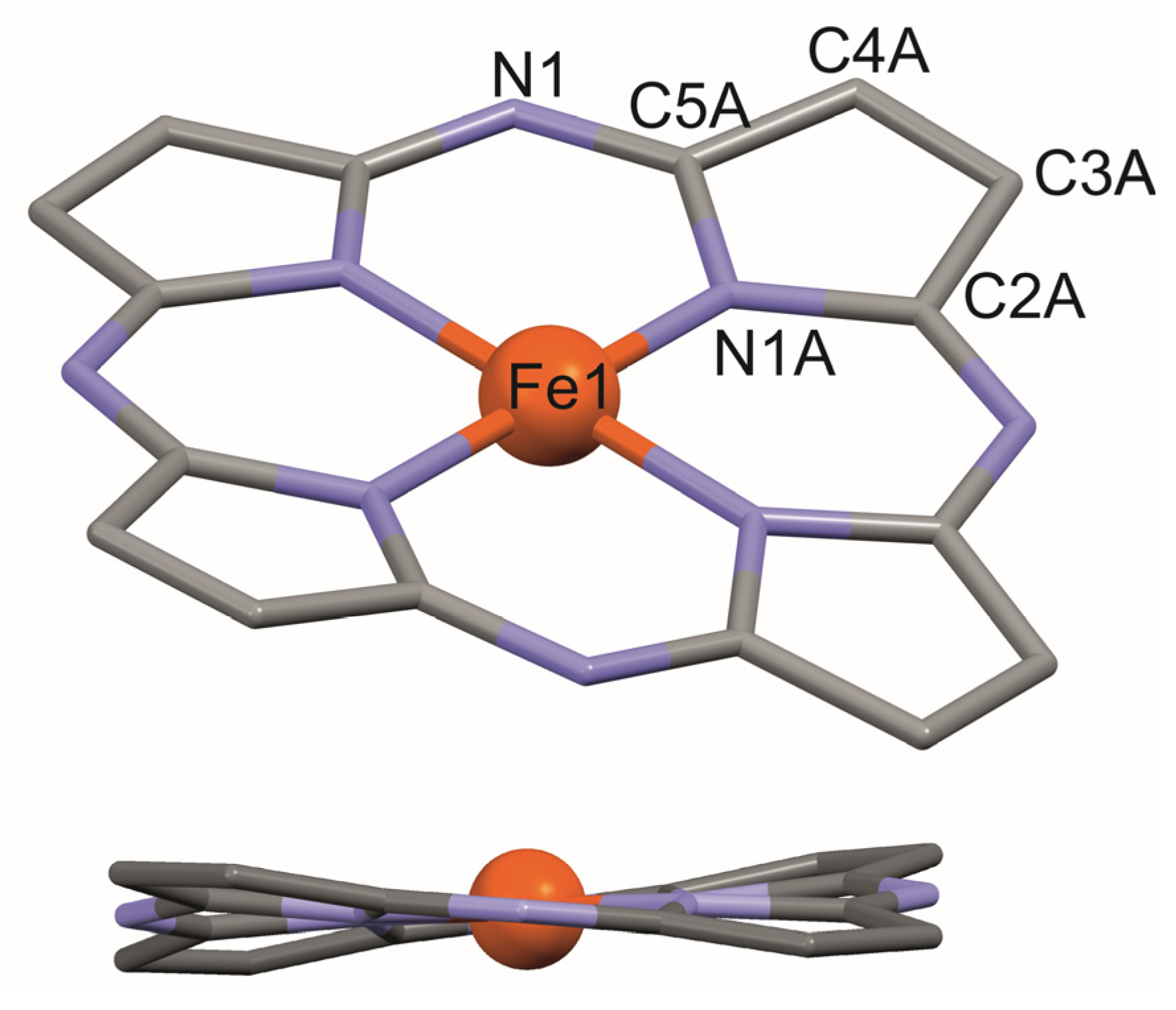
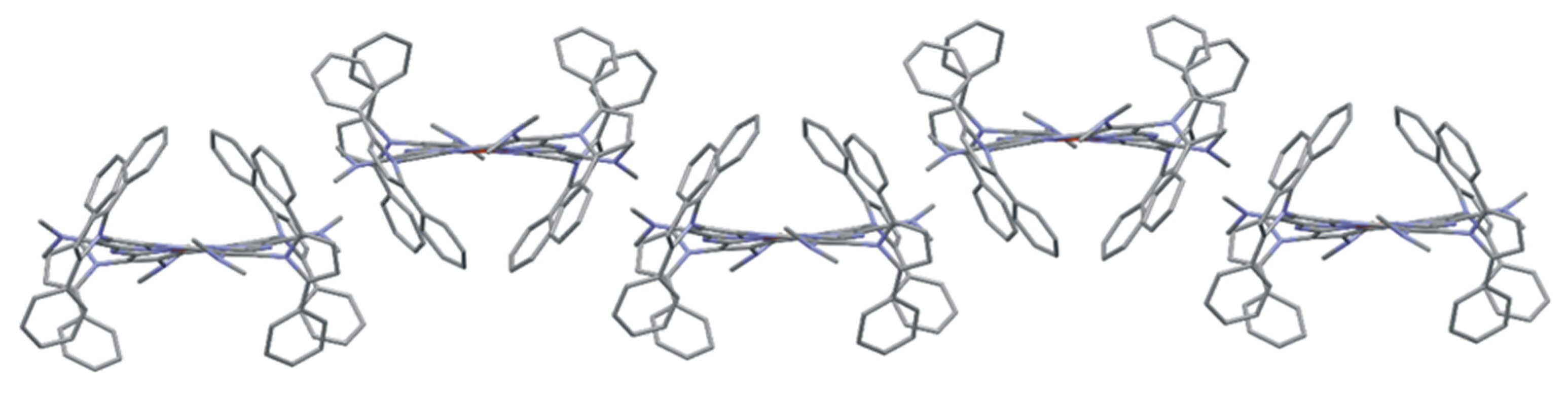
2.2. X-ray Study
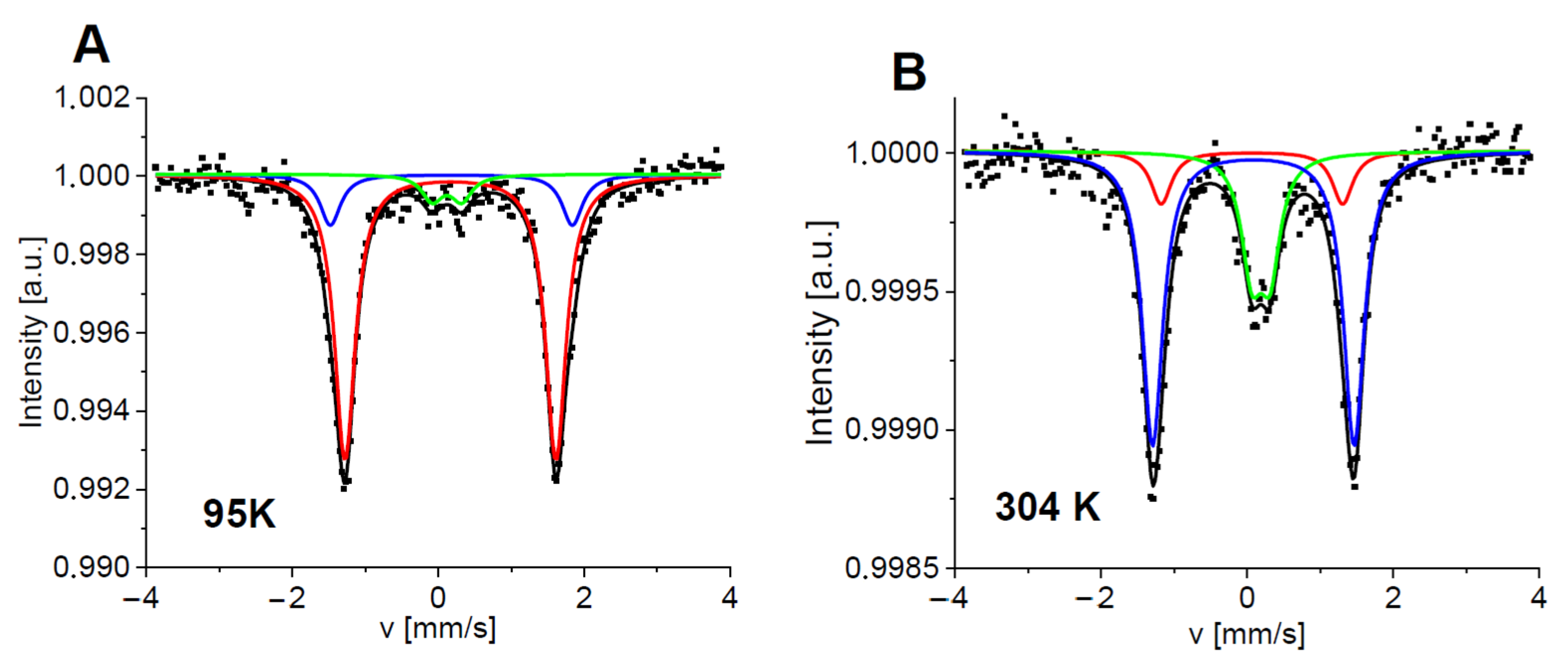
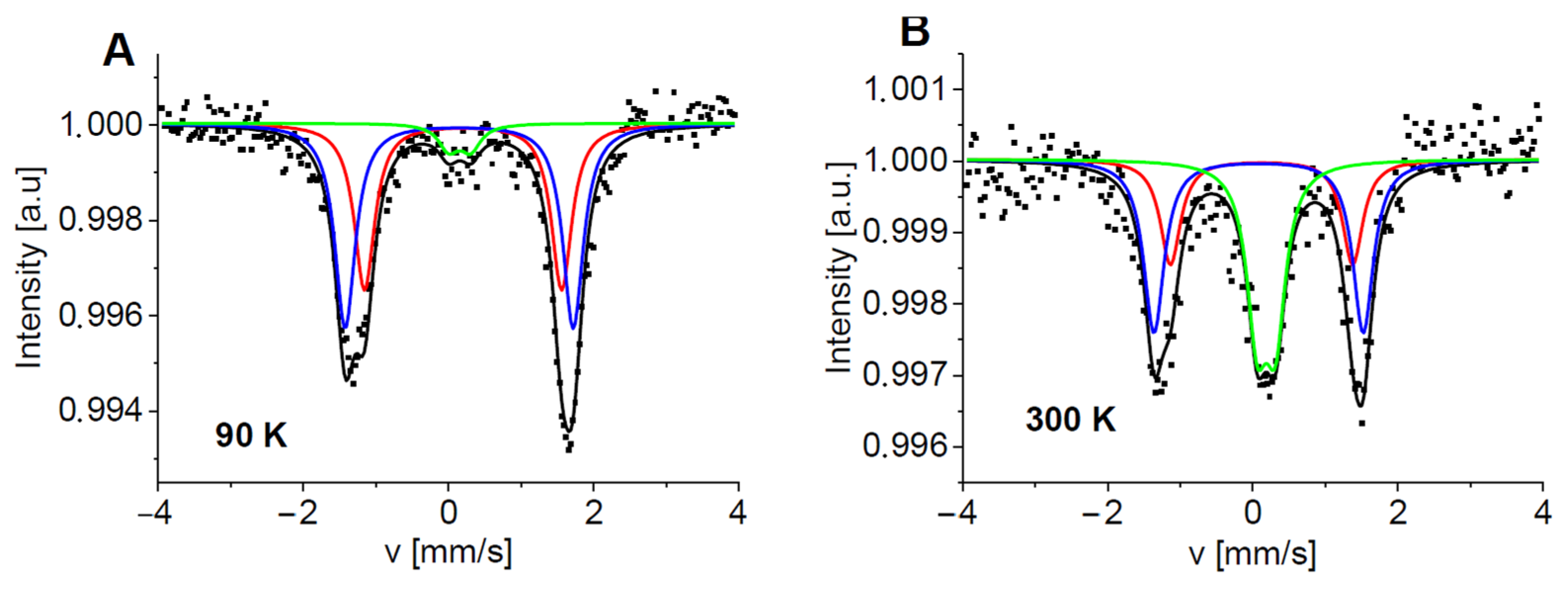
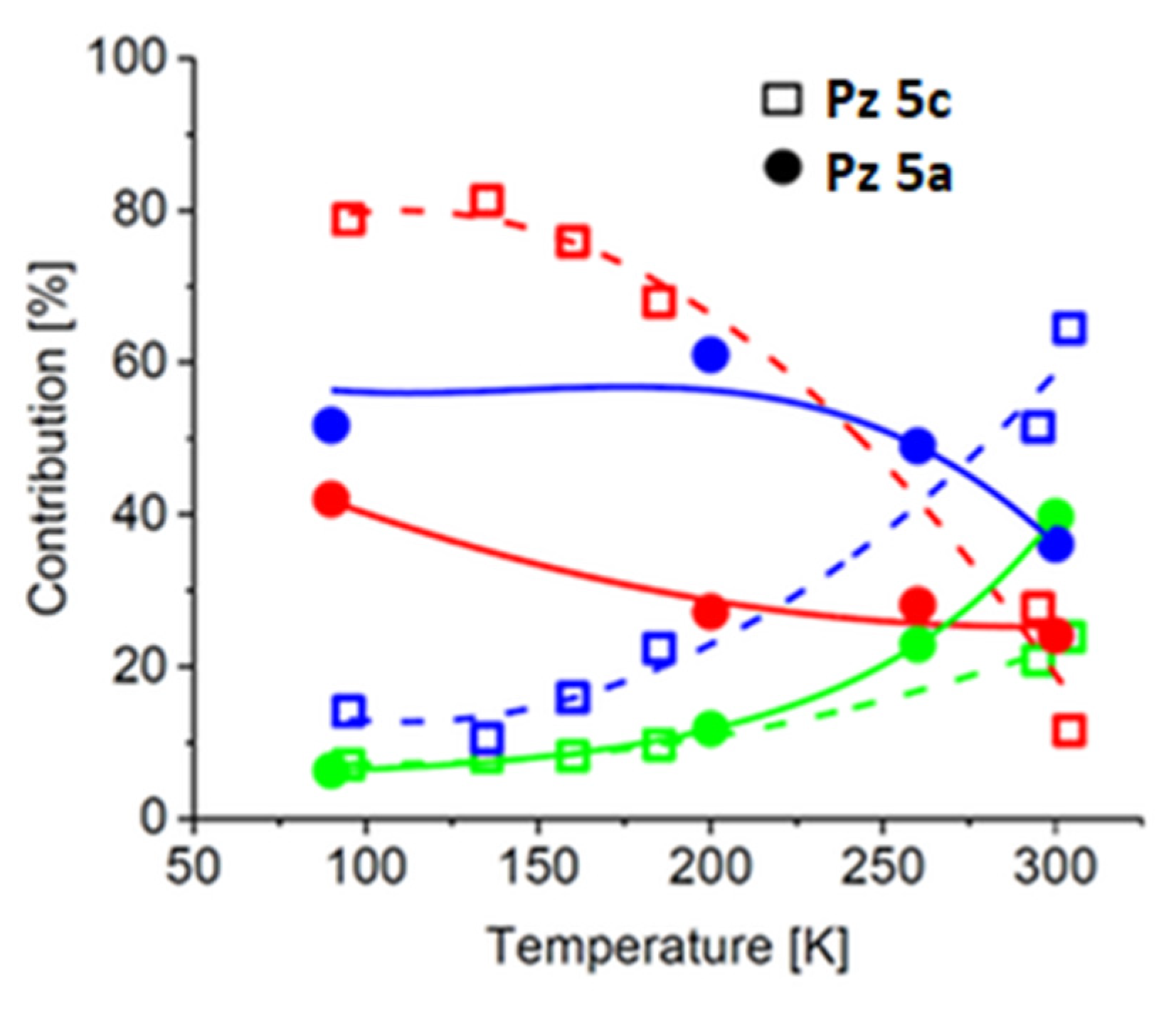
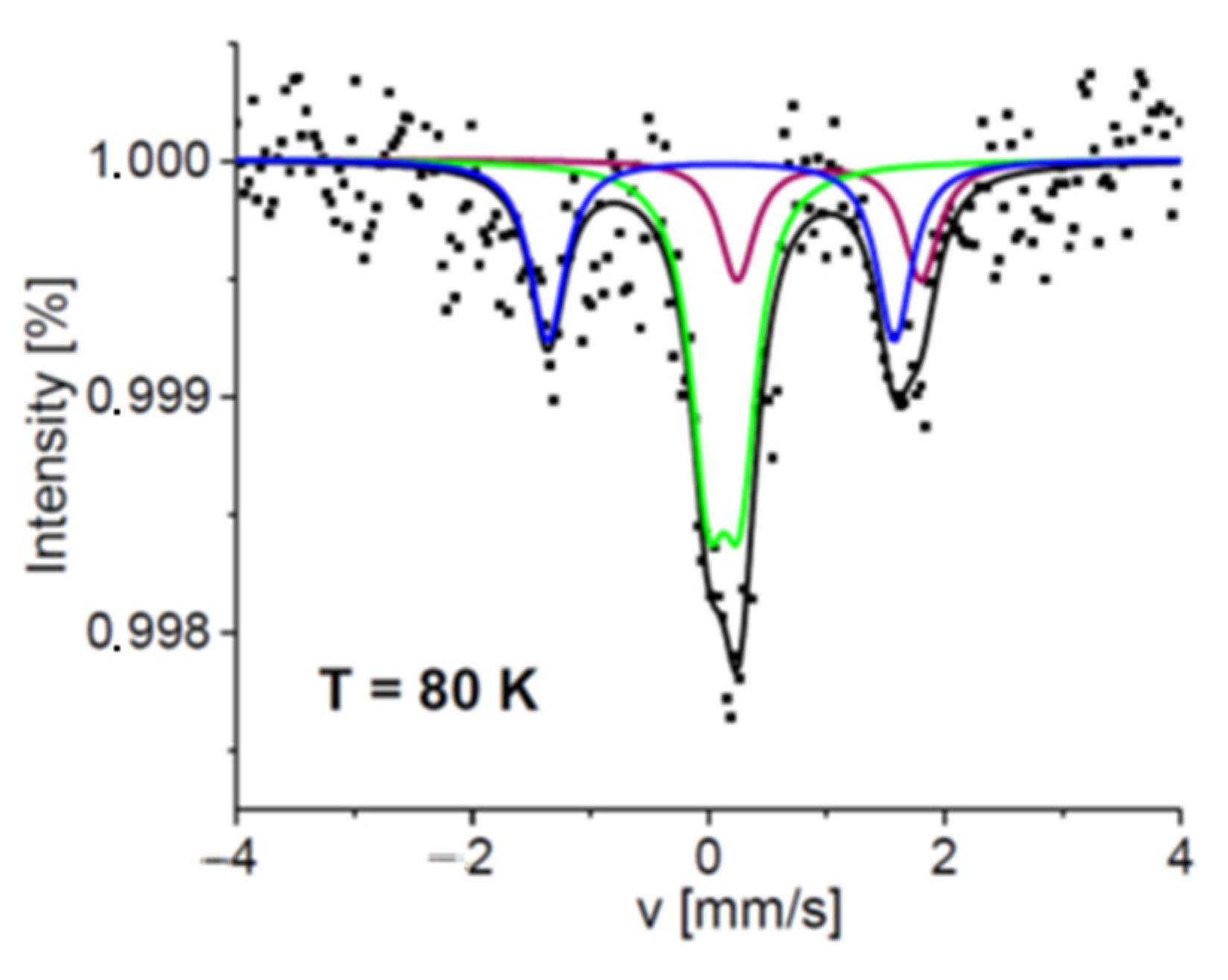
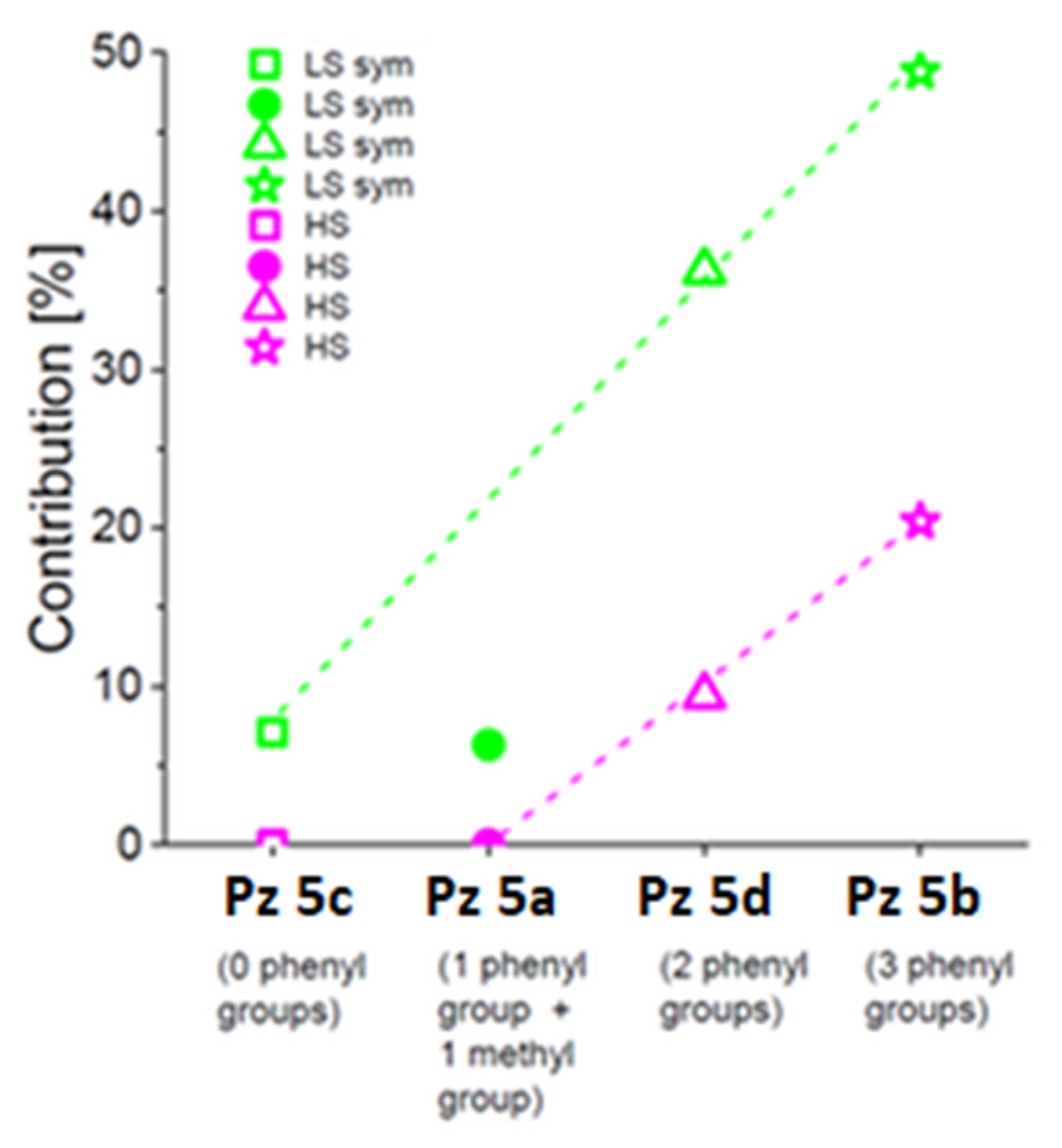
2.3. Mössbauer Spectroscopy Study
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis
General Synthetic Procedure
3.3. Solvation Study
3.4. Single-Crystal X-ray Structure Determination
3.5. NMR Study
3.6. Mössbauer Spectroscopy Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Montalban, A.G.; Jarrell, W.; Riguet, E.; Mccubbin, Q.J.; Anderson, M.E.; White, A.J.P.; Williams, D.J.; Barrett, A.G.M.; Hoffman, B.M. Bis(dimethylamino)porphyrazines: Synthetic, Structural, and Spectroscopic Investigations. J. Org. Chem. 2000, 65, 2472–2478. [Google Scholar] [CrossRef] [PubMed]
- Fuchter, M.J.; Zhong, C.; Zong, H.; Hoffman, B.M.; Barrett, A.G.M. Porphyrazines: Designer Macrocycles by Peripheral Substituent Change. Aust. J. Chem. 2008, 61, 235–255. [Google Scholar] [CrossRef]
- Trivedi, E.R.; Vesper, B.J.; Weitman, H.; Ehrenberg, B.; Barrett, A.G.M.; Radosevich, J.A.; Hoffman, B.M. Chiral Bis-Acetal Porphyrazines as near-Infrared Optical Agents for Detection and Treatment of Cancer. Photochem. Photobiol. 2010, 86, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Manet, I.; Manoli, F.; Donzello, M.P.; Viola, E.; Masi, A.; Andreano, G.; Ricciardi, G.; Rosa, A.; Cellai, L.; Ercolani, C.; et al. Pyrazinoporphyrazines with Externally Appended Pyridine Rings. 13. Structure, UV-Visible Spectral Features, and Noncovalent Interaction with DNA of a Positively Charged Binuclear (ZnII/PtII) Macrocycle with Multimodal Anticancer Potentialities. Inorg. Chem. 2013, 52, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Morgade, M.S.; Stuzhin, P.A. The Chemistry of Porphyrazines: An Overview. J. Porphyr. Phthalocyanines 2004, 8, 1129–1165. [Google Scholar] [CrossRef]
- Michel, S.L.J.; Hoffman, B.M.; Baum, S.M.; Barrett, A.G.M. Peripherally Functionalized Porphyrazines: Novel Metallomacrocycles with Broad, Untapped Potential. In Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; Volume 50, ISBN 0471435104. [Google Scholar]
- Güzel, E.; Yarasir, M.N.; Özkaya, A.R. Low Symmetry Solitaire- and Trans-Functional Porphyrazine/Phthalocyanine Hybrid Complexes: Synthesis, Isolation, Characterization, and Electrochemical and in-Situ Spectroelectrochemical Properties. Synth. Met. 2020, 262, 116331. [Google Scholar] [CrossRef]
- Liao, M.; Watts, J.D.; Huang, M.; Box, P.O.; State, J.; Uni, V. Fe II in Different Macrocycles: Electronic Structures and Properties. J. Phys. Chem. A 2005, 109, 7988–8000. [Google Scholar] [CrossRef]
- Theodoridis, A.; Maigut, J.; Puchta, R.; Kudrik, E.V.; Van Eldik, R. Novel Iron(III) Porphyrazine Complex. Complex Speciation and Reactions with NO and H2O2. Inorg. Chem. 2008, 47, 2994–3013. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, Q.; Sun, J.; Fang, L.; Deng, K. Enhancement of Catalytic Activities of a Biomimetic Catalyst FePz(DtnCl2)4 for the Wet Oxidation of Brilliant Red X3B through the Synergetic Effect of Heat and Light Irradiation. Ind. Eng. Chem. Res. 2013, 52, 13342–13349. [Google Scholar] [CrossRef]
- Koczorowski, T.; Szczolko, W.; Goslinski, T. Physicochemical Properties and Catalytic Applications of Iron Porphyrazines and Phthalocyanines. In Recent Progress in Organometallic Chemistry; InTech Open: Rijeka, Croatia, 2017; pp. 101–121. [Google Scholar]
- Koczorowski, T.; Cerbin-Koczorowska, M.; Rębiś, T. Azaporphyrins Embedded on Carbon-Based Nanomaterials for Potential Use in Electrochemical Sensing—A Review. Nanomaterials 2021, 11, 2861. [Google Scholar] [CrossRef]
- Nakamura, M. Electronic Structures of Highly Deformed Iron(III) Porphyrin Complexes. Coord. Chem. Rev. 2006, 250, 2271–2294. [Google Scholar] [CrossRef]
- Stuzhin, P.A. Iron Complexes of Octaphenyltetraazaporphine. Macroheterocycles 2009, 2, 114–129. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Mahmud, H.; Ulrich, Z. Iron octaphenyltetraazaporphyrins: Synthesis and characterization of the five-coordinate complexes of iron (III)(XFeIIIOPTAP; X= F, Cl, Br, I, HSO4). Inorganica Chimica Acta 1995, 236, 131–139. [Google Scholar] [CrossRef]
- Luo, Q.; Cheng, S.; Tian, H. Synthesis and Photochromism of a New Binuclear Porphyrazinato Magnesium(II). Tetrahedron Lett. 2004, 45, 7737–7740. [Google Scholar] [CrossRef]
- Goslinski, T.; White, A.J.P. Synthesis, Characterization and Spectroscopic Properties of Novel Periphery—Functionalized Unsymmetrical Porphyrazines Containing Mixed Dithienylpyrrolyl and Dimethylamino Groups. Polyhedron 2009, 28, 2579–2584. [Google Scholar] [CrossRef]
- Szczolko, W.; Koczorowski, T.; Wicher, B.; Sobotta, L.; Gdaniec, M.; Teubert, A.; Mielcarek, J.; Korecki, J.; Burda, K.; Tykarska, E.; et al. X-ray and NMR Structural Studies of the Series of Porphyrazines with Peripheral Pyrrolyl Groups. Inorg. Chim. Acta 2019, 484, 368–374. [Google Scholar] [CrossRef]
- Szczolko, W.; Koczorowski, T.; Wicher, B.; Kryjewski, M.; Krakowska, Z.; Tykarska, E.; Goslinski, T. Porphyrazines with Bulky Peripheral Pyrrolyl Substituents—Synthesis via Microwave-Assisted Suzuki-Miyaura Cross-Coupling Reaction, Optical and Electrochemical Properties. Dye. Pigment. 2022, 206, 110607. [Google Scholar] [CrossRef]
- Szczolko, W.; Wzgarda, A.; Koczorowski, T.; Wicher, B.; Sobotta, L.; Gdaniec, Z.; Gdaniec, M.; Mielcarek, J.; Tykarska, E.; Goslinski, T. The Suzuki Cross-Coupling Reaction for the Synthesis of Porphyrazine Possessing Bulky 2,5-(Biphenyl-4-Yl)Pyrrol-1-Yl Substituents in the Periphery. Polyhedron 2015, 102, 462–468. [Google Scholar] [CrossRef]
- Kryjewski, M.; Tykarska, E.; Rebis, T.; Dlugaszewska, J.; Ratajczak, M.; Teubert, A.; Gapiński, J.; Patkowski, A.; Piskorz, J.; Milczarek, G.; et al. Porphyrazine with Bulky 2-(1-Adamantyl)-5-Phenylpyrrol-1-Yl Periphery Tuning Its Spectral and Electrochemical Properties. Polyhedron 2015, 98, 217–223. [Google Scholar] [CrossRef]
- Koczorowski, T.; Szczolko, W.; Burda, K.; Nowak, M.; Dawidowska, M.; Teubert, A.; Sobotta, L.; Gdaniec, M.; Korecki, J.; Mielcarek, J.; et al. Influence of Bulky Pyrrolyl Substitent on the Physicochemical Properties of Porphyrazines. Dye. Pigment. 2015, 112, 138–144. [Google Scholar] [CrossRef]
- Szczolko, W.; Sobotta, L.; Fita, P.; Koczorowski, T.; Mikus, M.; Gdaniec, M.; Orzechowska, A.; Burda, K.; Sobiak, S.; Wierzchowski, M.; et al. Synthesis, Characteristics and Photochemical Studies of Novel Porphyrazines Possessing Peripheral 2,5-Dimethylpyrrol-1-Yl and Dimethylamino Groups. Tetrahedron Lett. 2012, 53, 2040–2044. [Google Scholar] [CrossRef]
- Begland, R.W.; Hartter, D.R.; Jones, F.N.; Sam, D.J.; Sheppard, W.A.; Webster, O.W.; Weigert, F.J. Hydrogen Cyanide Chemistry. VIII. New Chemistry of Diaminomaleonitrile. Heterocyclic Synthesis. J. Org. Chem. 1974, 39, 2341–2350. [Google Scholar] [CrossRef]
- Beall, L.S.; Mani, N.S.; White, A.J.P.; Williams, D.J.; Barrett, A.G.M.; Hoffman, B.M. Porphyrazines and Norphthalocyanines Bearing Nitrogen Donor Pockets: Metal Sensor Properties. J. Org. Chem. 1998, 63, 5806–5817. [Google Scholar] [CrossRef] [PubMed]
- Decréau, R.; Chanon, M.; Julliard, M. Synthesis and Characterization of a Series of Hexadecachlorinated Phthalocyanines. Inorg. Chim. Acta 1999, 293, 80–87. [Google Scholar] [CrossRef]
- Goslinski, T.; Zhong, C.; Fuchter, M.J.; Stern, C.L.; White, A.J.P.; Barrett, A.G.M.; Hoffman, B.M. Porphyrazines as Molecular Scaffolds: Flexible Syntheses of Novel Multimetallic Complexes. Inorg. Chem. 2006, 45, 3686–3694. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, C.; Bavoso, A.; Bencini, A.; Rosa, A.; Lelj, F.; Bonosi, F. Synthesis, Structure, Magnetic, Spectroscopic and Electrochemical Behaviour of Chloro-Iron(III) and -Manganese(III) Complexes of 2,3,7,8,12,13,17,18-Octakis(Ethylsulfanyl)-5,10,15,20-Tetraazaporphyrin. Dalton Trans. 1996, 2799–2807. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Nefedov, S.E.; Kumeev, R.S.; Ul-Haq, A.; Minin, V.V.; Ivanova, S.S. Effects of Solvation on the Spin State of Iron(III) in 2,8,12,18-Tetrabutyl-3,7,13,17-Tetramethyl-5,10-Diazaporphyrinatoiron(III) Chloride. Inorg. Chem. 2010, 49, 4802–4813. [Google Scholar] [CrossRef]
- Colomban, C.; Kudrik, E.V.; Tyurin, D.V.; Albrieux, F.; Nefedov, S.E.; Afanasiev, P.; Sorokin, A.B. Synthesis and Characterization of μ-Nitrido, μ-Carbido and μ-Oxo Dimers of Iron Octapropylporphyrazine. Dalton Trans. 2015, 44, 2240–2251. [Google Scholar] [CrossRef]
- Burda, K.; Hrynkiewicz, A.; Kołoczek, H.; Stanek, J.; Strzałka, K. Mixed Valence State in Ironporphyrin Aggregates. Biochim. Biophys. Acta 1995, 1244, 345–350. [Google Scholar] [CrossRef]
- Lang, G.; Marshali, W. Mössbauer Effect in Some Haemoglobin Compounds. Proc. Phys. Soc. 1966, 87, 3–34. [Google Scholar] [CrossRef]
- Fitzgerald, J.P.; Haggerty, B.S.; Rheingold, A.L.; May, L.; Brewer, G.A. Iron Octaethyltetraazaporphyrins—Synthesis, Characterization, Coordination Chemistry, and Comparisons to Related Iron Porphyrins and Phthalocyanines. Inorg. Chem. 1992, 31, 2006–2013. [Google Scholar] [CrossRef]
- Reimer, K.J.; Sibley, C.A. Electric Field Gradient at Iron in Dicarbonyl Complexes of (Tetraphenylporphyrinato)- and (Octamethyltetrabenzoporphyrinato)Iron(II). J. Am. Chem. Soc. 1983, 105, 5147–5149. [Google Scholar] [CrossRef]
- Kaczmarska, M.; Fornal, M.; Messerli, F.H.; Korecki, J.; Grodzicki, T.; Burda, K. Erythrocyte Membrane Properties in Patients with Essential Hypertension. Cell Biochem. Biophys. 2013, 67, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Noll, B.C.; Schulz, C.E.; Scheidt, W.R. Four-Coordinate Iron(II) Porphyrinates: Electronic Configuration Change by Intermolecular Interaction. Inorg. Chem. 2007, 46, 619–621. [Google Scholar] [CrossRef][Green Version]
- Burda, K.; Hrynkiewicz, A.; Kołoczek, H.; Stanek, J.; Strzałka, K. Molecular Dynamics and Local Electronic States of Sn and Fe in Metallocytochrome and Metalloporphyrin. Hyperfine Interact. 1994, 91, 891–897. [Google Scholar] [CrossRef]
- Hayami, S.; Komatsu, Y.; Shimizu, T.; Kamihata, H.; Lee, Y.H. Spin-Crossover in Cobalt(II) Compounds Containing Terpyridine and Its Derivatives. Coord. Chem. Rev. 2011, 255, 1981–1990. [Google Scholar] [CrossRef]
- Jahn, H.A.; Teller, E. Stability of Polyatomic Molecules in Degenerate Electronic States I. Orbital Degeneracy. Proc. R. Soc. Lond. 1937, 161, 220–235. [Google Scholar]
- Schläfer, H.L.; Gliemann, G. (Eds.) Basic Principles of Ligand Field Theory; Wiley-Interscience of John Wiley & Sons Ltd.: London, UK; New York, NY, USA; Sydney, Australia; Toronto, ON, Canada, 1969. [Google Scholar]
- Hałas, A.; Orzechowska, A.; Derrien, V.; Chumakov, A.I.; Sebban, P.; Fiedor, J.; Lipińska, M.; Zaja̧c, M.; Ślězak, T.; Strzałka, K.; et al. The Dynamics of the Non-Heme Iron in Bacterial Reaction Centers from Rhodobacter Sphaeroides. Biochim. Biophys. Acta—Bioenerg. 2012, 1817, 2095–2102. [Google Scholar] [CrossRef]
- Sobotta, L.; Fita, P.; Szczolko, W.; Wrotynski, M.; Wierzchowski, M.; Goslinski, T.; Mielcarek, J. Functional Singlet Oxygen Generators Based on Porphyrazines with Peripheral 2,5-Dimethylpyrrol-1-Yl and Dimethylamino Groups. J. Photochem. Photobiol. A Chem. 2013, 269, 9–16. [Google Scholar] [CrossRef]
- Agilent Technologies. CrysAlis PRO Software v38; Agilent Technologies: Yarnton, UK, 2009. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An Improved Tool for Crystal Structure Determination and Refinement. J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, D.G.; Ping, J.Y. Voigt-Based Methods for Arbitrary-Shape Static Hyperfine Parameter Distributions in Mössbauer Spectroscopy. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1991, 58, 85–97. [Google Scholar] [CrossRef]
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Cross over in Transition Metal Compounds; Springer: Berlin/Heidelberg, Germany, 2004; Volume 233–235. [Google Scholar]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koczorowski, T.; Szczolko, W.; Bakun, P.; Wicher, B.; Sobotta, L.; Gdaniec, M.; Teubert, A.; Mielcarek, J.; Tykarska, E.; Korecki, J.; et al. The Valence and Spin State Tuning of Iron(II/III) Porphyrazines with Bulky Pyrrolyl Periphery in Solution and Solid State. Molecules 2022, 27, 7820. https://doi.org/10.3390/molecules27227820
Koczorowski T, Szczolko W, Bakun P, Wicher B, Sobotta L, Gdaniec M, Teubert A, Mielcarek J, Tykarska E, Korecki J, et al. The Valence and Spin State Tuning of Iron(II/III) Porphyrazines with Bulky Pyrrolyl Periphery in Solution and Solid State. Molecules. 2022; 27(22):7820. https://doi.org/10.3390/molecules27227820
Chicago/Turabian StyleKoczorowski, Tomasz, Wojciech Szczolko, Pawel Bakun, Barbara Wicher, Lukasz Sobotta, Maria Gdaniec, Anna Teubert, Jadwiga Mielcarek, Ewa Tykarska, Jozef Korecki, and et al. 2022. "The Valence and Spin State Tuning of Iron(II/III) Porphyrazines with Bulky Pyrrolyl Periphery in Solution and Solid State" Molecules 27, no. 22: 7820. https://doi.org/10.3390/molecules27227820
APA StyleKoczorowski, T., Szczolko, W., Bakun, P., Wicher, B., Sobotta, L., Gdaniec, M., Teubert, A., Mielcarek, J., Tykarska, E., Korecki, J., Burda, K., & Goslinski, T. (2022). The Valence and Spin State Tuning of Iron(II/III) Porphyrazines with Bulky Pyrrolyl Periphery in Solution and Solid State. Molecules, 27(22), 7820. https://doi.org/10.3390/molecules27227820