
In Silico Screening of Quorum Sensing Inhibitor Candidates Obtained by Chemical Similarity Search


Abstract
:1. Introduction
2. Results
2.1. Generation of AI2 Inhibitor Candidate Library
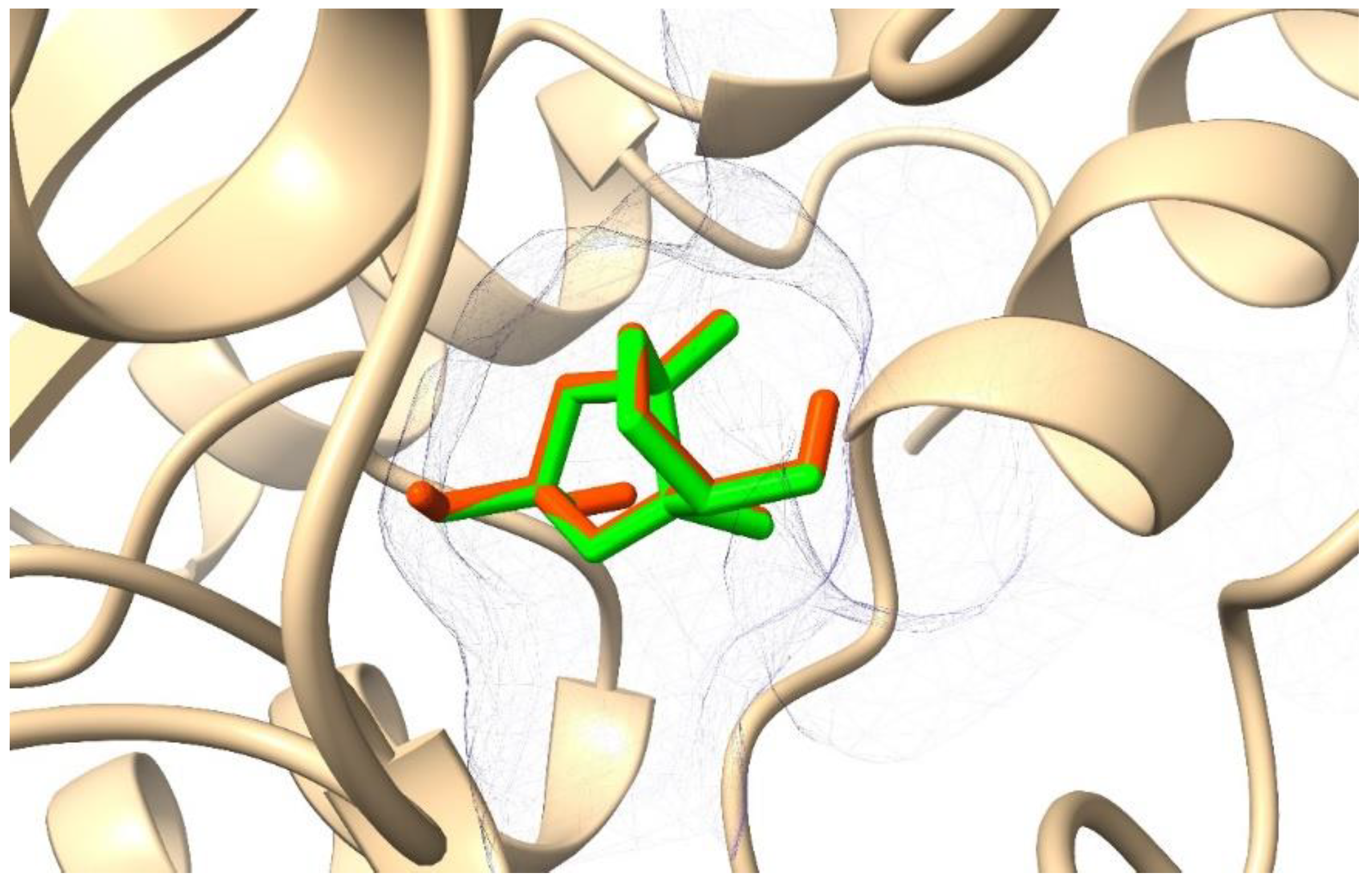
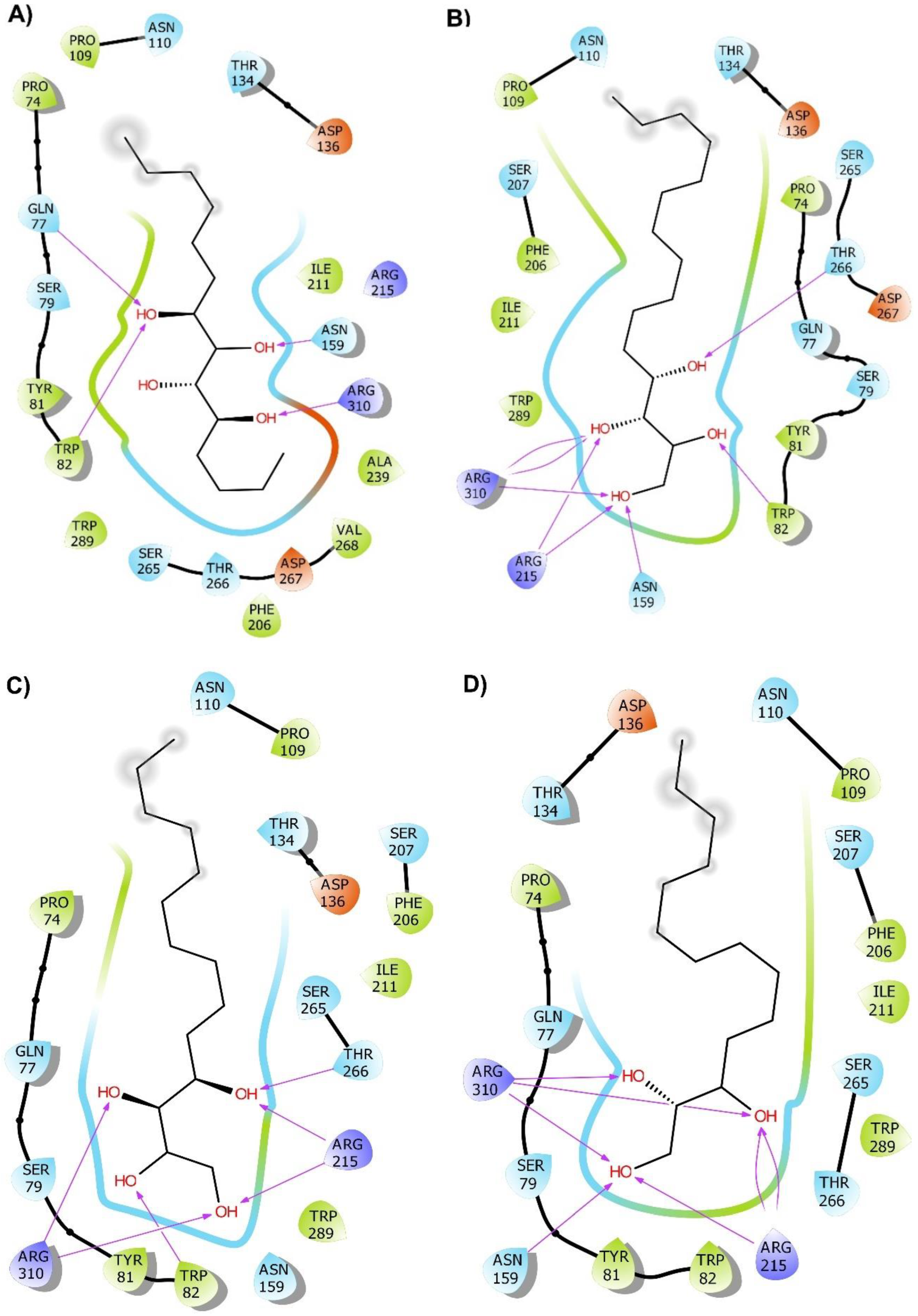
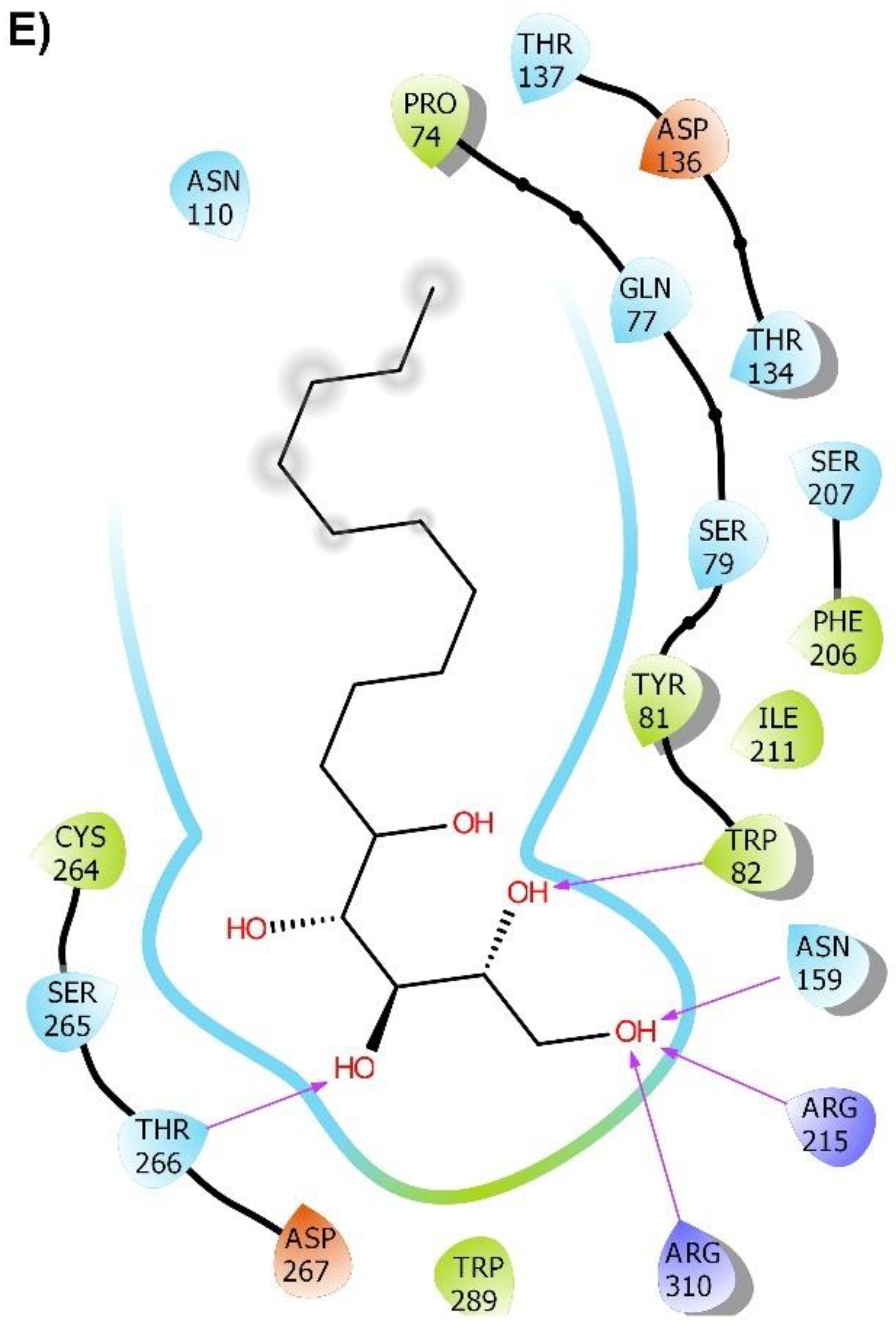
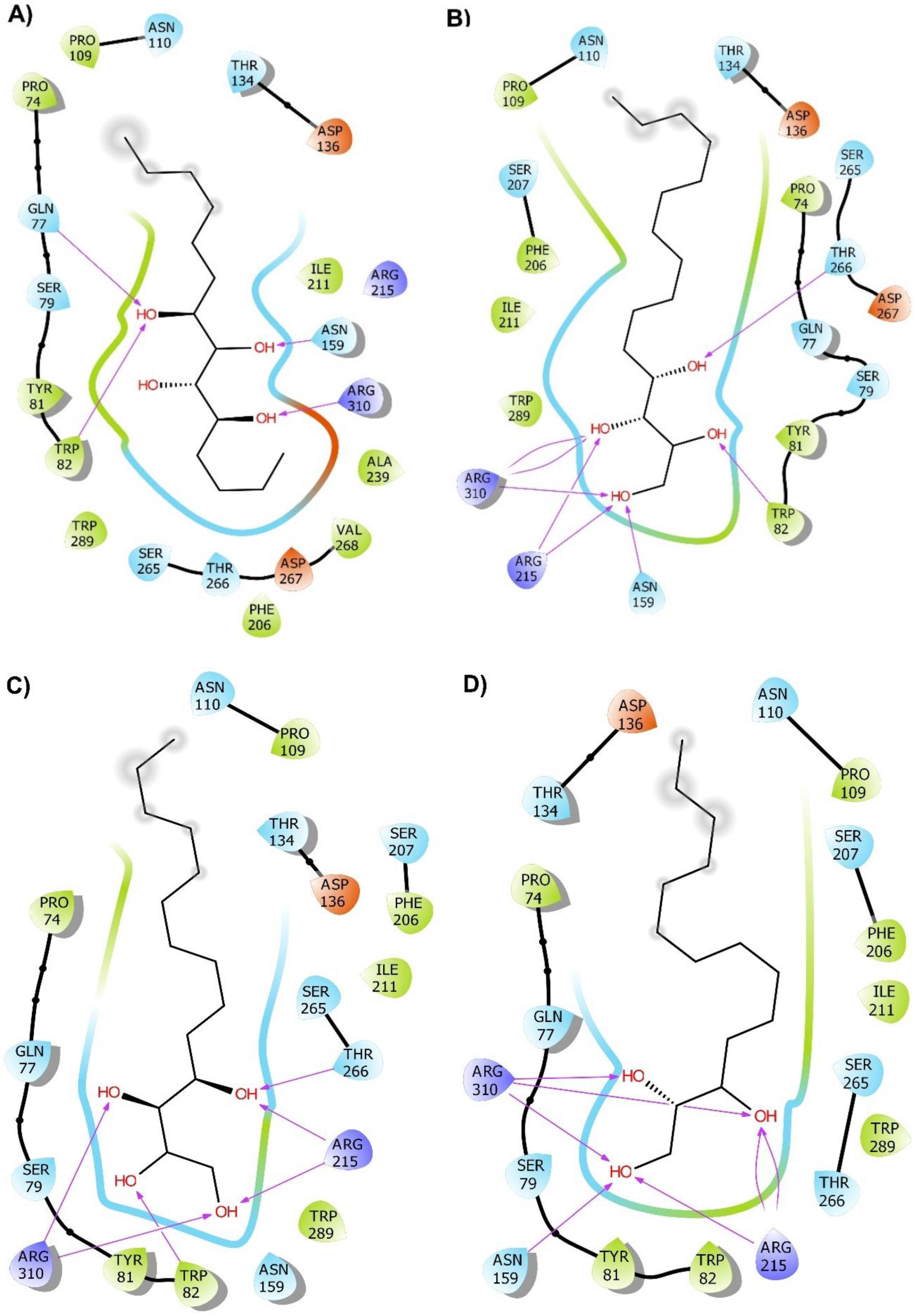
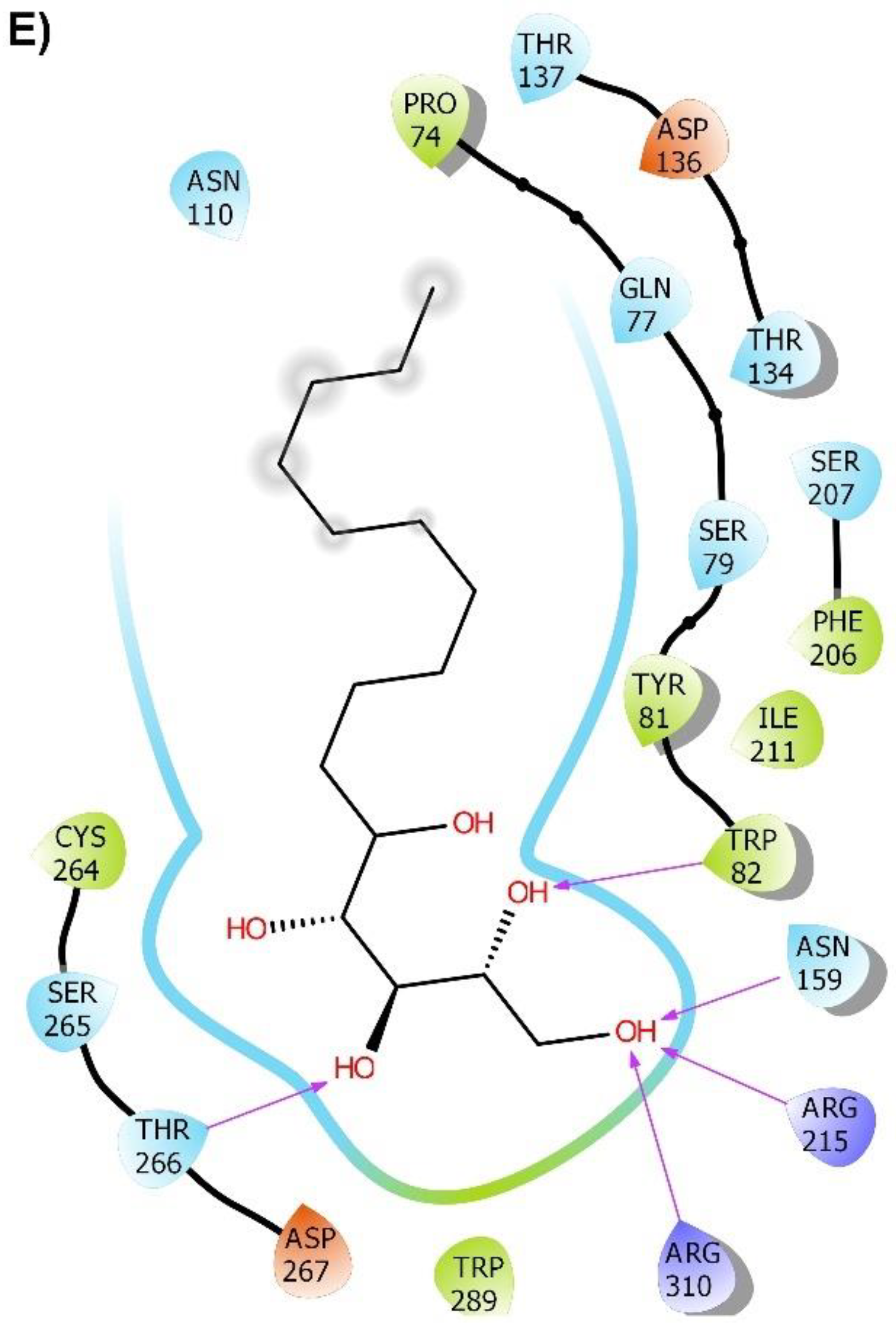
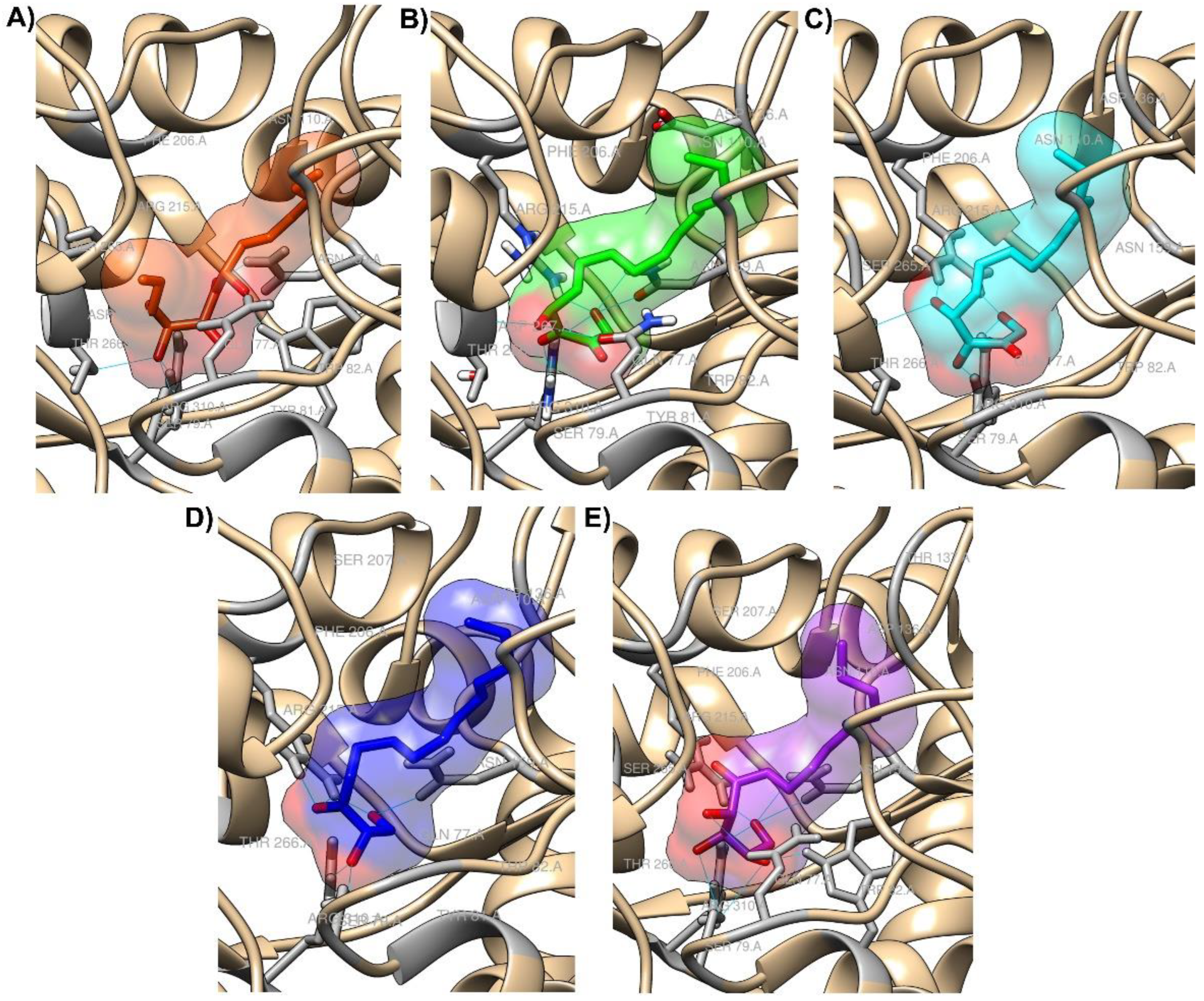
2.2. Molecular Docking
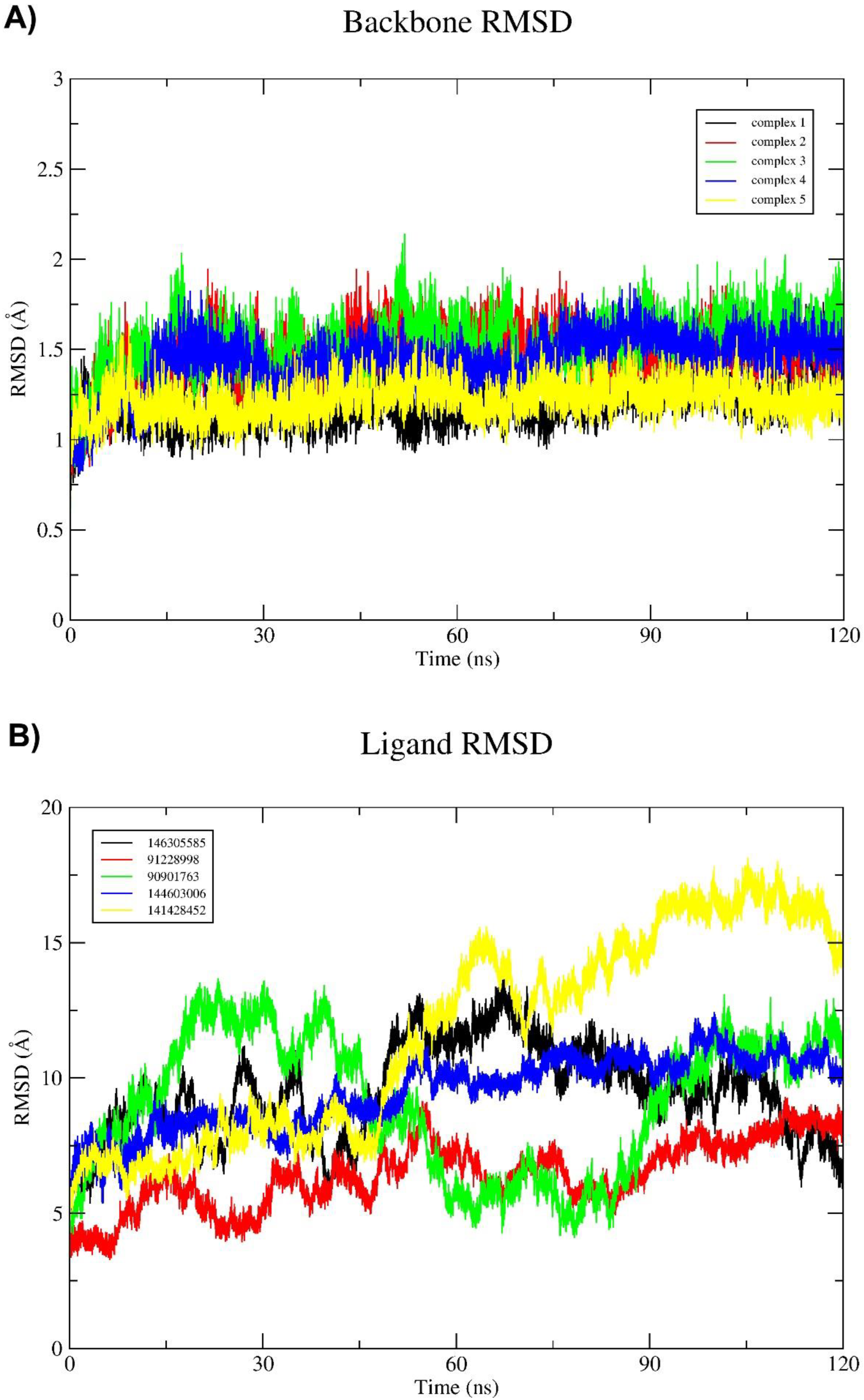
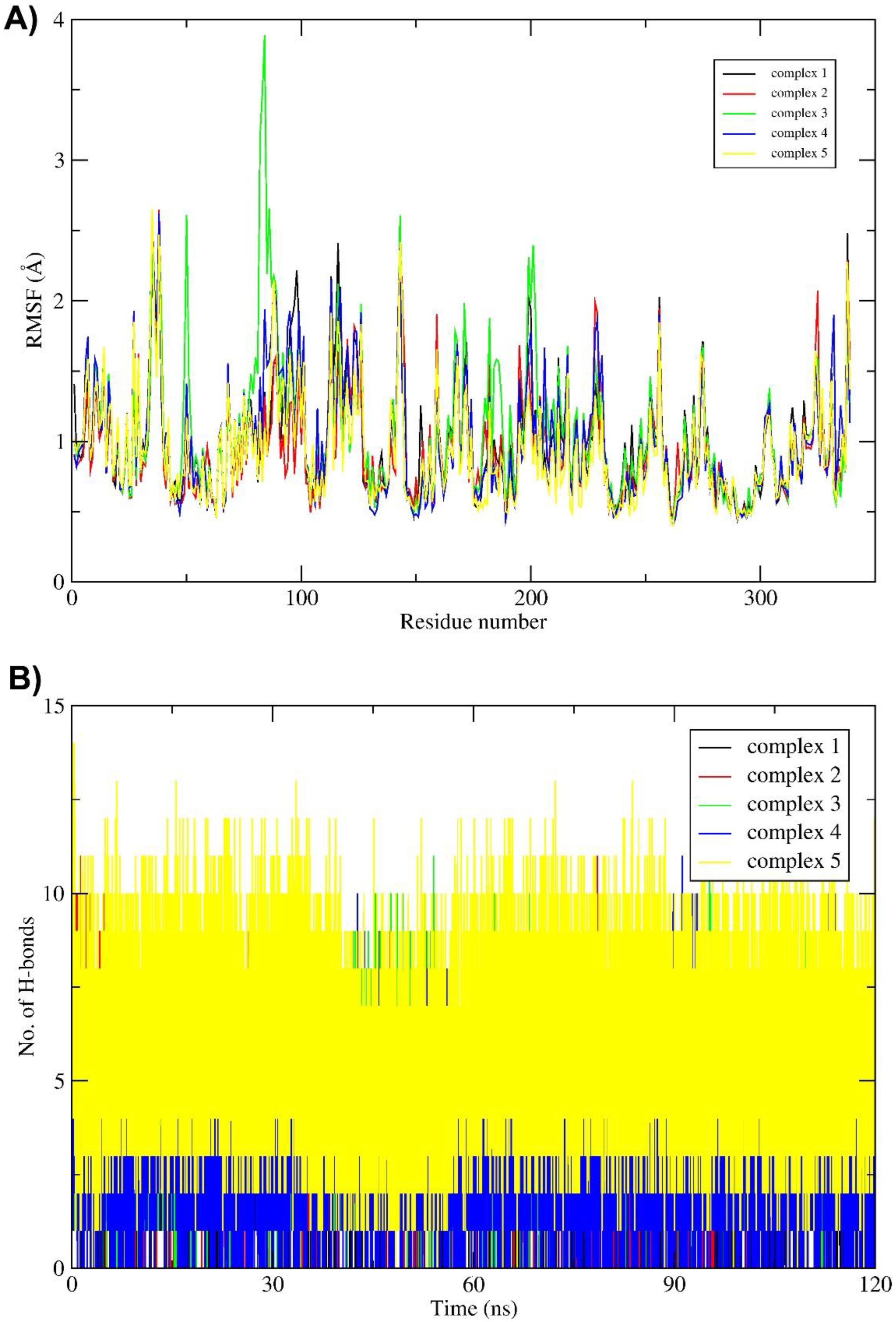
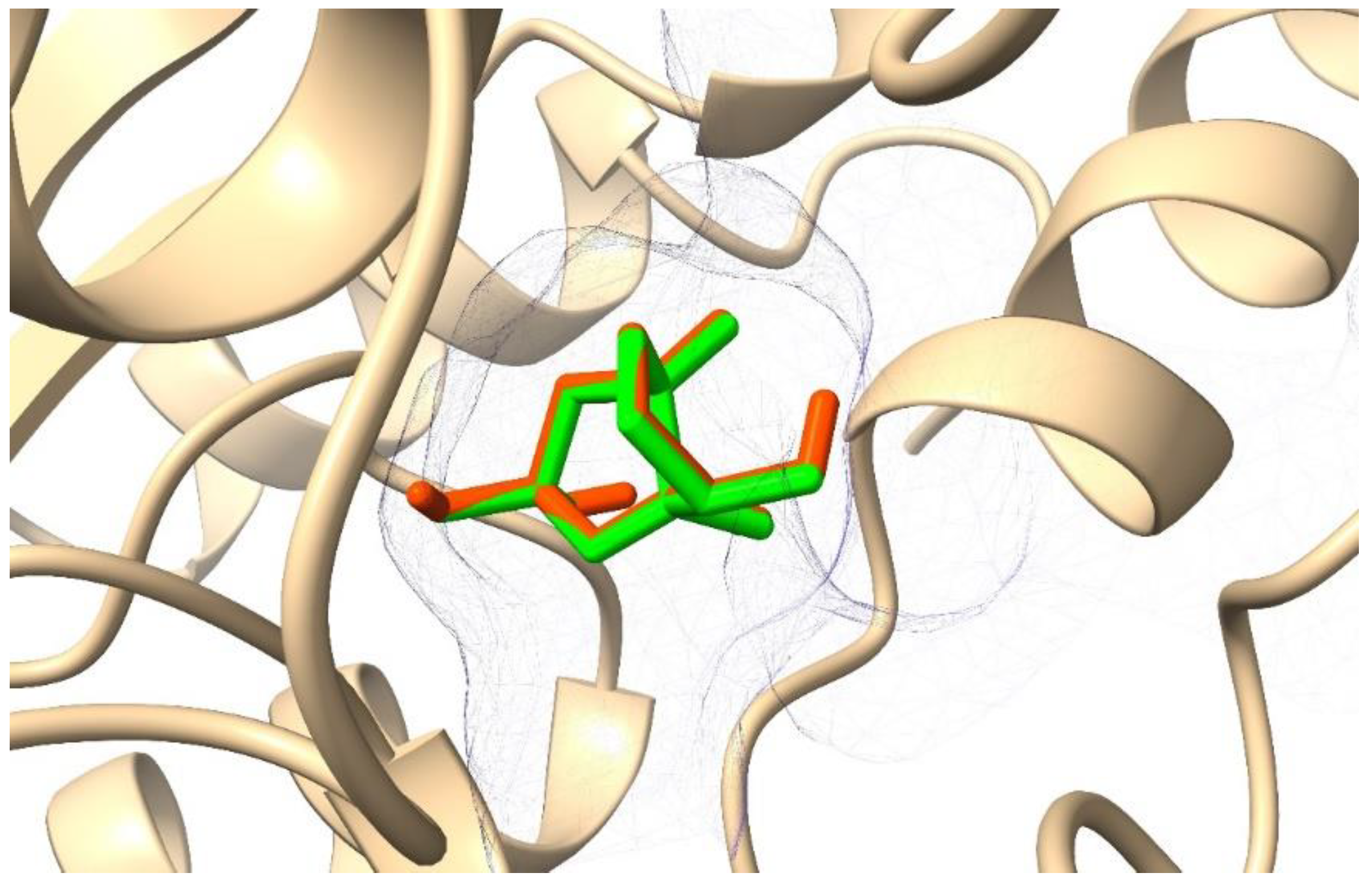
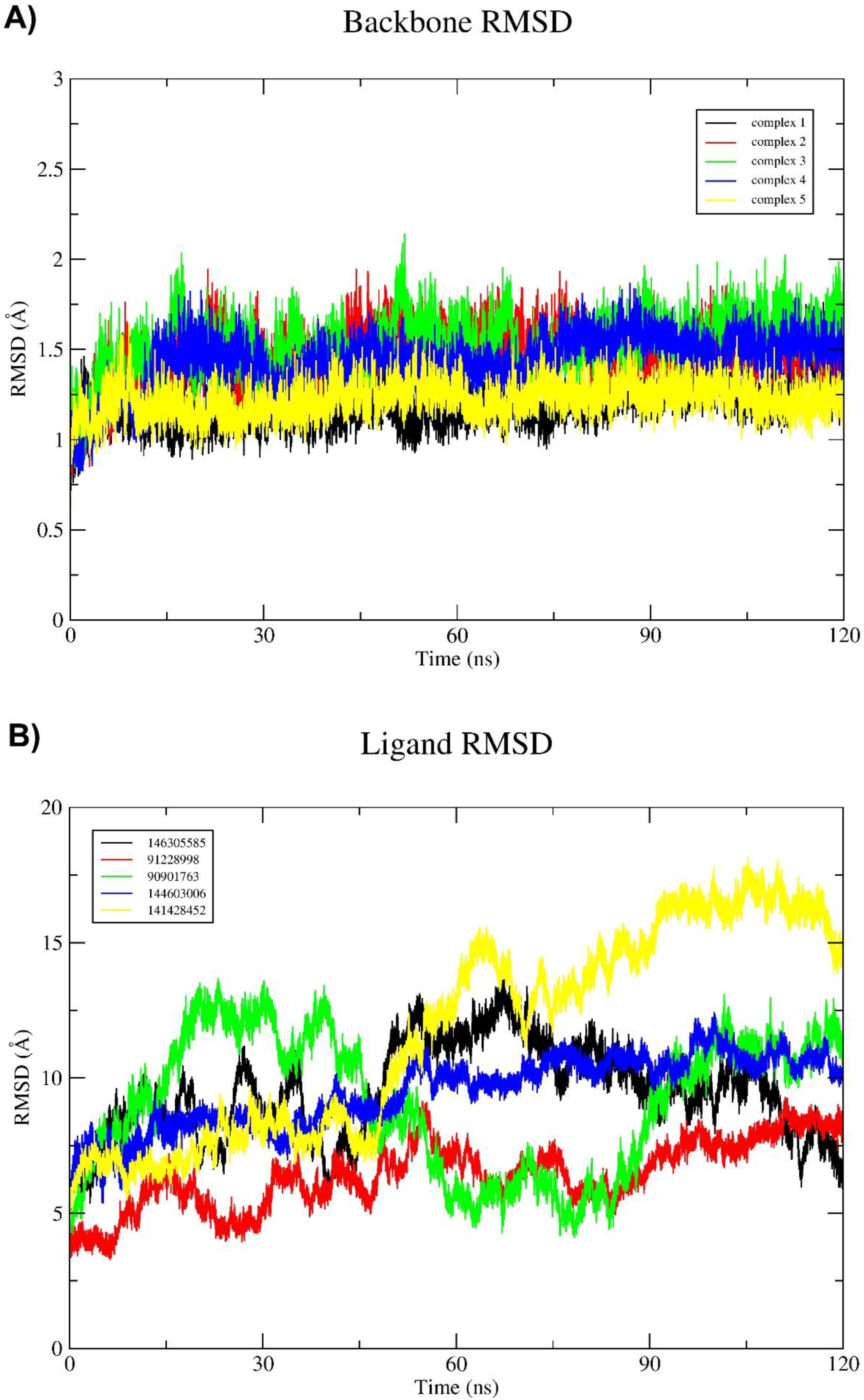
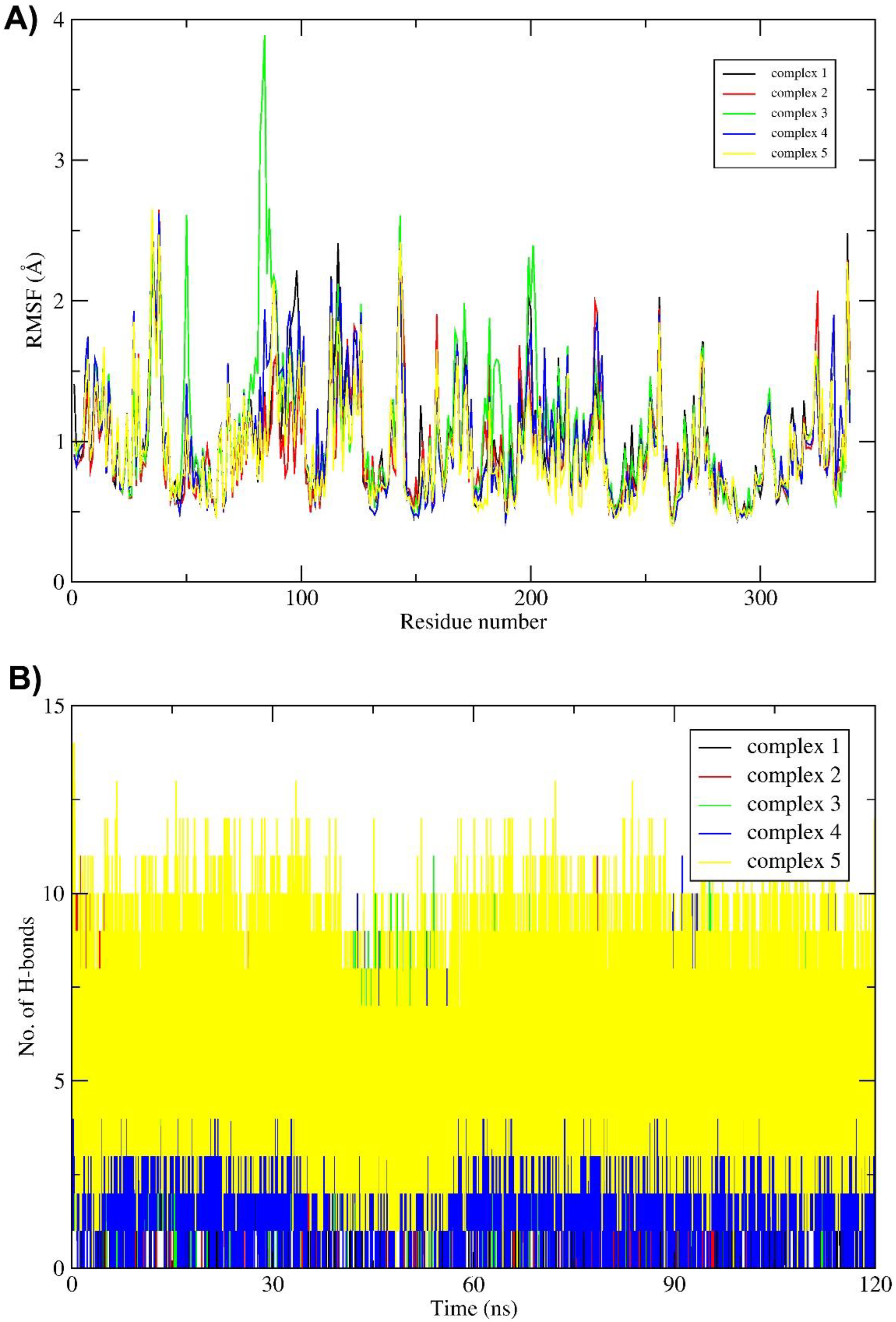
2.3. Molecular Dynamics (MD) Simulation and Binding Free Energy Calculation
3. Discussion
4. Materials and Methods
4.1. Preparation of the Ligand-Based Compound Library
4.1.1. Molecular Fingerprinting Analysis
4.1.2. Compound Filtering
4.2. Protein Preparation
4.3. Molecular Docking and Rescoring Calculation
4.4. Molecular Dynamic (MD) Simulations
4.5. Binding Free Energy Calculations and Decomposition
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Taga, M.E.; Bassler, B.L. Chemical Communication among Bacteria. Proc. Natl. Acad. Sci. USA 2003, 100, 14549–14554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, G.J.; Novick, R.P. Peptide Signaling in Staphylococcus Aureus and Other Gram-Positive Bacteria. Peptides 2004, 25, 1389–1403. [Google Scholar] [CrossRef]
- Kleerebezem, M.; Quadri, L.E.N.; Kuipers, O.P.; de Vos, W.M. Quorum Sensing by Peptide Pheromones and Two-component Signal-transduction Systems in Gram-positive Bacteria. Mol. Microbiol. 1997, 24, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Eberl, L. N-Acyl Homoserinelactone-Mediated Gene Regulation in Gram-Negative Bacteria. Syst. Appl. Microbiol. 1999, 22, 493–506. [Google Scholar] [CrossRef]
- Fuqua, C.; Parsek, M.R.; Greenberg, E.P. Regulation of gene expression by cell-to-cell communication: Acyl-homoserine lactone quorum sensing. Annu. Rev. Genet. 2001, 35, 439–468. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Daniel, R.; Wagner-Döbler, I.; Zeng, A.-P. Is Autoinducer-2 a Universal Signal for Interspecies Communication: A Comparative Genomic and Phylogenetic Analysis of the Synthesis and Signal Transduction Pathways. BMC Evol. Biol. 2004, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendeville, A.; Winzer, K.; Heurlier, K.; Tang, C.M.; Hardie, K.R. Making “sense” of Metabolism: Autoinducer-2, LUXS and Pathogenic Bacteria. Nat. Rev. Microbiol. 2005, 3, 383–396. [Google Scholar] [CrossRef]
- Xavier, K.B.; Bassler, B.L. LuxS Quorum Sensing: More than Just a Numbers Game. Curr. Opin. Microbiol. 2003, 6, 191–197. [Google Scholar] [CrossRef]
- Surette, M.G.; Miller, M.B.; Bassler, B.L. Quorum Sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: A New Family of Genes Responsible for Autoinducer Production. Proc. Natl. Acad. Sci. USA 1999, 96, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Keller, L.; Surette, M.G. Communication in Bacteria: An Ecological and Evolutionary Perspective. Nat. Rev. Microbiol. 2006, 4, 249–258. [Google Scholar] [CrossRef]
- Saini, R.; Saini, S.; Sharma, S. Biofilm: A Dental Microbial Infection. J. Nat. Sci. Biol. Med. 2011, 2, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Ryu, E.-J.; Li, L.; Choi, B.-K.; Kim, B.M. New Bicyclic Brominated Furanones as Potent Autoinducer-2 Quorum-Sensing Inhibitors against Bacterial Biofilm Formation. Eur. J. Med. Chem. 2017, 137, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Ben Amara, H.; Song, H.Y.; Ryu, E.; Park, J.S.; Schwarz, F.; Kim, B.M.; Choi, B.-K.; Koo, K.-T. Effects of Quorum-Sensing Inhibition on Experimental Periodontitis Induced by Mixed Infection in Mice. Eur. J. Oral Sci. 2018, 126, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Choi, Y.J.; Lee, S.H.; Jun, H.K.; Choi, B.K. Autoinducer 2 of Fusobacterium nucleatum as a Target Molecule to Inhibit Biofilm Formation of Periodontopathogens. Arch. Oral Biol. 2013, 58, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Ryu, E.J.; Sim, J.; Sim, J.; Lee, J.; Choi, B.K. D-Galactose as an Autoinducer 2 Inhibitor to Control the Biofilm Formation of Periodontopathogens. J. Microbiol. 2016, 54, 632–637. [Google Scholar] [CrossRef]
- Choi, B.K.; Ryu, E.J.; Sim, J.H.; Kim, B.M.; Lee, J.; Sim, J. Method of Inhibiting Quorum Sensing Using D-Galactose. U.S. Patent 10,292,993 B2, 21 May 2019. pp. 1–21. [Google Scholar]
- Brackman, G.; al Quntar, A.A.A.; Enk, C.D.; Karalic, I.; Nelis, H.J.; van Calenbergh, S.; Srebnik, M.; Coenye, T. Synthesis and Evaluation of Thiazolidinedione and Dioxazaborocane Analogues as Inhibitors of AI-2 Quorum Sensing in Vibrio harveyi. Bioorganic Med. Chem. 2013, 21, 660–667. [Google Scholar] [CrossRef]
- Karnjana, K.; Nobsathian, S.; Soowannayan, C.; Zhao, W.; Tang, Y.J.; Wongprasert, K. Purification and Evaluation of N-Benzyl Cinnamamide from Red Seaweed Gracilaria Fisheri as an Inhibitor of Vibrio harveyi AI-2 Quorum Sensing. Mar. Drugs 2020, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Helmy, Y.A.; Kathayat, D.; Deblais, L.; Srivastava, V.; Closs, G.; Tokarski, R.J.; Ayinde, O.; Fuchs, J.R.; Rajashekara, G. Evaluation of Novel Quorum Sensing Inhibitors Targeting Auto-Inducer 2 (AI-2) for the Control of Avian Pathogenic Escherichia Coli Infections in Chickens. Microbiol. Spectr. 2022, 10. [Google Scholar] [CrossRef]
- Fernandes, S.; Borges, A.; Gomes, I.B.; Sousa, S.F.; Simões, M. In Silico Screening and In Vitro Validation of Natural-Based LuxS Inhibitors. Med. Sci. Forum 2022, 2022, 2029564. [Google Scholar] [CrossRef]
- Ali, F.; Yao, Z.; Li, W.; Sun, L.; Lin, W.; Lin, X. In-Silico Prediction and Modeling of the Quorum Sensing Luxs Protein and Inhibition of AI-2 Biosynthesis in Aeromonas Hydrophila. Molecules 2018, 23, 2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byeon, J.Y.; Sim, J.; Ryu, E.J.; Sim, J.; Lee, H.; Cho, K.H.; Choi, B.K.; Lee, J. In Silico Development of Quorum-Sensing Inhibitors. Bull. Korean Chem. Soc. 2017, 38, 728–734. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Witek, J.; Landrum, G.A.; Riniker, S. Improving Conformer Generation for Small Rings and Macrocycles Based on Distance Geometry and Experimental Torsional-Angle Preferences. J. Chem. Inf. Modeling 2020, 60, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Coop, A.; Polli, J.E.; MacKerell, A.D. Recent Advances in Ligand-Based Drug Design: Relevance and Utility of the Conformationally Sampled Pharmacophore Approach. Curr. Comput. Aided-Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2022-2. Maestro, Schrödinger, LLC.: 2021. Available online: https://www.schrodinger.com/citations (accessed on 12 May 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Rastelli, G.; Rio, A.d.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2009, 31, 797–810. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Available online: http://www.Rdkit.Org (accessed on 12 May 2022).
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riniker, S.; Landrum, G.A. Better Informed Distance Geometry: Using What We Know to Improve Conformation Generation. J. Chem. Inf. Modeling 2015, 55, 2562–2574. [Google Scholar] [CrossRef]
- Sud, M. MayaChemTools: An Open Source Package for Computational Drug Discovery. J. Chem. Inf. Modeling 2016, 56, 2292–2297. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Modeling 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M. Local Alignment of Ligand Binding Sites in Proteins for Polypharmacology and Drug Repositioning. Methods Mol. Biol. 2017, 1611, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of Machine-Learning Scoring Functions in Structure-Based Virtual Screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. Improving AutoDock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Mol. Inform. 2015, 34, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.R.; Pantano, S. Split the Charge Difference in Two! A Rule of Thumb for Adding Proper Amounts of Ions in MD Simulations. J. Chem. Theory Comput. 2020, 16, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016. University of California; San Francisco: 2016. Available online: https://ambermd.org/doc12/Amber16.pdf (accessed on 12 May 2022).
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1998, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Ferenczy, G.G. Thermodynamics and Kinetics of Drug Binding; Keserü, G.M., Swinney, D.C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; ISBN 9783527673025. [Google Scholar]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial Number | Compound Name (PubChem CID) | Structure of the Compounds | Smina (Binding Energy in kcal/mol) | RF_Score (Binding Affinity in pKd) |
|---|---|---|---|---|
| 1 | 5,8-dihydroxytetradecane-6,7-dione (146305585) |  | −7.7 | 6.76 |
| 2 | 1,4-dihydroxypentadecane-2,3-dione (91228998) | 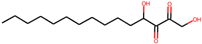 | −9.3 | 7.10 |
| 3 | 1,4-dihydroxytetradecane-2,3-dione(90901763) | 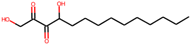 | −9.1 | 6.70 |
| 4 | 1,2-dihydroxytetradecan-3-one(144603006) | 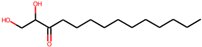 | −8.6 | 6.39 |
| 5 | (2R,3S,4R)-1,2,3,4-tetrahydroxypentadecan-5-one(141428452) | 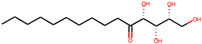 | −9.0 | 6.84 |
| Serial No. | Compound Name (PubChem CID) | H-Bonds | ΔGBIND * (kcal/mol) | |
|---|---|---|---|---|
| Mean | s.d. | |||
| 1 | 5,8-dihydroxytetradecane-6,7-dione (146305585) | 2.52 | 1.28 | −38.17 |
| 2 | 1,4-dihydroxypentadecane-2,3-dione (91228998) | 3.43 | 1.32 | −44.09 |
| 3 | 1,4-dihydroxytetradecane-2,3-dione (90901763) | 3.6 | 1.36 | −40.66 |
| 4 | 1,2-dihydroxytetradecan-3-one (144603006) | 2.99 | 1.51 | −41.77 |
| 5 | (2R,3S,4R)-1,2,3,4-tetrahydroxypentadecan-5-one (141428452) | 5.64 | 1.74 | −49.43 |
| Complex 1 | Complex 2 | Complex 3 | Complex 4 | Complex 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | ΔGBIND | Residue | ∆GBIND | Residue | ∆GBIND | Residue | ∆GBIND | Residue | ∆GBIND |
| Val 57 | −9.75 | Glu 37 | −8.74 | Glu 37 | −6 | Asp 36 | −8.24 | Pro 58 | −9.5 |
| Ser 60 | −8.5 | Ala 54 | −6.69 | Glu 40 | −7.99 | Pro 58 | −6 | Lys 61 | −8.49 |
| Leu 96 | −8.24 | Thr 63 | −6.49 | Glu 50 | −6.25 | Ser 60 | −5 | Gln 64 | −5.5 |
| Ile 98 | −7.75 | Gln 64 | −5.99 | Val 57 | −4.25 | Thr 63 | −7.74 | Leu 96 | −7 |
| Thr 199 | −7.5 | Phe 194 | −9.74 | Pro 58 | −5.49 | Leu 96 | −4.74 | Asn 97 | −7.1 |
| His 255 | −7.75 | Asp 257 | −8.5 | Leu 59 | −6.5 | Asn 97 | −3.74 | Leu 148 | −5.25 |
| Asp 257 | −9.49 | Ala 277 | −6.74 | Pro 66 | −4.49 | Ile 98 | −4 | Pro 256 | −8.49 |
| Asp 259 | −7.49 | Leu 279 | −8.5 | Ile 98 | −9.25 | Asn 99 | −5.74 | MET 311 | −7.49 |
| Leu 279 | −9.25 | Leu 327 | −8.99 | Asn 99 | −6.99 | Phe 194 | −3.49 | Ile 323 | −7.49 |
| Leu 355 | −9.5 | Asp 329 | −5.99 | Val 223 | −5.5 | Lys 197 | −8.99 | GLY 338 | −5.75 |
| Complex 1 | Complex 2 | Complex 3 | Complex 4 | Complex 5 | |
|---|---|---|---|---|---|
| ΔEvdW | −42.01 | −46.27 | −39.85 | −44.43 | −45.26 |
| ΔEelectrostatic | −27.42 | −27.32 | −35.39 | −19.003 | −45.47 |
| ΔGGB | 37.83 | 36.64 | 41.02 | 28.28 | 48.57 |
| ΔGSA | −6.56 | −7.14 | −6.44 | −6.62 | −7.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shekarappa, S.B.; Rimac, H.; Lee, J. In Silico Screening of Quorum Sensing Inhibitor Candidates Obtained by Chemical Similarity Search. Molecules 2022, 27, 4887. https://doi.org/10.3390/molecules27154887
Shekarappa SB, Rimac H, Lee J. In Silico Screening of Quorum Sensing Inhibitor Candidates Obtained by Chemical Similarity Search. Molecules. 2022; 27(15):4887. https://doi.org/10.3390/molecules27154887
Chicago/Turabian StyleShekarappa, Sharath Belenahalli, Hrvoje Rimac, and Julian Lee. 2022. "In Silico Screening of Quorum Sensing Inhibitor Candidates Obtained by Chemical Similarity Search" Molecules 27, no. 15: 4887. https://doi.org/10.3390/molecules27154887
APA StyleShekarappa, S. B., Rimac, H., & Lee, J. (2022). In Silico Screening of Quorum Sensing Inhibitor Candidates Obtained by Chemical Similarity Search. Molecules, 27(15), 4887. https://doi.org/10.3390/molecules27154887