
Biochemical Evolution of a Potent Target of Mosquito Larvicide, 3-Hydroxykynurenine Transaminase

 ,
,  , and
, and 
Abstract

1. Introduction
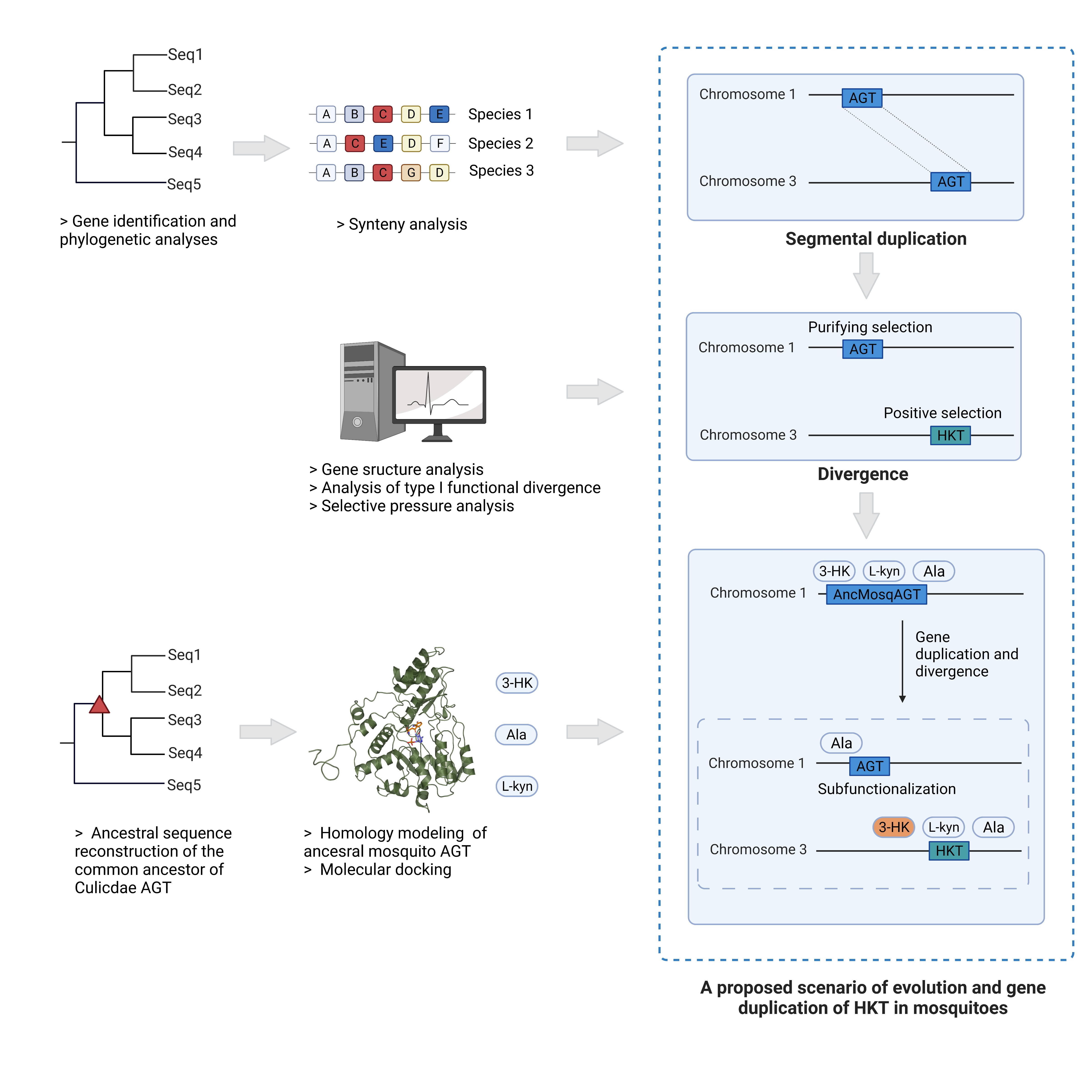
2. Results
2.1. HKT Phylogeny and Mosquito-Specific Duplication
2.2. Micro-Synteny Analyses
2.3. Gene Structure Divergence between HKT and AGT in Mosquitoes
2.4. Functional Divergence between AGT and HKT
2.5. Detection of Positive Selection in HKT Genes
2.6. Positive Selection Sites Affect Substrate Binding
2.7. Ancestral Sequence Reconstruction of AGT in Mosquitoes
2.8. Homology Modeling, Molecular Docking, and Dynamics Simulation of AncMosqAGT
3. Discussion
4. Materials and Methods
4.1. Gene Identification and Phylogenetic Analyses
4.2. Conserved Micro-Synteny Analysis
4.3. Sequence Structure Analysis
4.4. Analyses of Type I Functional Divergence
4.5. Selective Pressure Analyses
4.6. Ancestral Sequence Reconstruction of the Common Ancestor of Culicdae AGT
4.7. Homology Modeling, Molecular Docking, and Molecular Dynamic Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J. Neurochem. 2010, 70, 299–307. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Kotanen, S.; Huybrechts, J.; Cerstiaens, A.; Zoltan, K.; Daloze, D.; Baggerman, G.; Forgo, P.; Loof, A.D.; Schoofs, L. Identification of tryptophan and β-carboline as paralysins in larvae of the yellow mealworm, Tenebrio molitor. Biochem. Biophys. Res. Commun. 2003, 310, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Cerstiaens, A.; Huybrechts, J.; Kotanen, S.; Lebeau, I.; Meylaers, K.; Loof, A.D.; Schoofs, L. Neurotoxic and neurobehavioral effects of kynurenines in adult insects. Biochem. Biophys. Res. Commun. 2003, 312, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Peng, Y.; Wen, H.; Song, X.; An, Y.; Tang, H.; Wang, J. Microbial tryptophan catabolism affects the vector competence of Anopheles. bioRxiv 2021. [Google Scholar] [CrossRef]
- Feng, Y.; Peng, Y.; Song, X.; Wen, H.; An, Y.; Tang, H.; Wang, J. Anopheline mosquitoes are protected against parasite infection by tryptophan catabolism in gut microbiota. Nat. Microbiol. 2022, 7, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Tearle, R. Tissue specific effects of ommochrome pathway mutations in Drosophila melanogaster. Genet. Res. 1991, 57, 257–266. [Google Scholar] [CrossRef]
- Li, J.; Beerntsen, B.T.; James, A.A. Oxidation of 3-hydroxykynurenine to produce xanthommatin for eye pigmentation: A major branch pathway of tryptophan catabolism during pupal development in the yellow fever mosquito, Aedes aegypti. Insect Biochem. Mol. Biol. 1999, 29, 329–338. [Google Scholar] [CrossRef]
- Yuan, L.; Huang, X.-Y.; Liu, Z.-Y.; Zhang, F.; Zhu, X.-L.; Yu, J.-Y.; Ji, X.; Xu, Y.-P.; Li, G.; Li, C.; et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Fang, J.; Li, J. 3-Hydroxykynurenine transaminase identity with alanine glyoxylate transaminase. A probable detoxification protein in Aedes aegypti. J. Biol. Chem. 2002, 277, 15781–15787. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Beerntsen, B.T.; Li, J.Y. The tryptophan oxidation pathway in mosquitoes with emphasis on xanthurenic acid biosynthesis. J. Insect Physiol. 2007, 53, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, G. Transamination of 3-hydroxykynurenine to produce xanthurenic acid: A major branch pathway of tryptophan metabolism in the mosquito, Aedes aegypti, during larval development. Insect Biochem. Mol. Biol. 1997, 27, 859. [Google Scholar] [CrossRef]
- Han, Q.; Li, J. Comparative characterization of Aedes 3-hydroxykynurenine transaminase/alanine glyoxylate transaminase and Drosophila serine pyruvate aminotransferase. FEBS Lett. 2002, 527, 199–204. [Google Scholar] [CrossRef]
- Romo, T.P. Discovery of 1,2,4-oxadiazole derivatives as a novel class of noncompetitive inhibitors of 3-hydroxykynurenine transaminase (HKT) from Aedes aegypti. Bioorg. Med. Chem. 2020, 28, 115252. [Google Scholar]
- Oliveira, V.S.; Pimenteira, C.; Silva-Alves, D.C.B.D.; Leal, L.; Soares, T.A. The enzyme 3-hydroxykynurenine transaminase as potential target for 1,2,4-oxadiazoles with larvicide activity against the dengue vector Aedes aegypti. Bioorg. Med. Chem. 2013, 21, 6996–7003. [Google Scholar] [CrossRef]
- Maciel, L.G.; Barbosa, A.D.S.; de Alencar-Filho, E.B.; Soares, T.A.; Dos Anjos, J.V. A second generation of 1,2,4-oxadiazole derivatives with enhanced solubility for inhibition of 3-hydroxykynurenine transaminase (HKT) from Aedes aegypti. RSC Med. Chem. 2021, 12, 222–236. [Google Scholar] [CrossRef]
- Rossi, F.; Garavaglia, S.; Giovenzana, G.B.; Arcà, B.; Li, J.; Rizzi, M. Crystal structure of the Anopheles gambiae 3-hydroxykynurenine transaminase. Proc. Natl. Acad. Sci. USA 2006, 103, 5711–5716. [Google Scholar] [CrossRef]
- Han, Q.; Kim, S.; Ding, H.; Li, J. Evolution of two alanine glyoxylate aminotransferases in mosquito. Biochem. J. 2006, 397, 473–481. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, S.; Krinsky, B.H.; Long, M. New genes as drivers of phenotypic evolution. Nat. Rev. Genet. 2013, 14, 645–660. [Google Scholar] [CrossRef]
- Yun, D.; Qi, Z.; Wen, W. Origins of New Genes and Evolution of Their Novel Functions. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 345–363. [Google Scholar]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Gao, Y.G.; Vogelaar, N.; Wilson, S.R.; Rizzi, M.; Li, J. Crystal structures of Aedes aegypti alanine glyoxylate aminotransferase. J. Biol. Chem. 2006, 281, 37175–37182. [Google Scholar] [CrossRef]
- Christen, P.; Mehta, P.K. From cofactor to enzymes. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Chem Rec 2001, 1, 436–447. [Google Scholar] [CrossRef]
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Vidhyanandhini, R.; Kumar, N.P. Characterization of the 3-HKT gene in important malaria vectors in India, viz: Anopheles culicifacies and Anopheles stephensi (Diptera: Culicidae). Mem. Inst. Oswaldo Cruz 2008, 103, 595–597. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, J.; Dean, A.M.; Brunet, F.; Long, M. Evolving protein functional diversity in new genes of Drosophila. Proc. Natl. Acad. Sci. USA 2004, 101, 16246–16250. [Google Scholar] [CrossRef]
- Lan, T.; Wang, X.-R.; Zeng, Q.-Y. Structural and functional evolution of positively selected sites in pine glutathione S-transferase enzyme family. J. Biol. Chem. 2013, 288, 24441–24451. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by gene duplication. Am. J. Hum. Genet. 1971, 23, 541. [Google Scholar]
- Bergthorsson, U.; Andersson, D.I.; Roth, J.R. Ohno’s dilemma: Evolution of new genes under continuous selection. Proc. Natl. Acad. Sci. USA 2007, 104, 17004–17009. [Google Scholar] [CrossRef]
- Copley, S.D. Evolution of new enzymes by gene duplication and divergence. Febs j 2020, 287, 1262–1283. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Cassimjee, K.E.; Himo, F. Quantum Chemical Study of Dual-Substrate Recognition in ω-Transaminase. ACS Omega 2017, 2, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Rausch, C.; Lerchner, A.; Schiefner, A.; Skerra, A. Crystal structure of the ω-aminotransferase from Paracoccus denitrificans and its phylogenetic relationship with other class III aminotransferases that have biotechnological potential. Proteins 2013, 81, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Farideh, C.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Marc, G.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2019, 48, D265–D268. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Quang, M.B. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Huelsenbeck, J.P.; Ronquist, F.R. MRBAYES: Bayesian inference of phylogeny. Biometrics 2001, 17, 754–755. [Google Scholar]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Nguyen, N.T.T.; Vincens, P.; Dufayard, J.F.; Roest Crollius, H.; Louis, A. Genomicus in 2022: Comparative tools for thousands of genomes and reconstructed ancestors. Nucleic Acids Res. 2022, 50, D1025–D1031. [Google Scholar] [CrossRef]
- Gu, X. Statistical methods for testing functional divergence after gene duplication. Mol. Biol. Evol. 1999, 16, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zou, Y.; Su, Z.; Huang, W.; Zhou, Z.; Arendsee, Z.; Zeng, Y. An Update of DIVERGE Software for Functional Divergence Analysis of Protein Family. Mol. Biol. Evol. 2013, 30, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Merkl, R.; Sterner, R. Ancestral protein reconstruction: Techniques and applications. Biol. Chem. 2016, 397, 1–21. [Google Scholar] [CrossRef]
- Zeng, B.; Zhou, Y.; Yi, Z.; Zhou, R.; Jin, W.; Zhang, G. Highly thermostable and promiscuous β-1,3-xylanasen designed by optimized ancestral sequence reconstruction. Bioresour. Technol. 2021, 340, 125732. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef]
- Zheng, W.; Wuyun, Q.; Zhou, X.; Li, Y.; Freddolino, P.L.; Zhang, Y. LOMETS3: Integrating deep learning and profile alignment for advanced protein template recognition and function annotation. Nucleic Acids Res. 2022, 50, W454–W464. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Zhang, Y.; Jeffrey, S. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.v.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coefficient θ ± SE (p) | |
|---|---|
| HKT/mosqAGT | 0.288849 ± 0.096070 (p < 0.01) |
| HKT/DAGT | 0.292432 ± 0.082008 (p < 0.01) |
| HKT/CAGT | 0.385146 ± 0.082927 (p < 0.01) |
| HKT/LAGT | 0.560887 ± 0.081839 (p < 0.01) |
| DAGT/LAGT | 0.533485 ± 0.063676 (p < 0.01) |
| DAGT/CAGT | 0.349754 ± 0.063676 (p < 0.01) |
| CAGT/LAGT | 0.400981 ± 0.057970 (p < 0.01) |
| Site Model | |||||||
|---|---|---|---|---|---|---|---|
| Foreground Branch | Models | −lnl | 2Δlnl | LRT Pairs | LRT p-Value | Estimates of Parameters | Positive Sites (BEB) |
| M0 | 45,733.91409 | 2469.602752 | M0/M3 | <0.01 | ω = 0.07927 | Not allowed | |
| M3 | 44,499.11272 | ω0 = 0.01224, ω1 = 0.07281, ω2 = 0.19935, p0 = 0.29651, p1 = 0.45743, p2 = 0.24606 | Not allowed | ||||
| M1a | 45,672.02204 | 0 | M1a/M2a | >0.05 | ω0 = 0.08631, ω1 = 1.0000, p0 = 0.96868, p1 = 0.0.03132 | Not allowed | |
| M2a | 45,672.02204 | ω0 = 0.8631, ω1 = 1.0000, ω2 = 1.0000, p0 = 0.96868, p1 = 0.01322, p2 = 0.01810 | None | ||||
| M7 | 44,510.89475 | 0.002114 | M7/M8 | >0.05 | P = 0.77106, q = 6.60910 | Not allowed | |
| M8 | 44,510.8958 | p0 = 0.99999, p = 0.77108, q = 6.60934, p1 = 0.00001, ω = 1.0000 | None | ||||
| Branch Model | |||||||
| Foreground Branch | Models | −lnl | 2Δlnl | LRT Pairs | LRT p-Value | Estimates of Parameters | Positive Sites (BEB) |
| M0 | 45,733.91409 | 658.98236 | M0/Free ratio model | <0.01 | ω = 0.07927 | Not allowed | |
| Free Ratio Model | 45,404.42291 | ωmosqAGT = 0.3319, ωHKT = 999.0000, (variable ω) | Not allowed | ||||
| HKT | Two Ratio Model | 45,723.40152 | 8.104116 | M0/Two ratio Model | <0.01 | ω0 = 0.08006, ω1 = 999.00000 | Not allowed |
| mosqAGT | Two Ratio Model | 45,728.8162 | 10.195778 | M0/Two ratio Model | <0.01 | ω0 = 0.07926, ω1 = 0.00163 | Not allowed |
| Branch-Site Model | |||||||
| Foreground Branch | Models | −lnl | 2Δlnl | LRT Pairs | LRT p-Value | Estimates of Parameters | Positive Sites (BEB) |
| HKT | M0 | 45,659.52254 | 8.718492 | M0/MA | <0.01 | ω0 = 0.08764, ω1 = 1.00000, ω2 = 1.00000 | Not allowed |
| MA | 45,655.1633 | ω0 = 0.08847, ω1 = 1.00, ω2 = 999.00 | 9S *, 16I *, 21M *, 31K *, 35A *, 40T *, 43S *, 44N *, 45F *, 47D *, 78A *, 105A *, 124G *, 127D *, 146C **, 180A *, 185C *, 190Y *, 218I *, 221K *, 240L *, 251D *, 252E *, 254K *, 260V *, 263N *, 266F *, 267A *, 286R *, 319C **, 321M *, 339F *, 344Q **, 354A *, 355W **, 357A *, 359I *, 363S *, 364S *, 367Q *, 372Y ** | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Bhowmick, B.; Tang, Y.; Lozano-Fernandez, J.; Han, Q. Biochemical Evolution of a Potent Target of Mosquito Larvicide, 3-Hydroxykynurenine Transaminase. Molecules 2022, 27, 4929. https://doi.org/10.3390/molecules27154929
Chen H, Bhowmick B, Tang Y, Lozano-Fernandez J, Han Q. Biochemical Evolution of a Potent Target of Mosquito Larvicide, 3-Hydroxykynurenine Transaminase. Molecules. 2022; 27(15):4929. https://doi.org/10.3390/molecules27154929
Chicago/Turabian StyleChen, Huaqing, Biswajit Bhowmick, Yu Tang, Jesus Lozano-Fernandez, and Qian Han. 2022. "Biochemical Evolution of a Potent Target of Mosquito Larvicide, 3-Hydroxykynurenine Transaminase" Molecules 27, no. 15: 4929. https://doi.org/10.3390/molecules27154929
APA StyleChen, H., Bhowmick, B., Tang, Y., Lozano-Fernandez, J., & Han, Q. (2022). Biochemical Evolution of a Potent Target of Mosquito Larvicide, 3-Hydroxykynurenine Transaminase. Molecules, 27(15), 4929. https://doi.org/10.3390/molecules27154929