
Marine Cyanobacteria as Sources of Lead Anticancer Compounds: A Review of Families of Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects

 ,
,  , ,
, ,  and
and
Abstract
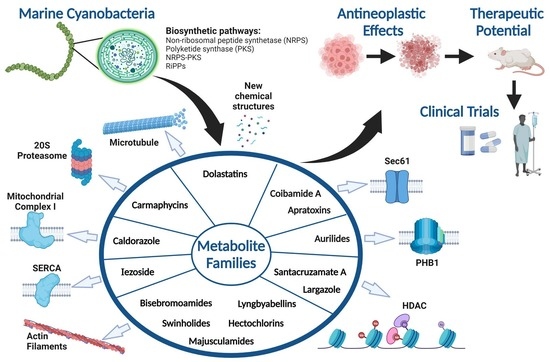
:
1. Introduction
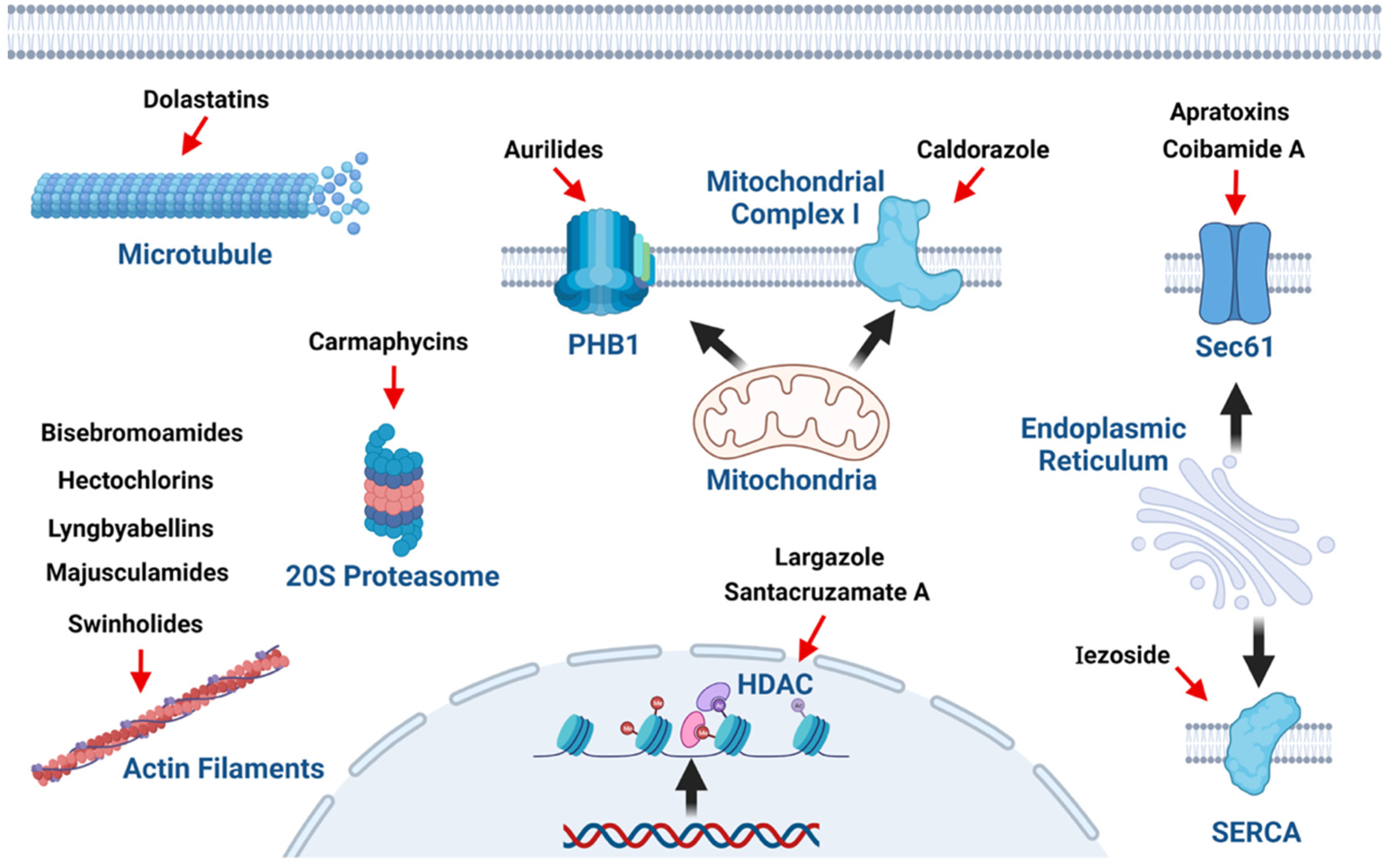
2. Marine Cyanobacterial Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects
2.1. Glycolipids
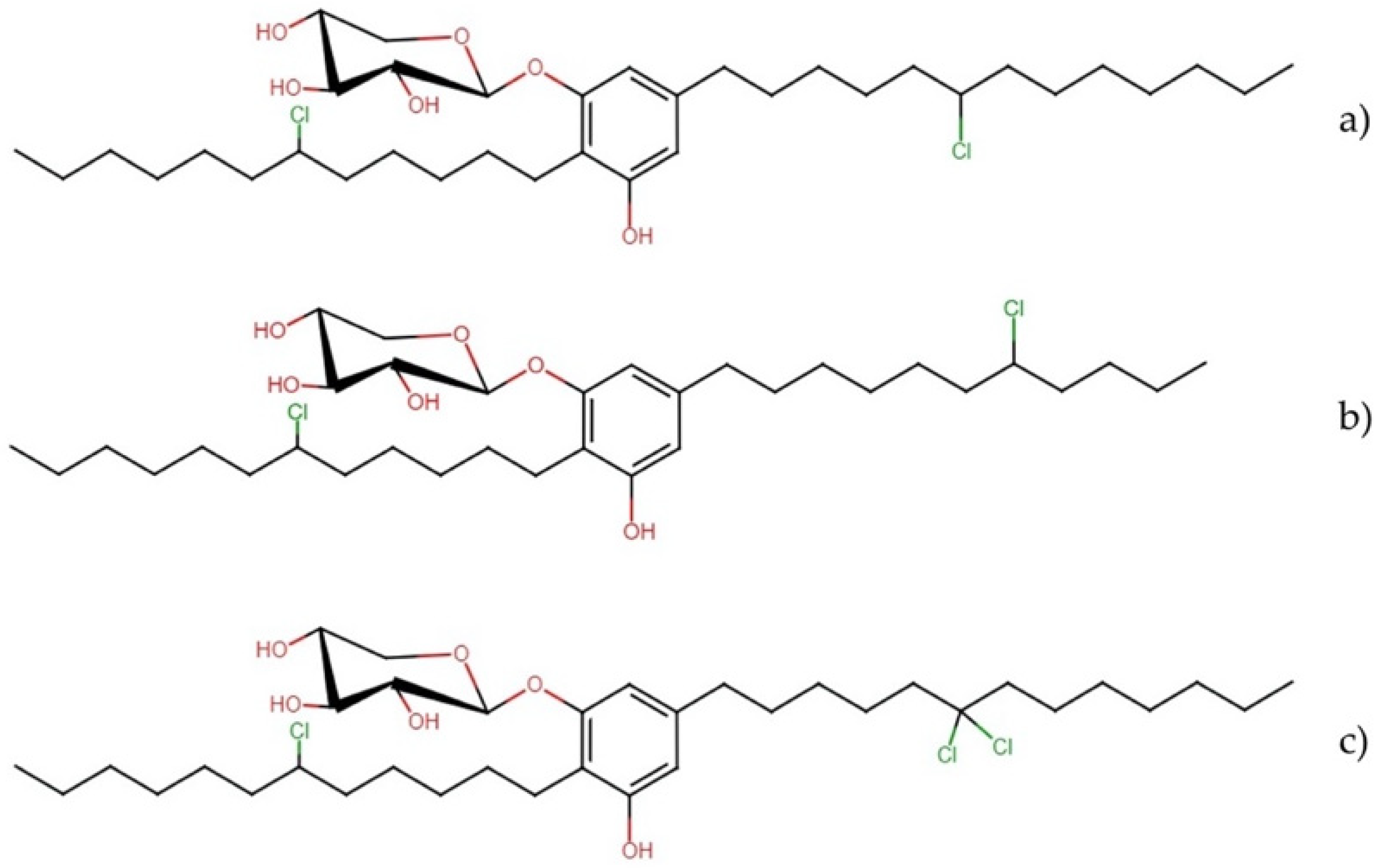
Bartolosides
2.2. Macrolides
2.2.1. Caylobolides
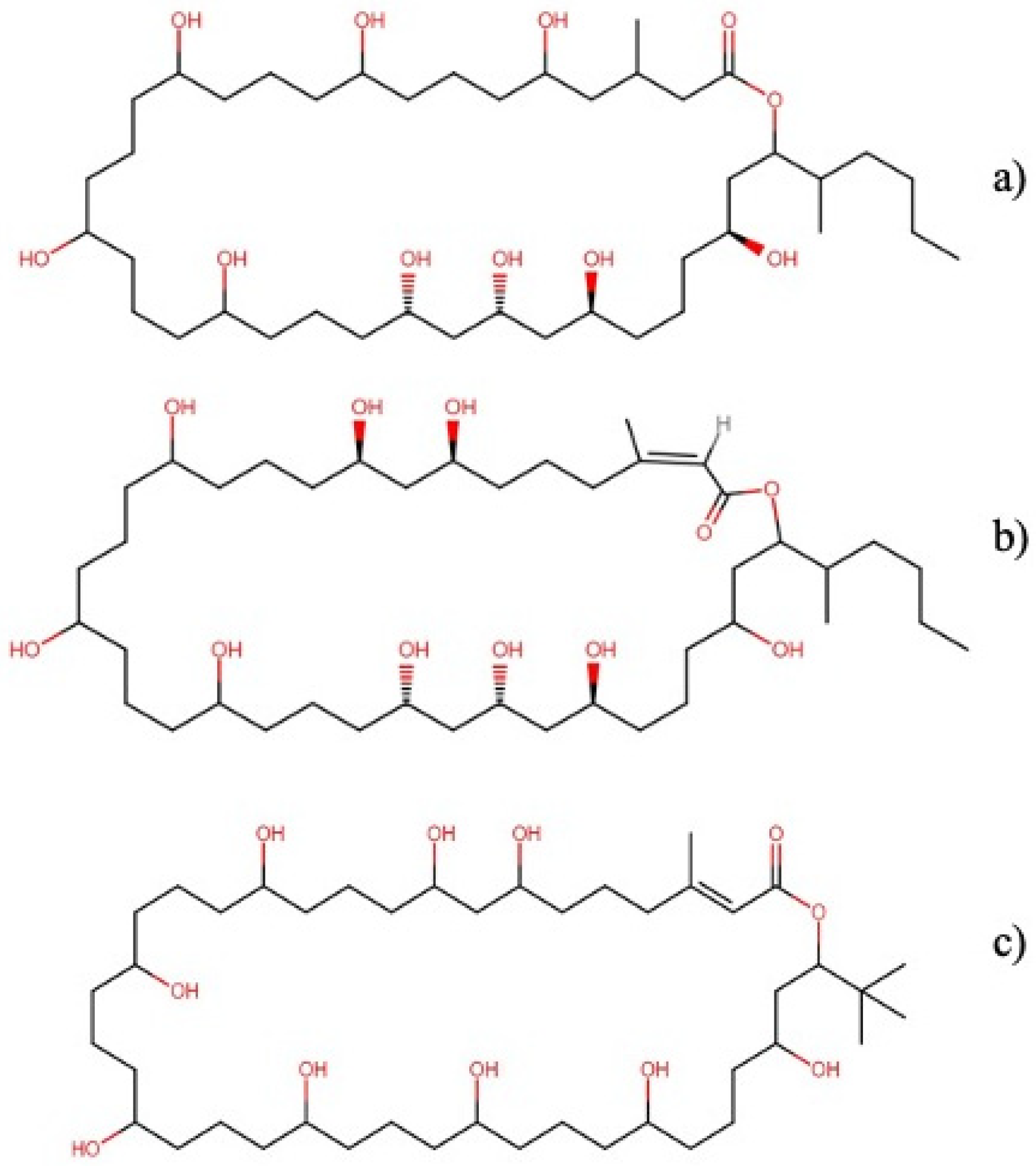
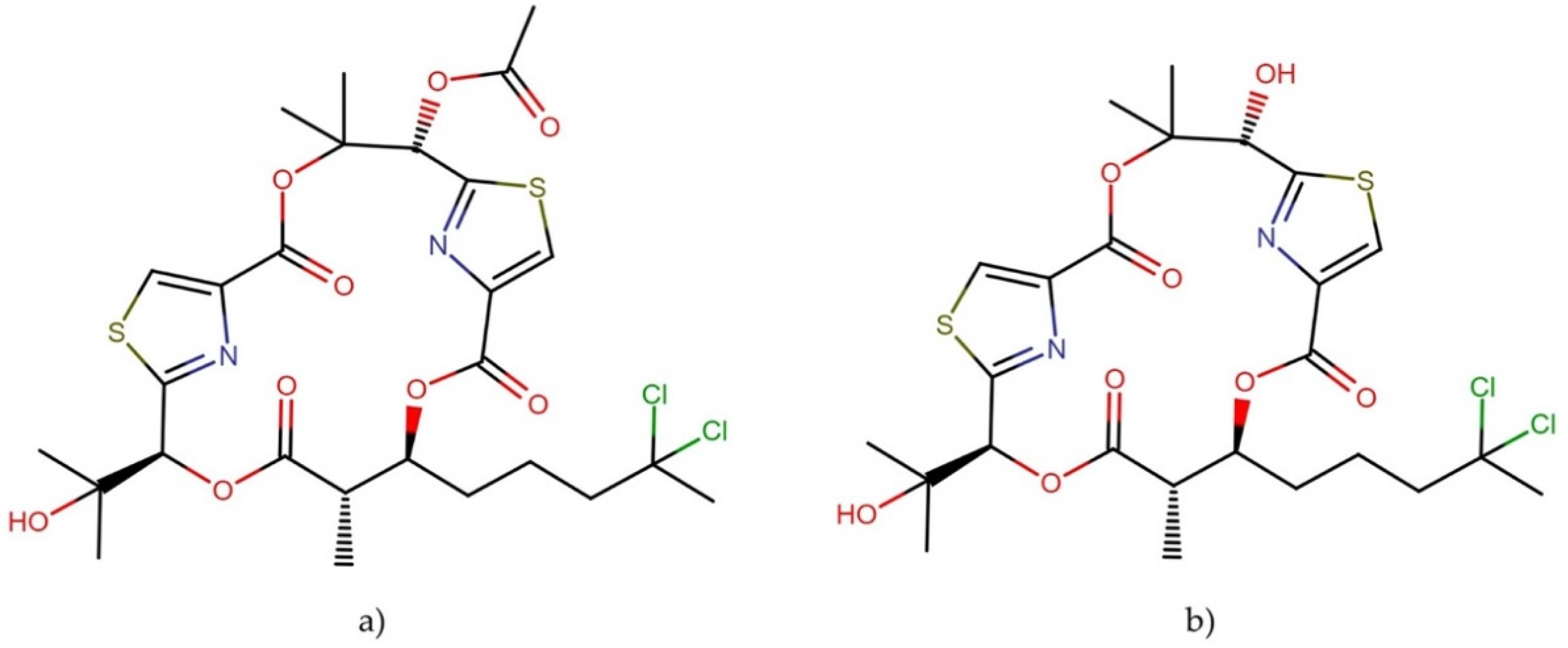
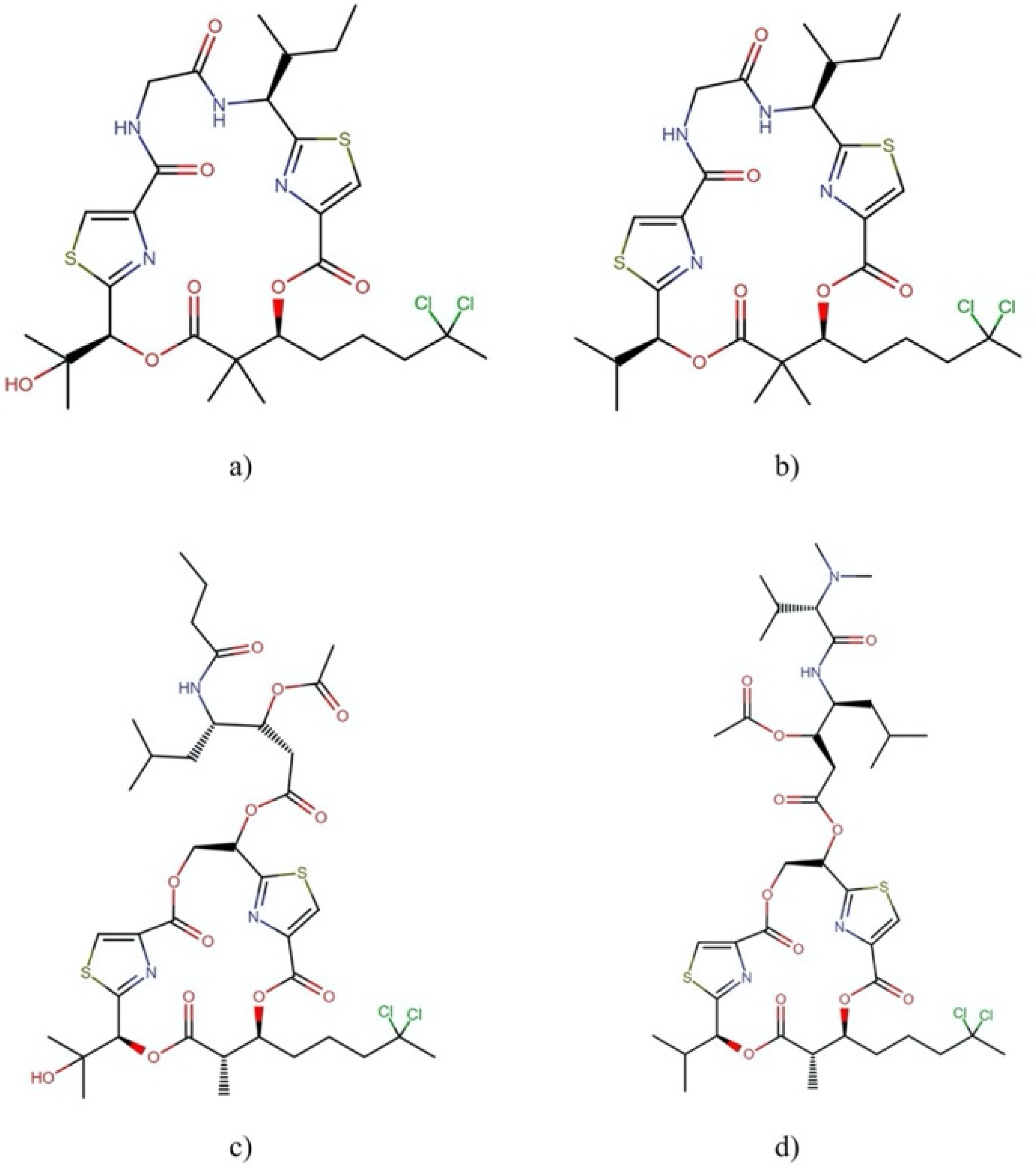
2.2.2. Swinholide-Type
2.3. Linear Peptides
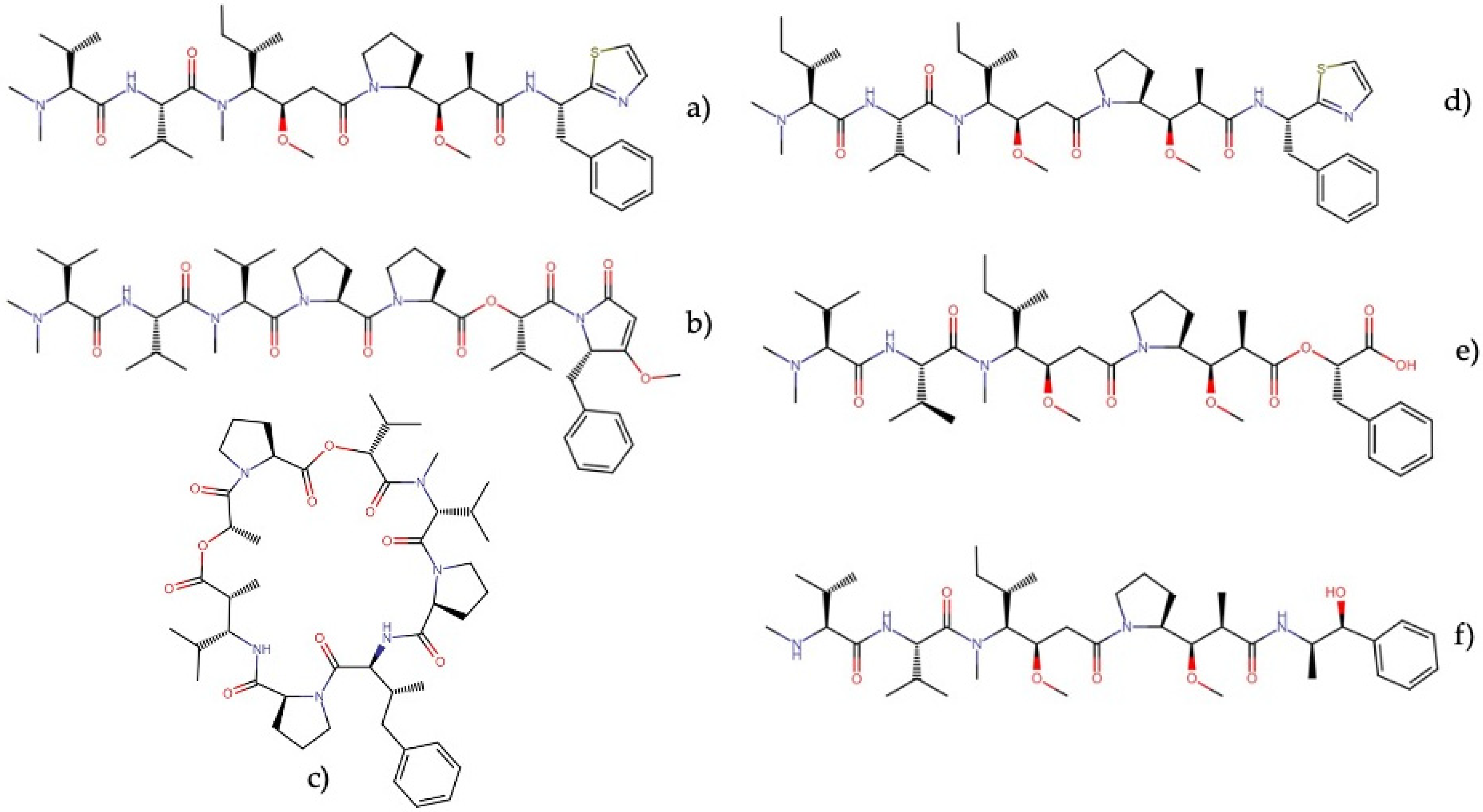
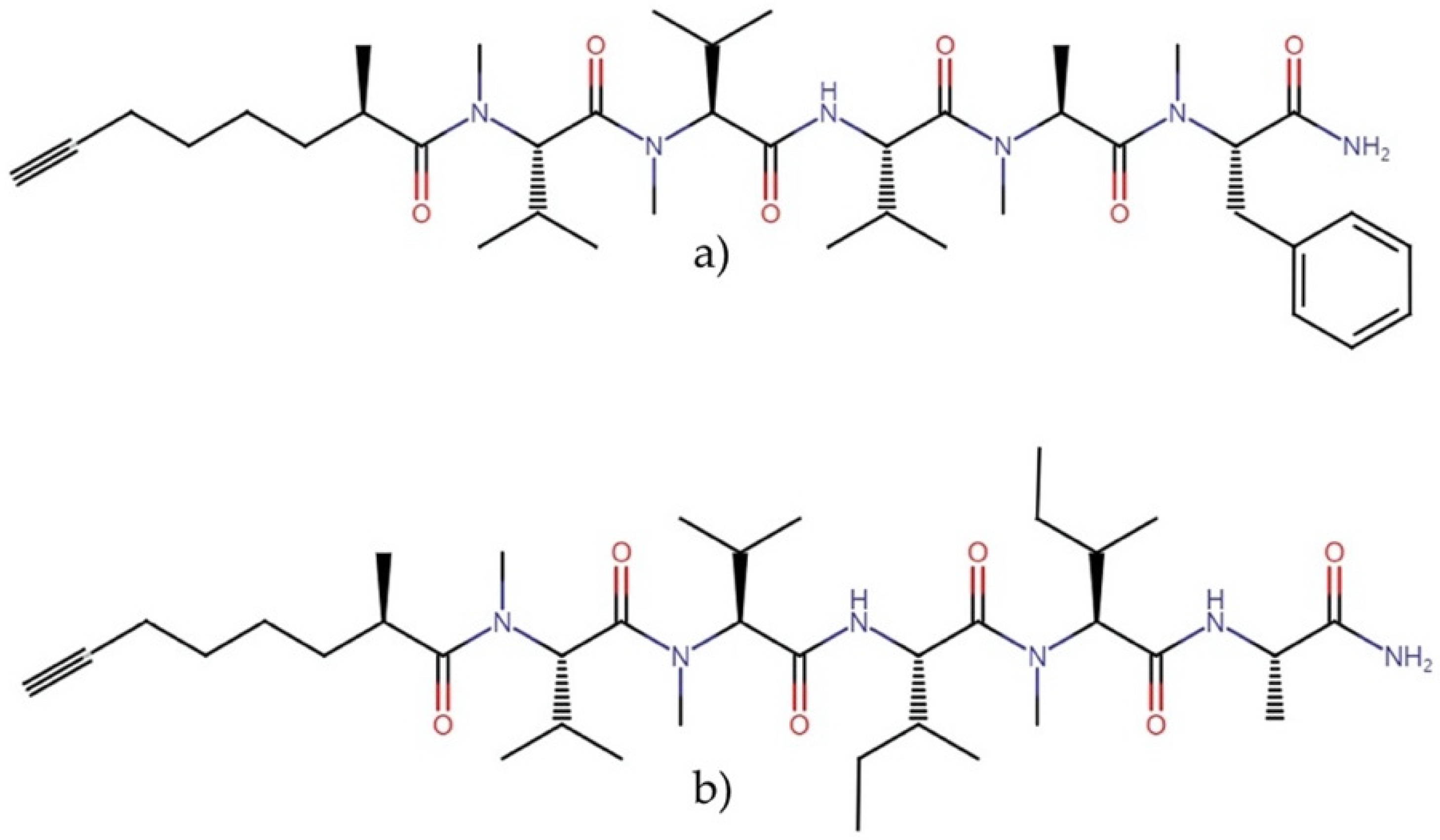
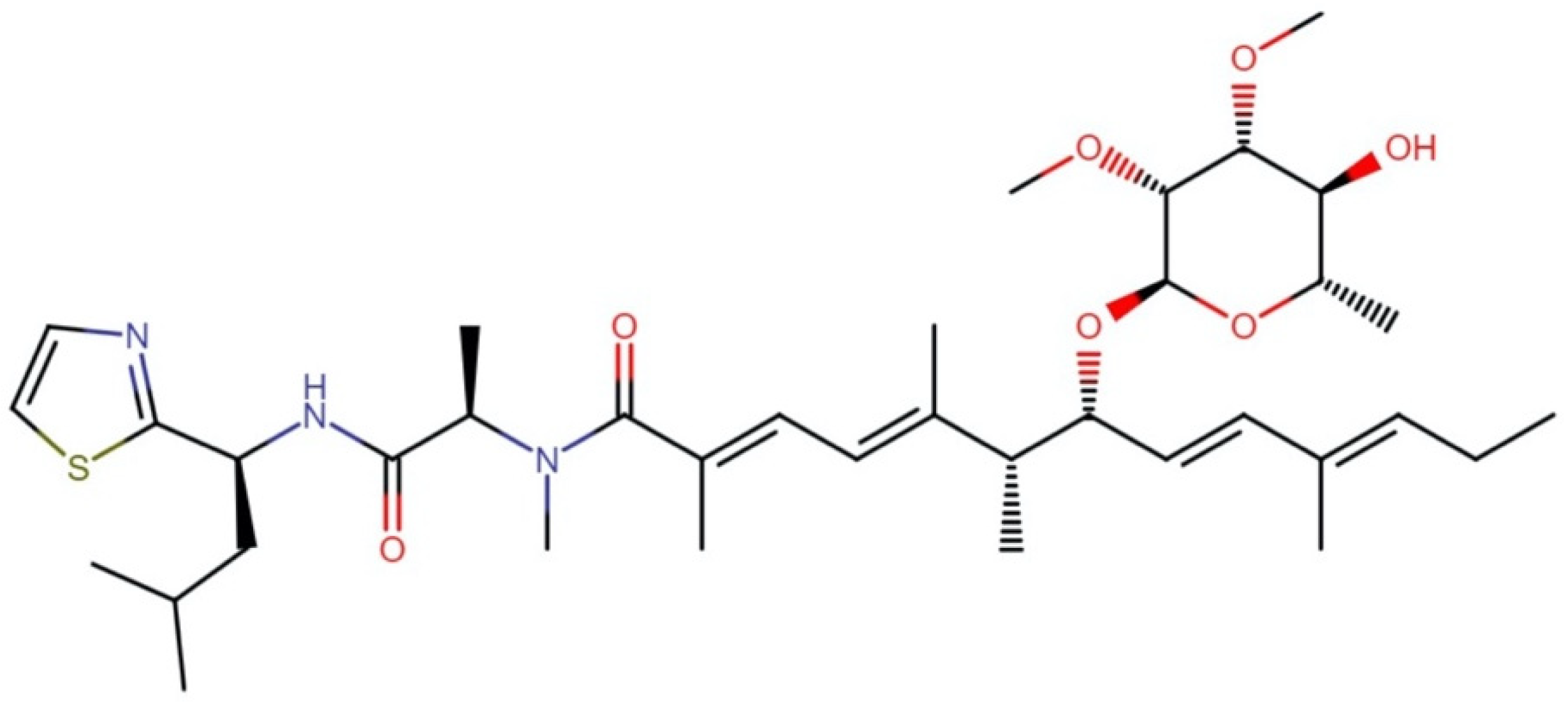
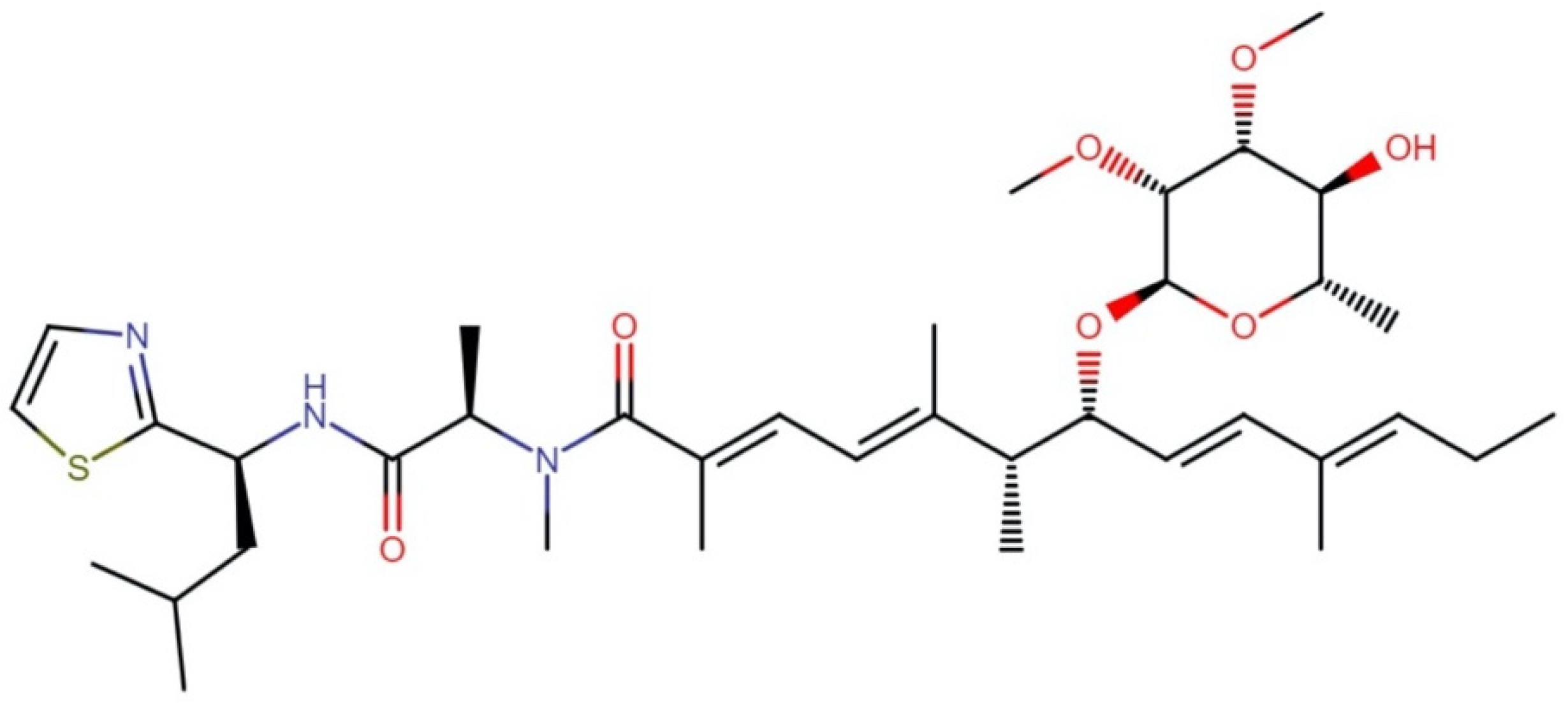
2.3.1. Bisebromoamides
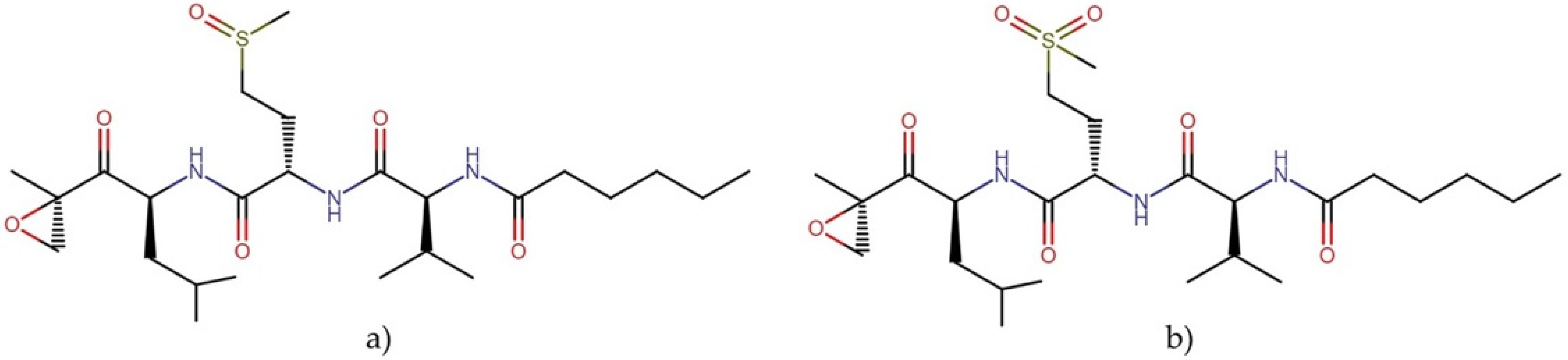
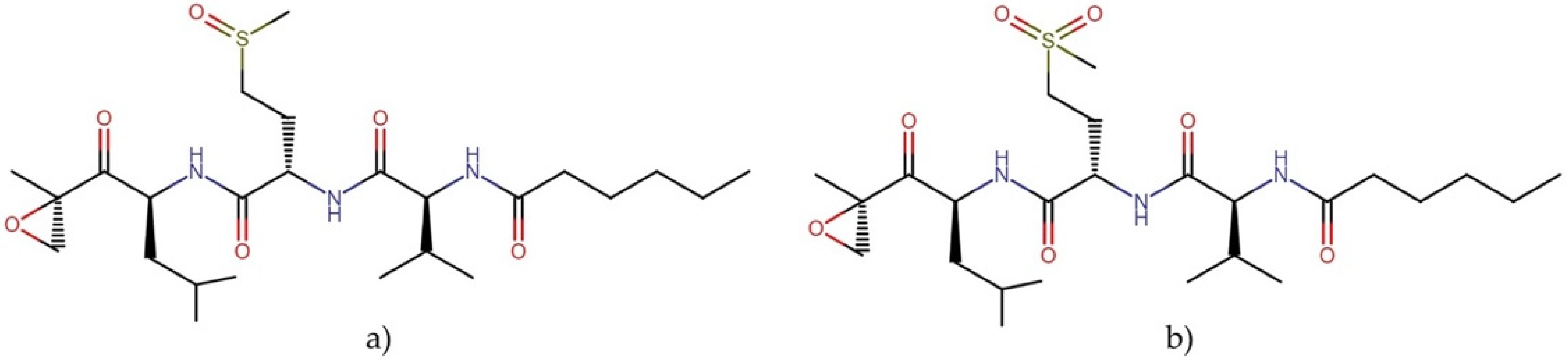
2.3.2. Carmaphycins
2.4. Depsipeptides
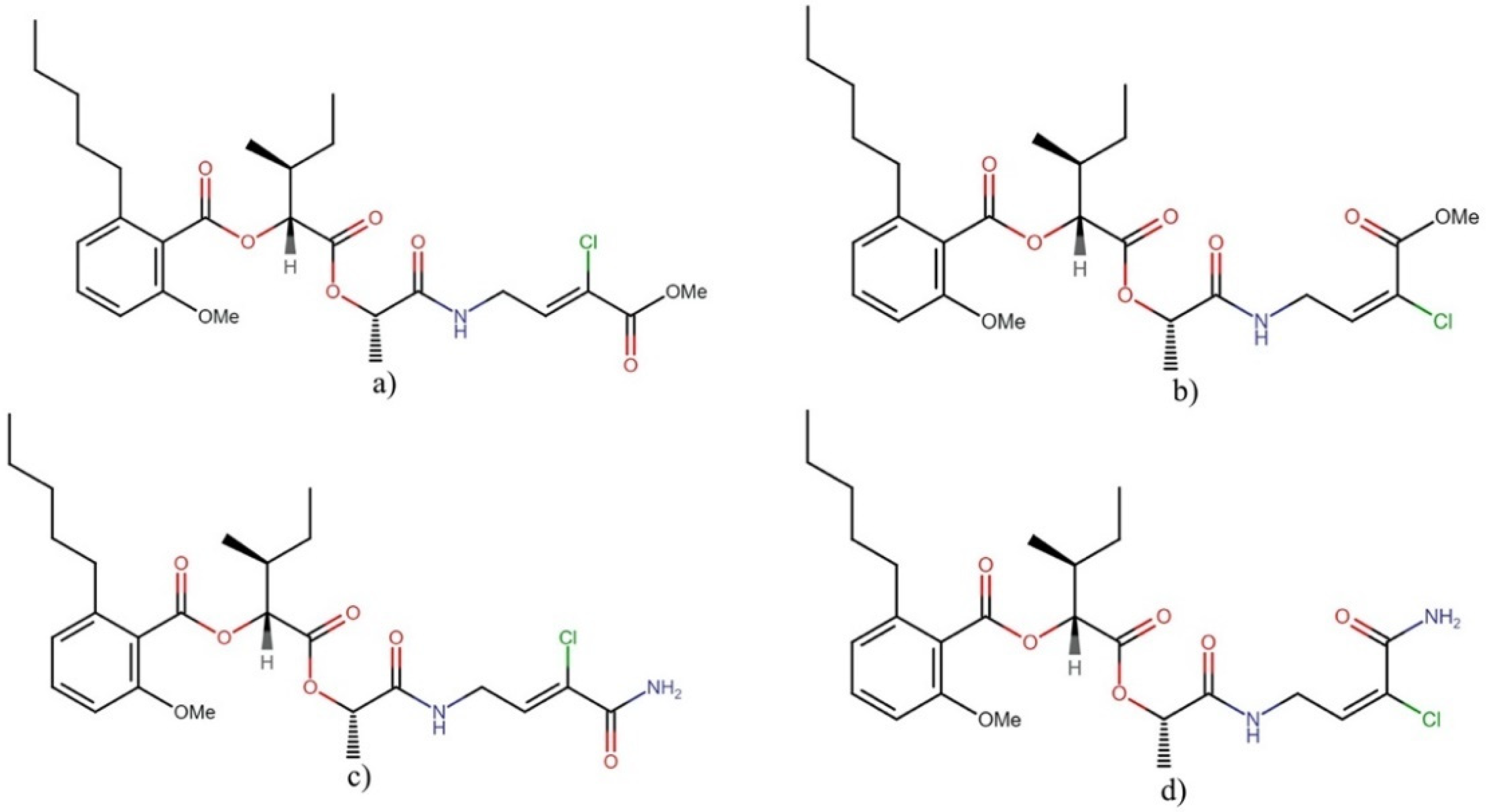
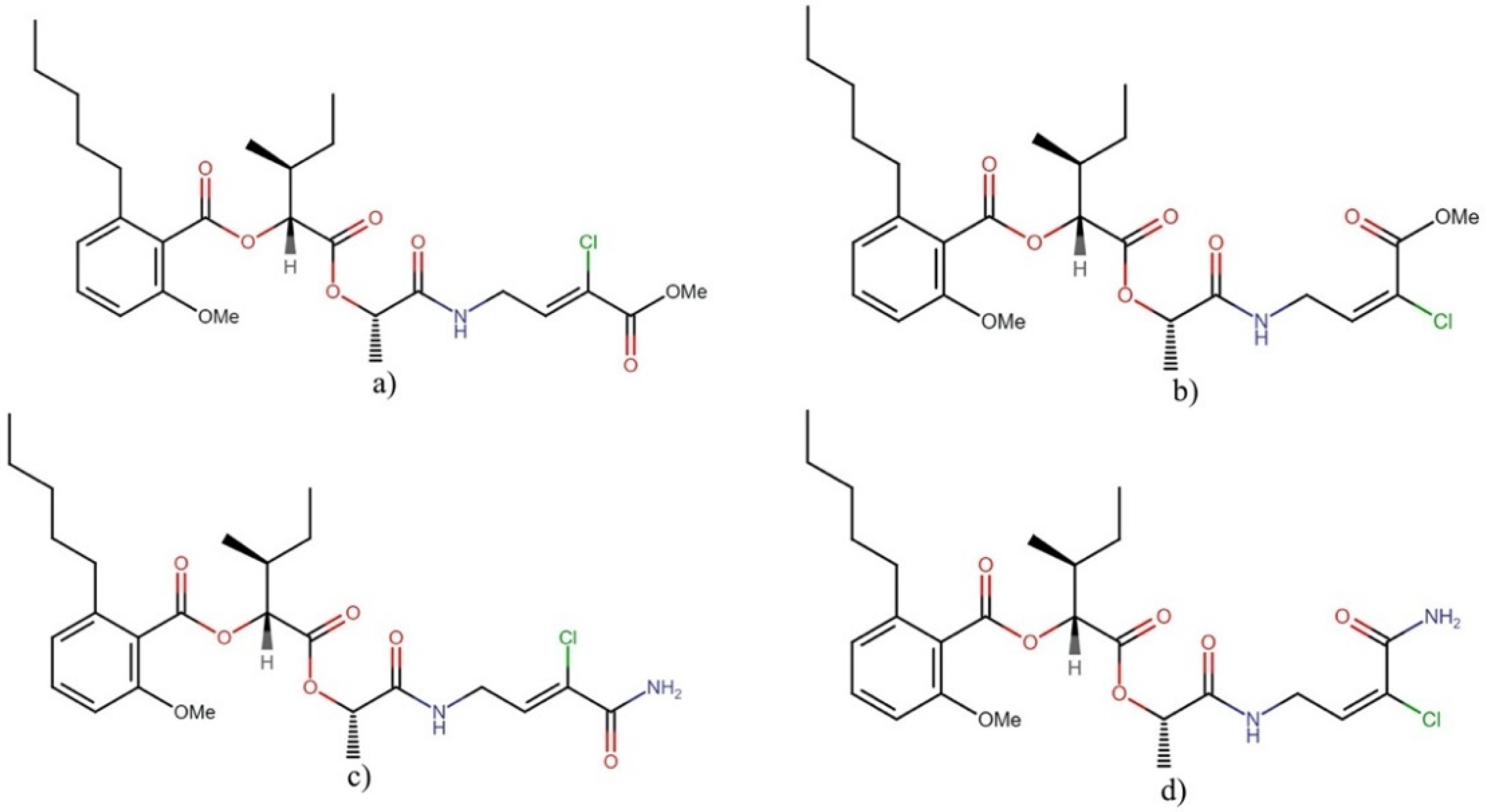
Anaenamides
2.5. Cyclic Depsipeptides
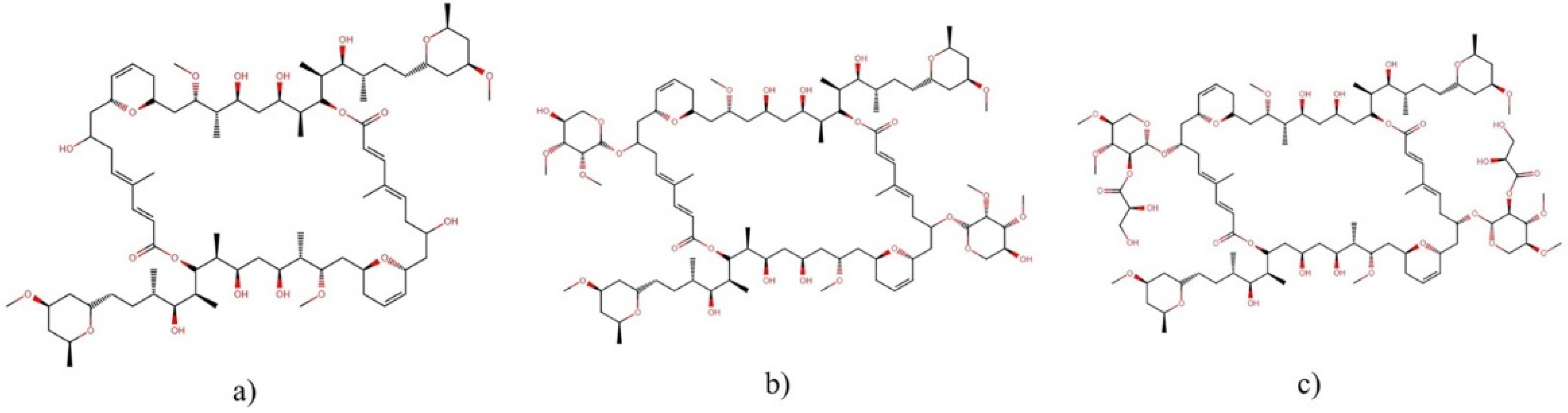
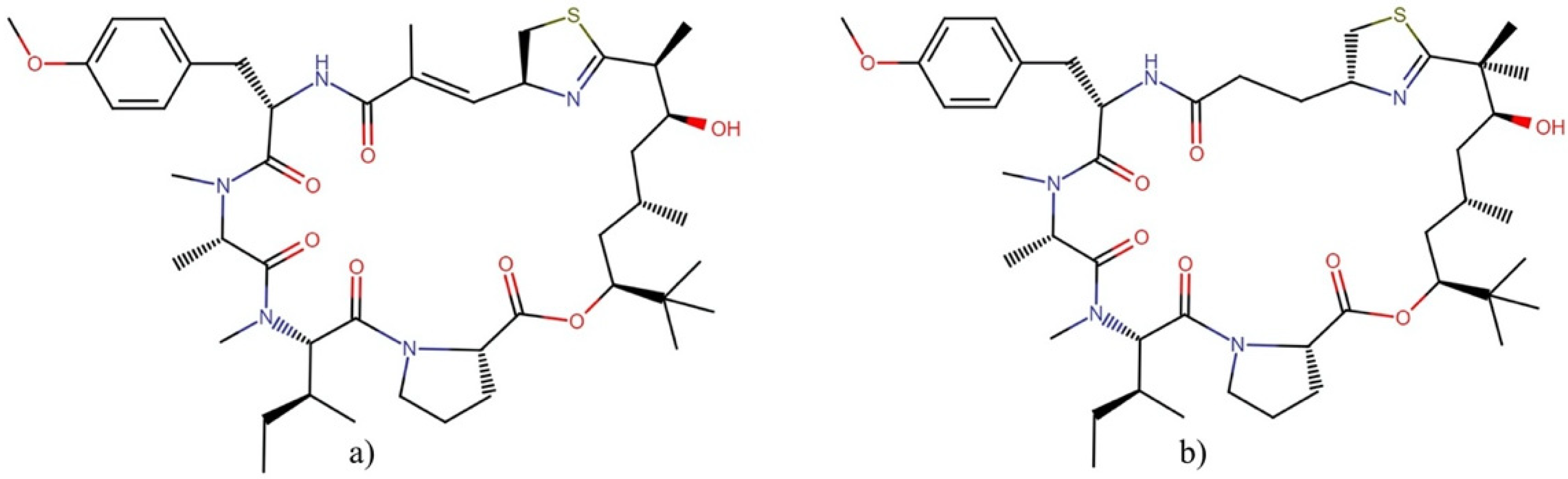
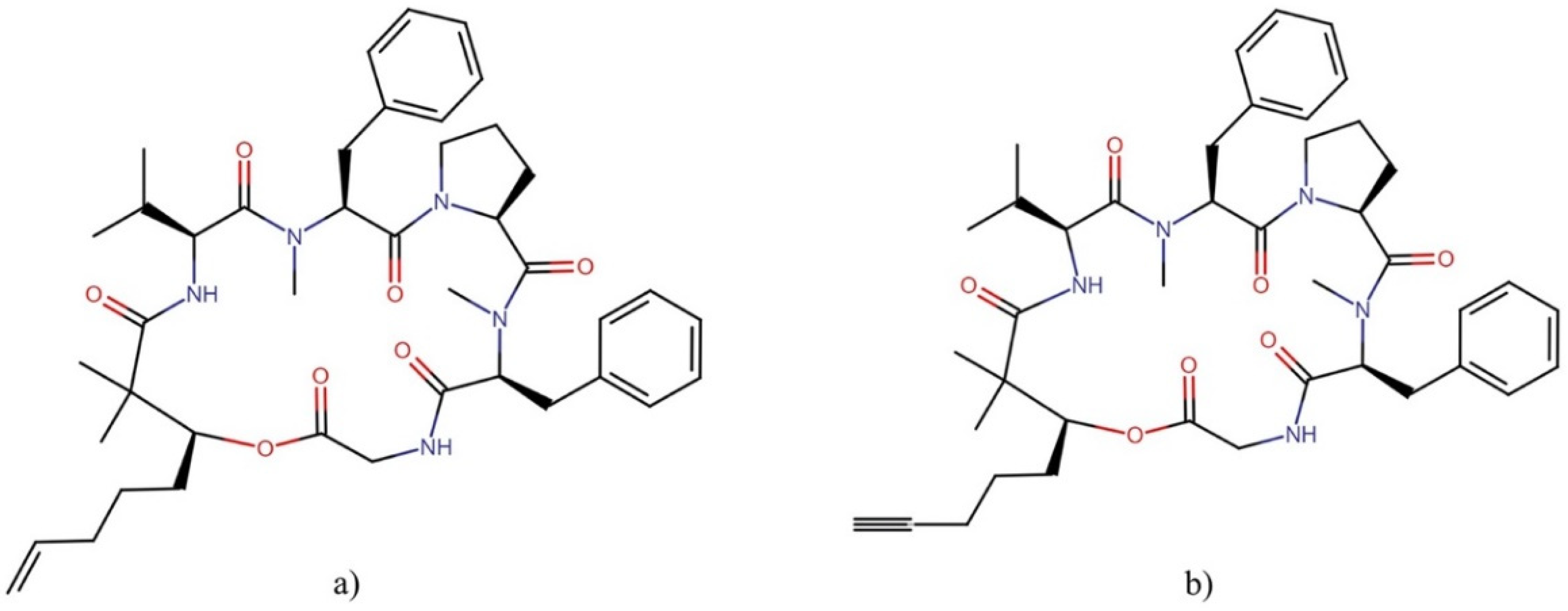
2.5.1. Apratoxins
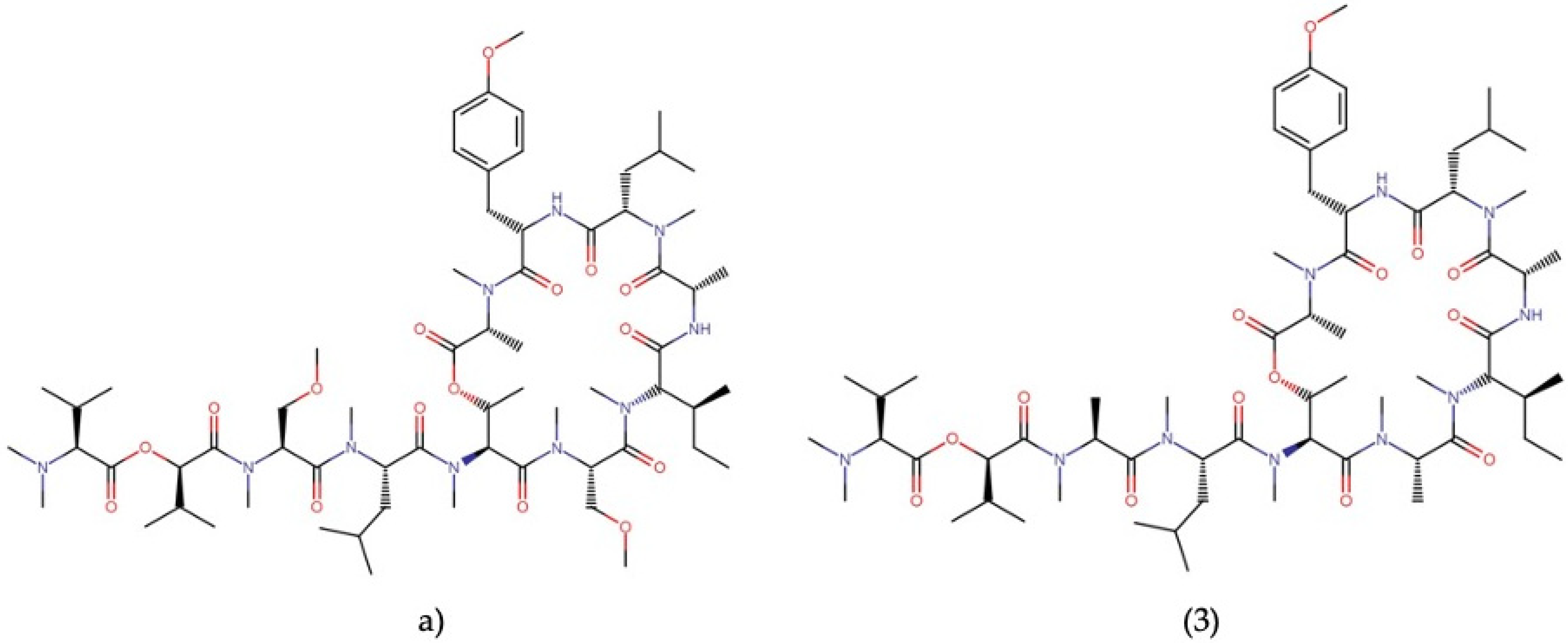
2.5.2. Aurilides
2.5.3. Cocosamides
2.5.4. Coibamide A
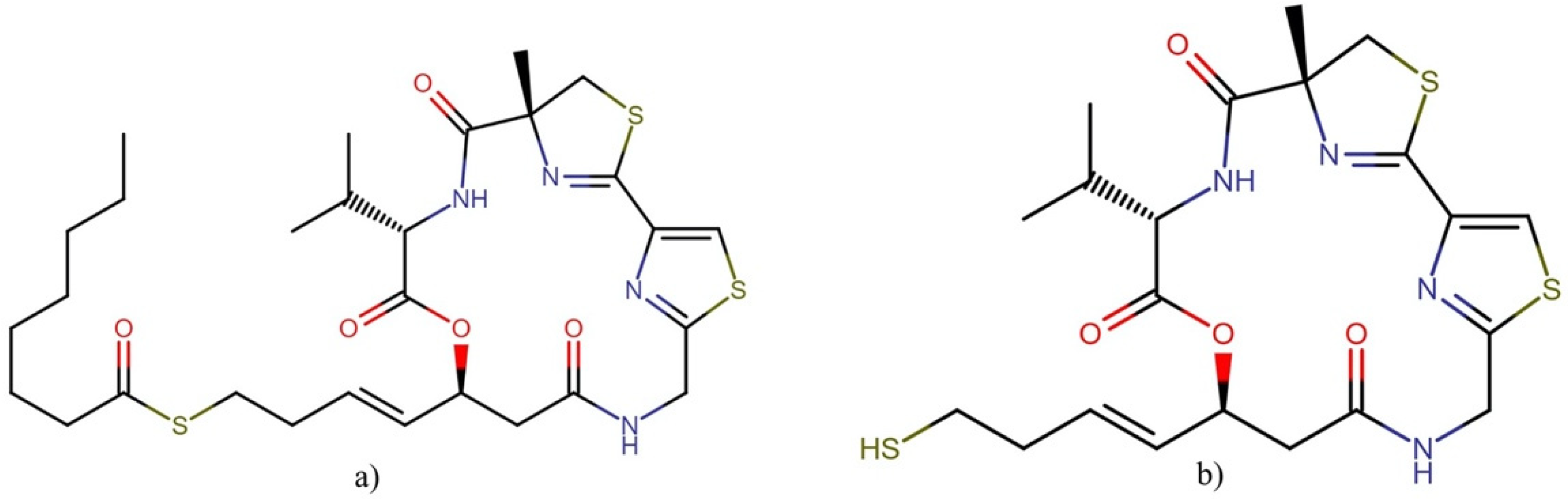
2.5.5. Largazole
2.6. Cyclic Peptides and Depsipeptides
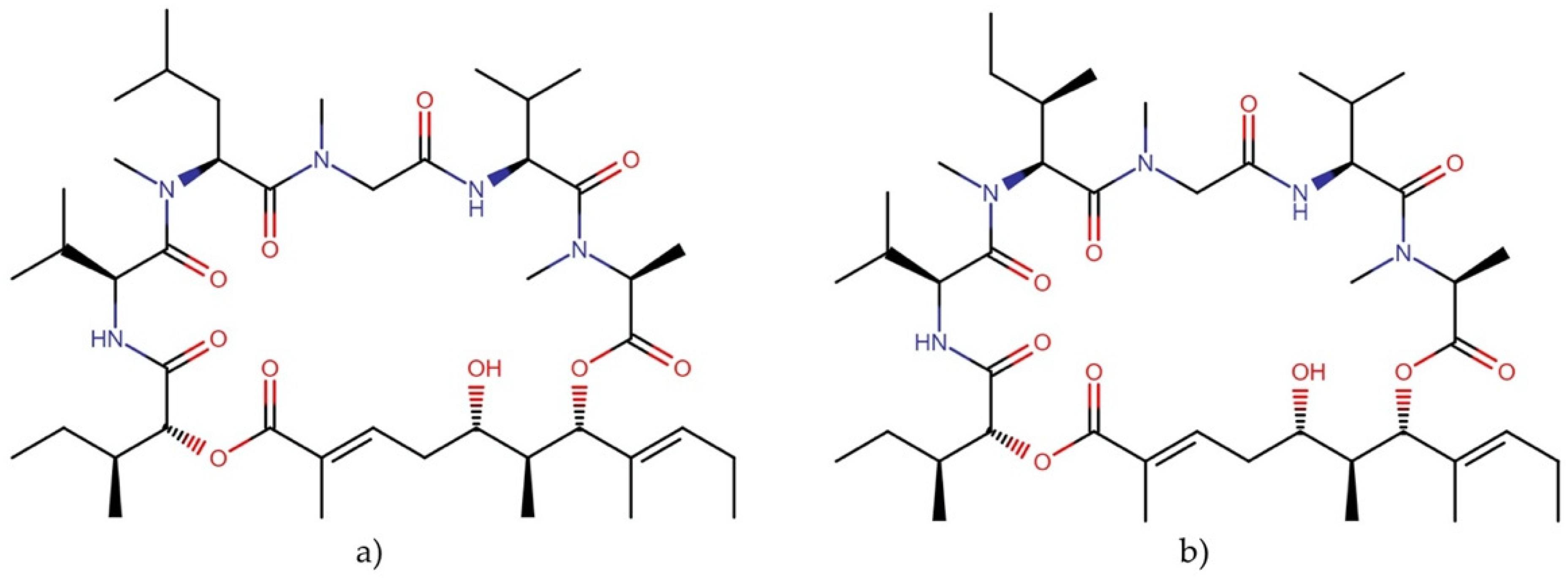
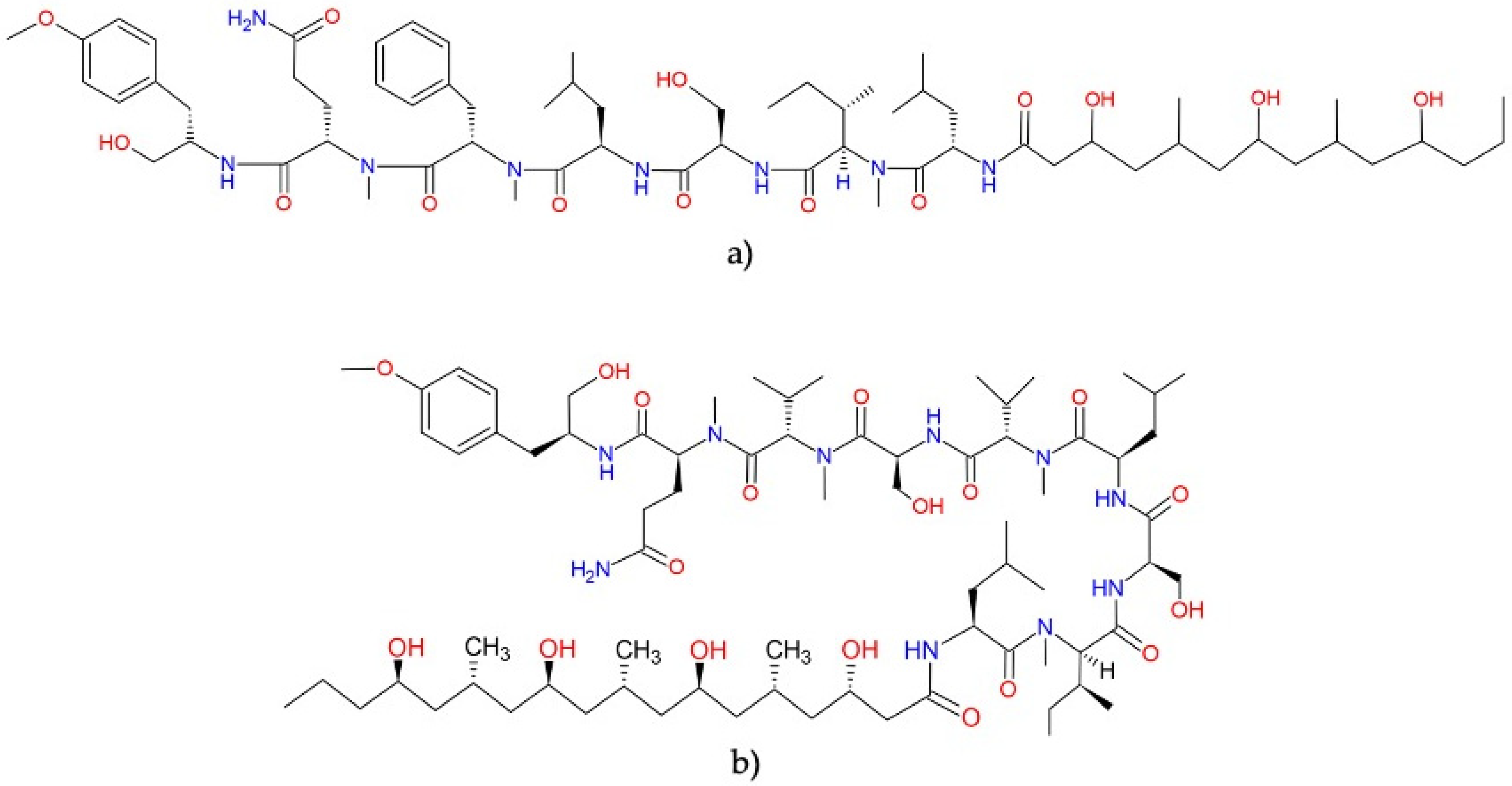
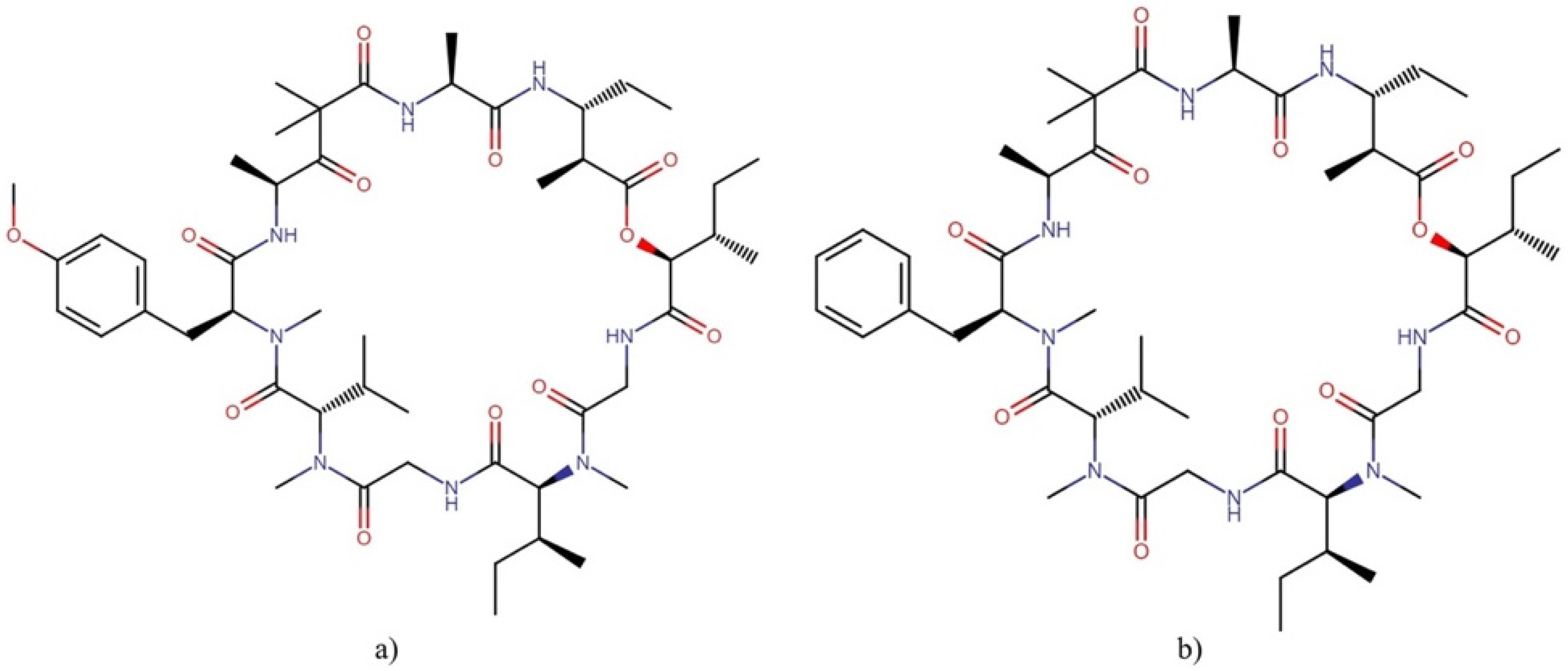
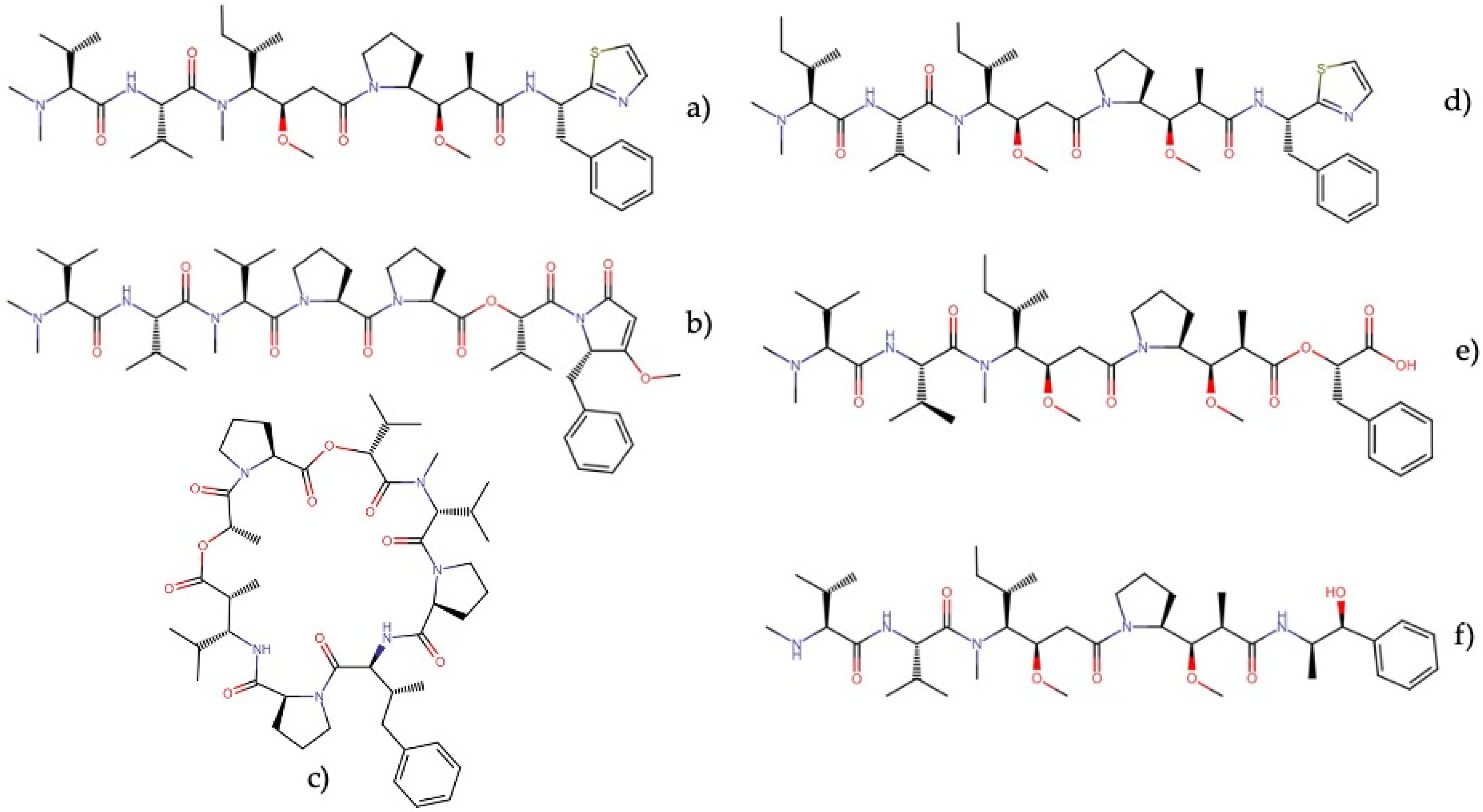
Dolastatins
2.7. Linear Lipopeptides
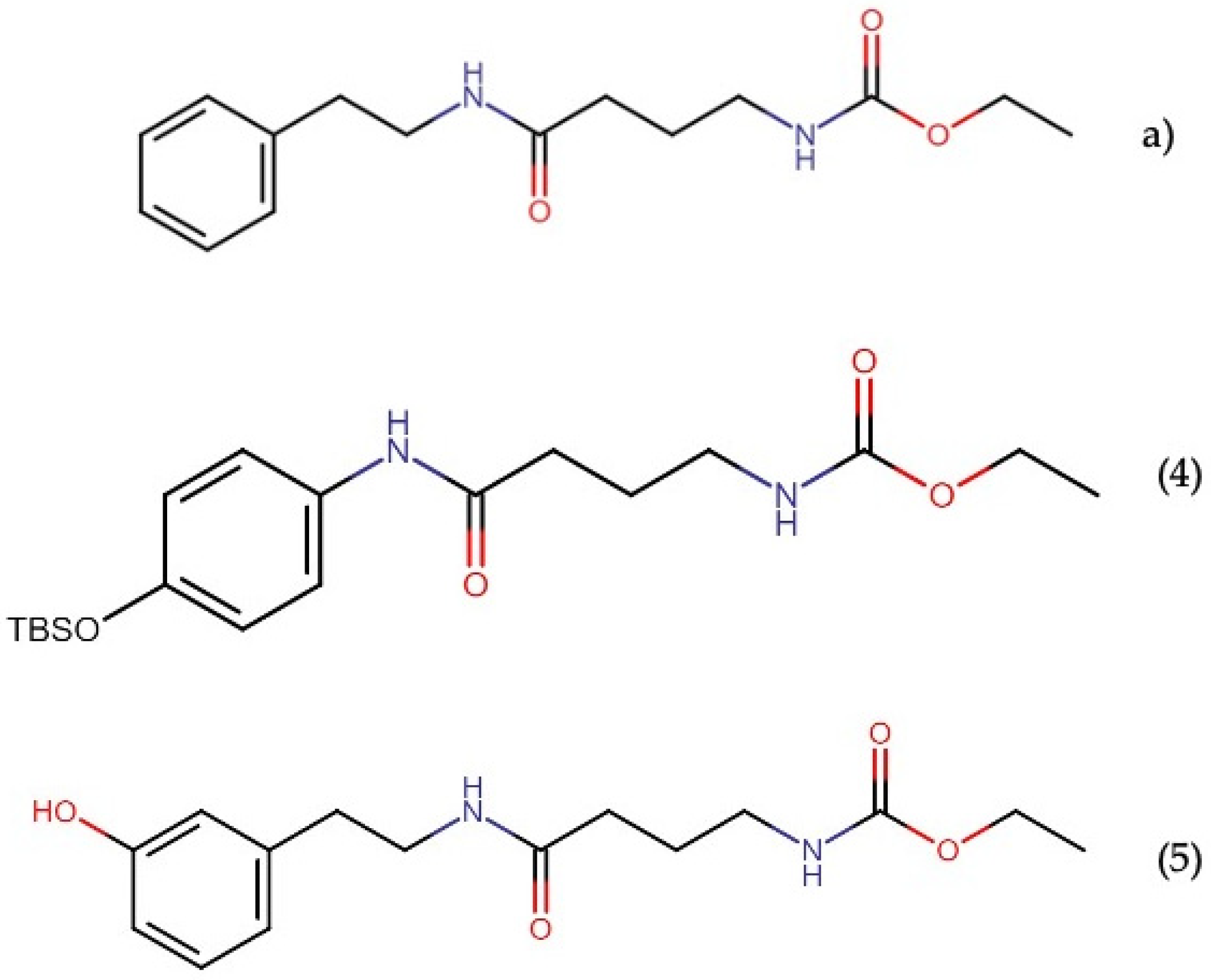
2.7.1. Almiramides
2.7.2. Microcolins
2.7.3. Wenchangamides
2.8. Cyclic Lipopeptides
Hectochlorins
2.9. Peptolides
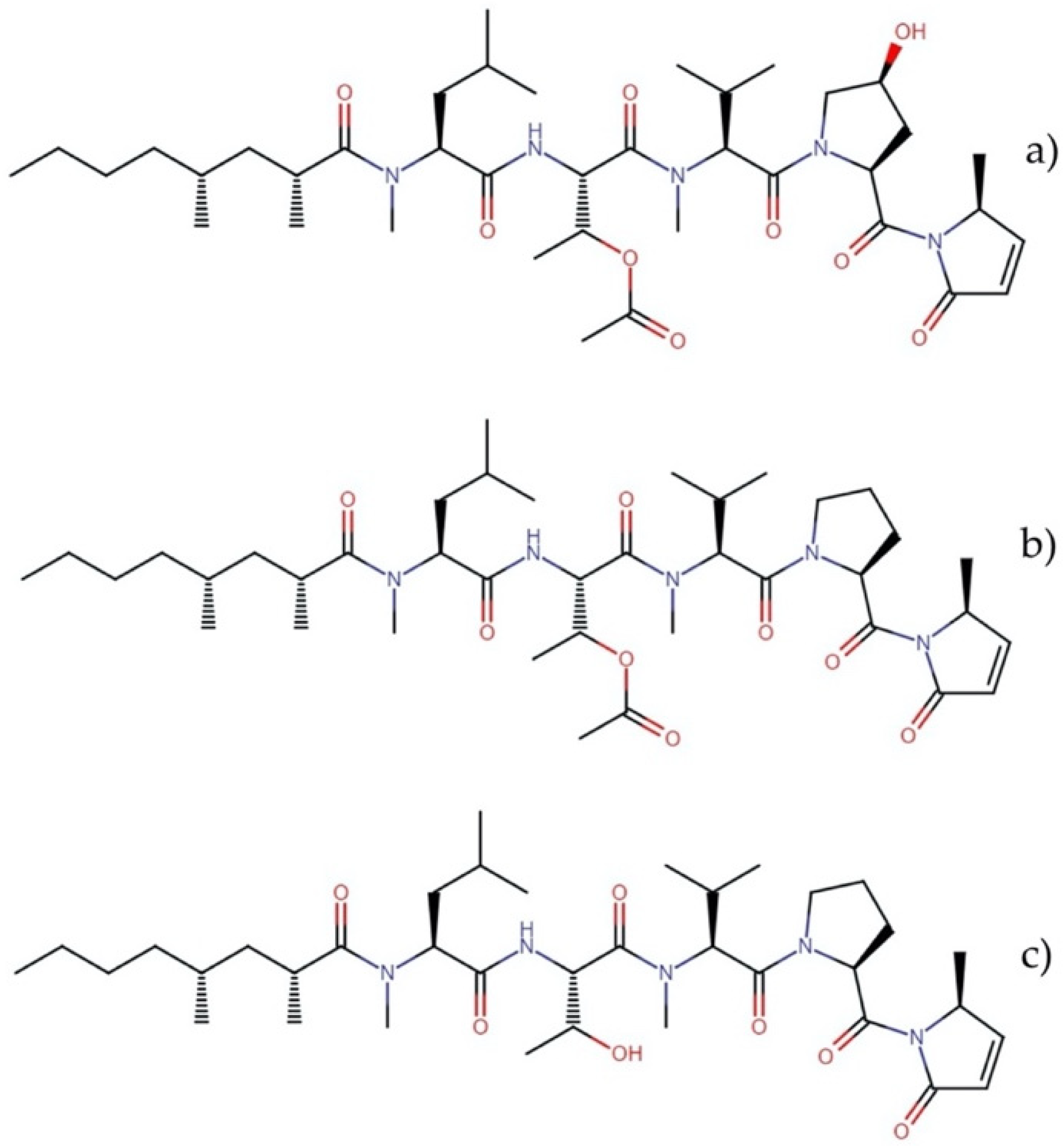
2.9.1. Lyngbyabellins
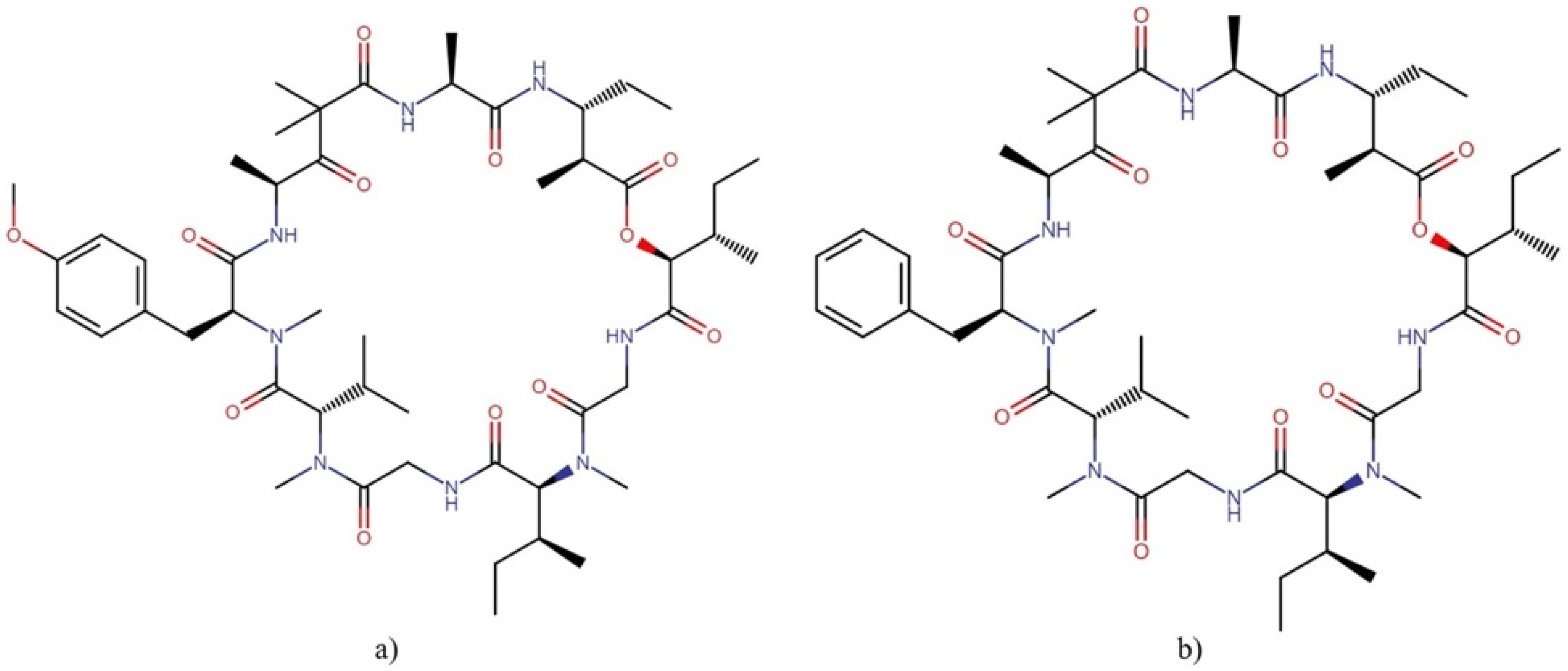
2.9.2. Majusculamides
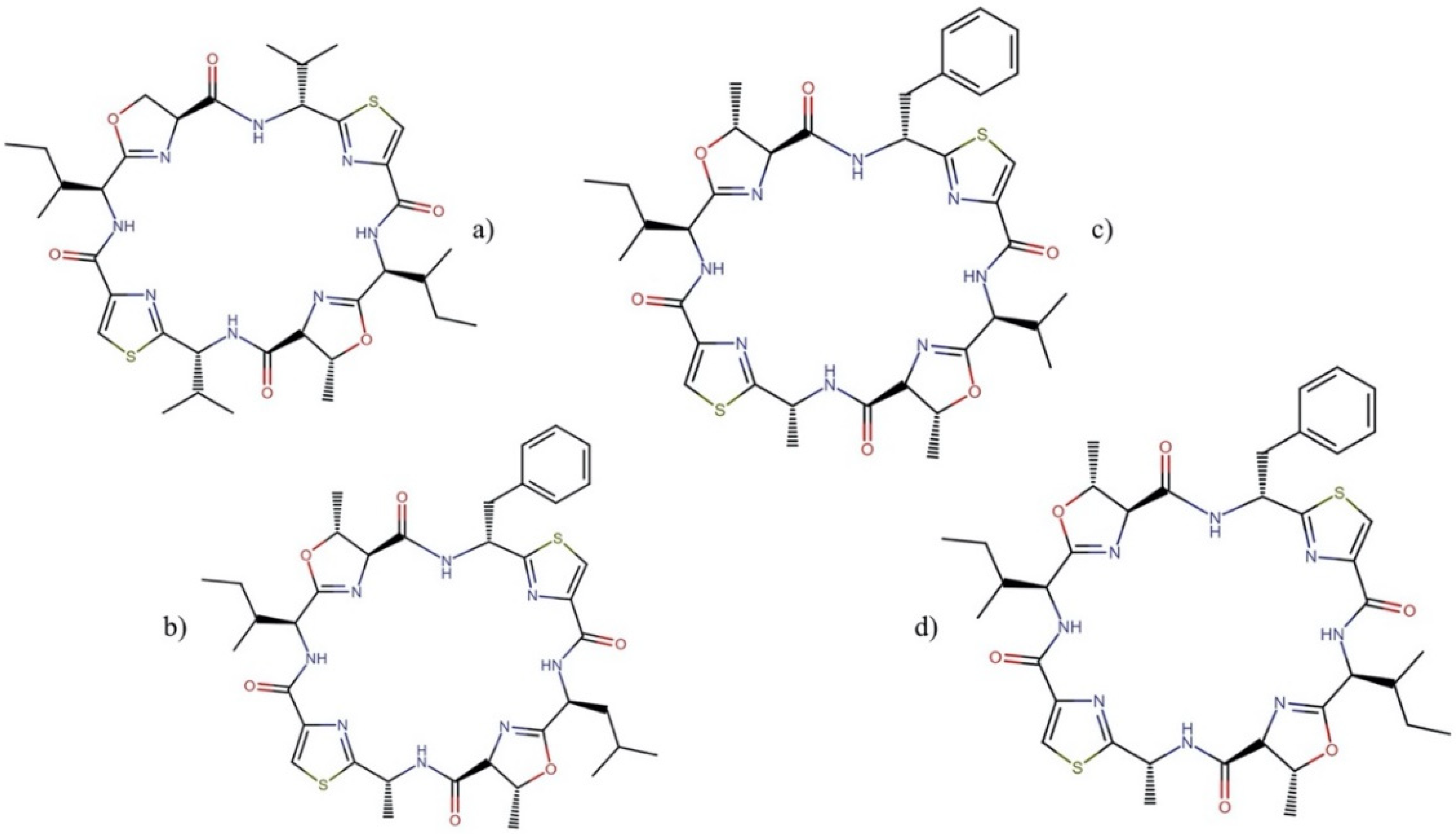
2.9.3. Patellamides
2.10. Polyketides
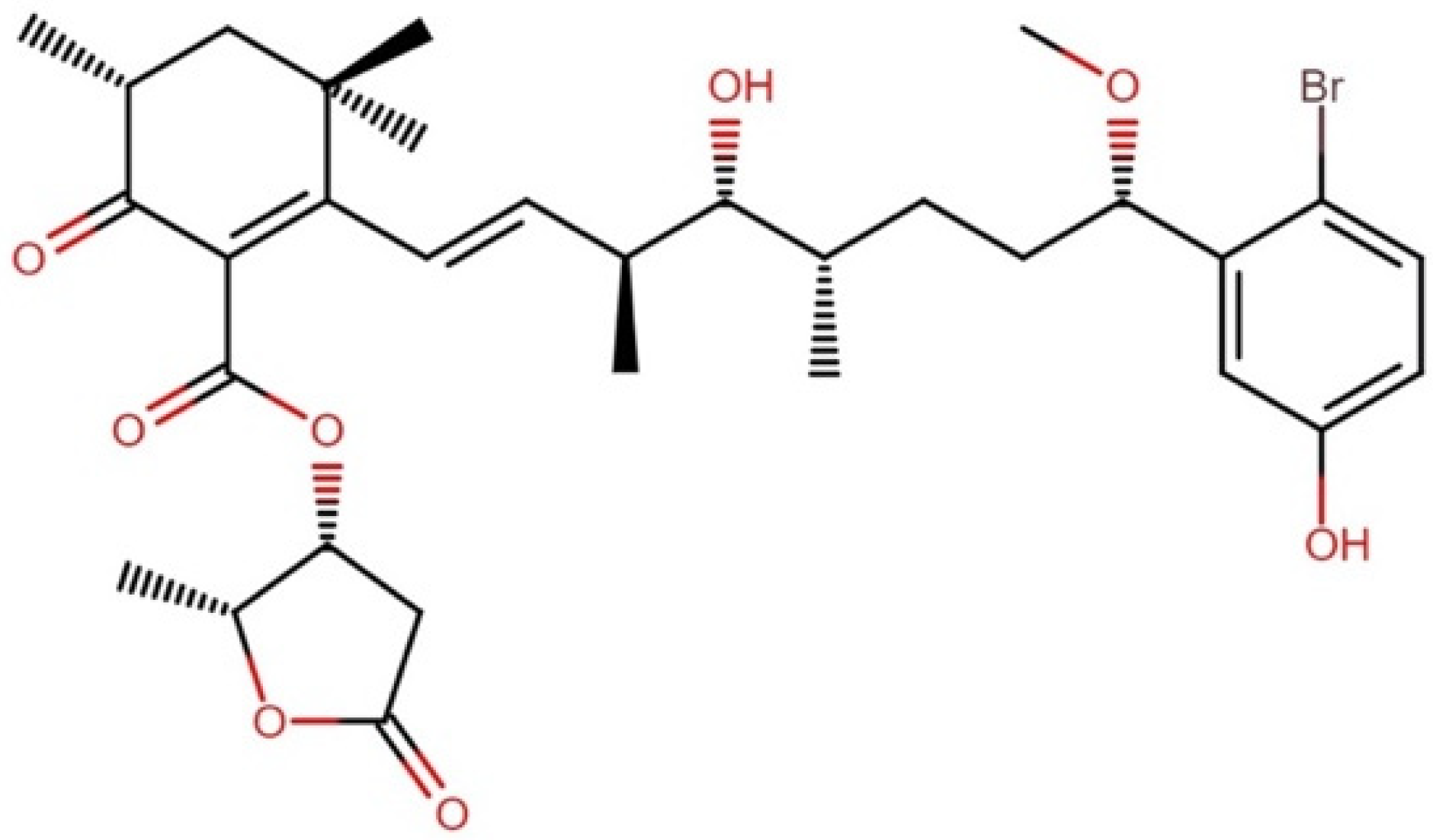
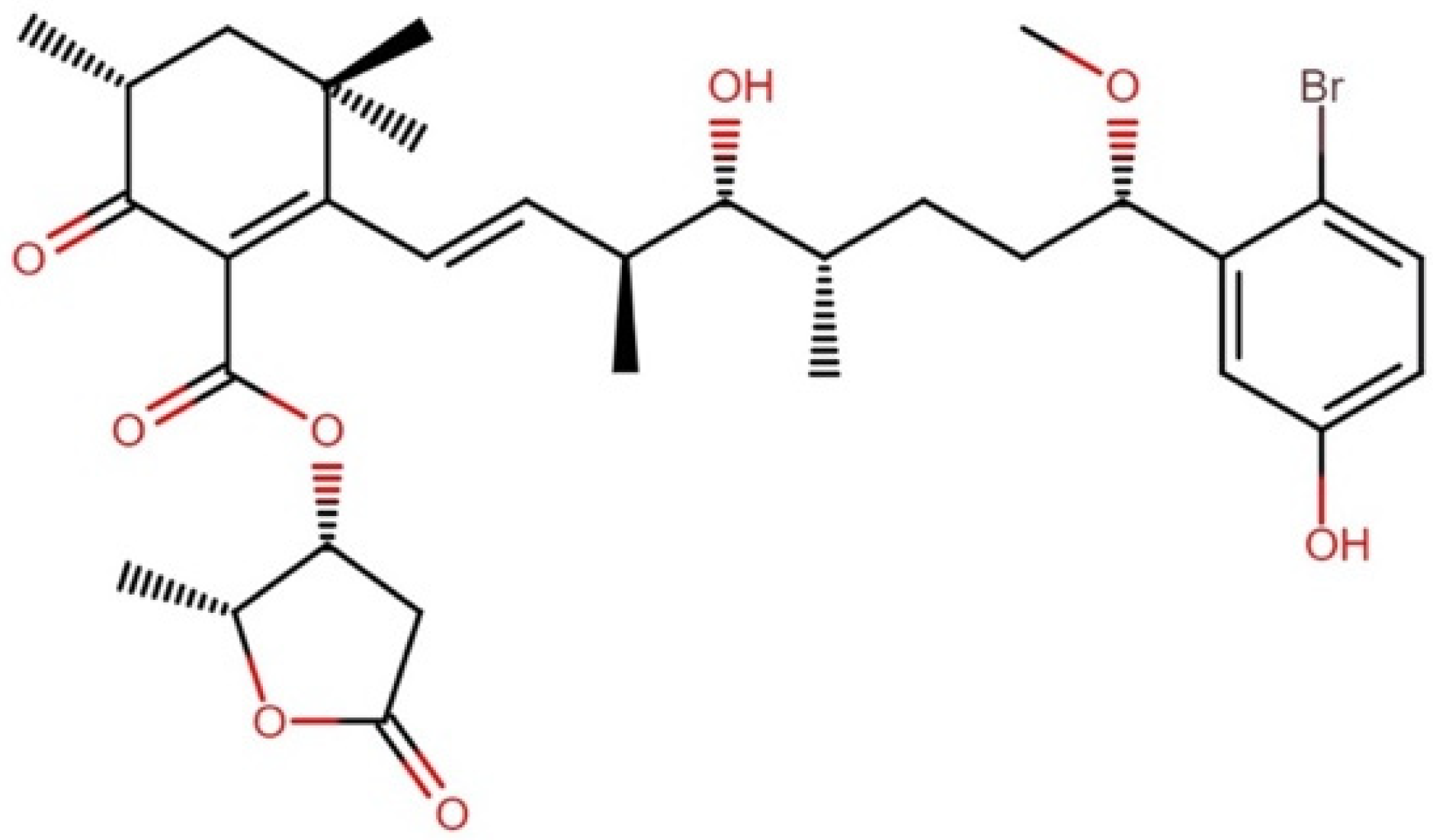
2.10.1. Aplysiatoxins
2.10.2. Caldorazole
2.11. Metabolites from Other Chemical Families
2.11.1. Iezoside
2.11.2. Santacruzamate A
3. Future Perspectives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Alessandra Gammone, M.; Riccioni, G.; Galvano, F.; Orazio, N. Novel therapeutic strategies against cancer: Marine-derived drugs may be the answer? Anti-Cancer Agents Med. Chem. 2016, 16, 1549–1557. [Google Scholar] [CrossRef]
- Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B. Natural products from cyanobacteria: Focus on beneficial activities. Mar. Drugs 2019, 17, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.C.; Monroe, E.A.; Eisman, E.B.; Gerwick, L.; Sherman, D.H.; Gerwick, W.H. The unique mechanistic transformations involved in the biosynthesis of modular natural products from marine cyanobacteria. Nat. Prod. Rep. 2010, 27, 1048–1065. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, D.; Chen, M.; Luesch, H.; Ding, Y. Heterologous production of cyanobacterial compounds. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab003. [Google Scholar] [CrossRef]
- Kleigrewe, K.; Gerwick, L.; Sherman, D.H.; Gerwick, W.H. Unique marine derived cyanobacterial biosynthetic genes for chemical diversity. Nat. Prod. Rep. 2016, 33, 348–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, A.; Bose, S.; Banerjee, S.; Patra, J.K.; Malik, J.; Mandal, S.K.; Kilpatrick, K.L.; Das, G.; Kerry, R.G.; Fimognari, C. Marine cyanobacteria and microalgae metabolites—A rich source of potential anticancer drugs. Mar. Drugs 2020, 18, 476. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.A.; Akhter, N.; Auckloo, B.N.; Khan, I.; Lu, Y.; Wang, K.; Wu, B.; Guo, Y.-W. Structural diversity, biological properties and applications of natural products from cyanobacteria. A review. Mar. Drugs 2017, 15, 354. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, S.A.; Shedid, E.S.; Saied, E.M.; Jassbi, A.R.; Jamebozorgi, F.H.; Rateb, M.; Du, M.; Abdel-Daim, M.M.; Kai, G.-Y.; Al-Hammady, M.A. Cyanobacteria—From the oceans to the potential biotechnological and biomedical applications. Mar. Drugs 2021, 19, 241. [Google Scholar] [CrossRef]
- Guiry, M.D.; Guiry, G.M. World-Wide Electronic Publication. National University of Ireland, Galway. Available online: http://www.algaebase.org (accessed on 5 January 2022).
- Harrigan, G.G.; Yoshida, W.Y.; Moore, R.E.; Nagle, D.G.; Park, P.U.; Biggs, J.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H.; Valeriote, F.A. Isolation, structure determination, and biological activity of dolastatin 12 and lyngbyastatin 1 from Lyngbya majuscula/Schizothrix calcicola cyanobacterial assemblages. J. Nat. Prod. 1998, 61, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Nogle, L.M.; Williamson, R.T.; Gerwick, W.H. Somamides A and B, two new depsipeptide analogues of dolastatin 13 from a Fijian cyanobacterial assemblage of Lyngbya majuscula and Schizothrix species. J. Nat. Prod. 2001, 64, 716–719. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Zhang, X.-K.; Si, R.-R.; Shen, S.-C.; Liang, T.-T.; Fan, T.-T.; Chen, W.; Xu, L.-H.; Han, B.-N. Chemical and biological study of novel aplysiatoxin derivatives from the marine cyanobacterium Lyngbya sp. Toxins 2020, 12, 733. [Google Scholar] [CrossRef]
- Andrianasolo, E.H.; Gross, H.; Goeger, D.; Musafija-Girt, M.; McPhail, K.; Leal, R.M.; Mooberry, S.L.; Gerwick, W.H. Isolation of swinholide A and related glycosylated derivatives from two field collections of marine cyanobacteria. Org. Lett. 2005, 7, 1375–1378. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Bar-Shalom, R.; Aharonovich, D.; Kurisawa, N.; Patial, G.; Li, S.; He, S.; Yan, X.; Iwasaki, A.; Suenaga, K. Metabolomic characterization of a cf. Neolyngbya cyanobacterium from the South China Sea reveals wenchangamide A, a lipopeptide with in vitro apoptotic potential in colon cancer cells. Mar. Drugs 2021, 19, 397. [Google Scholar] [CrossRef] [PubMed]
- Brumley, D.A.; Gunasekera, S.P.; Chen, Q.-Y.; Paul, V.J.; Luesch, H. Discovery, total synthesis, and SAR of anaenamides A and B: Anticancer cyanobacterial depsipeptides with a chlorinated pharmacophore. Org. Lett. 2020, 22, 4235–4239. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Rasko, D.A.; Sudek, S.; Eisen, J.A.; Haygood, M.G.; Ravel, J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. USA 2005, 102, 7315–7320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leão, P.N.; Nakamura, H.; Costa, M.; Pereira, A.R.; Martins, R.; Vasconcelos, V.; Gerwick, W.H.; Balskus, E.P. Biosynthesis-assisted structural elucidation of the bartolosides, chlorinated aromatic glycolipids from cyanobacteria. Angew. Chem. Int. Ed. Engl. 2015, 54, 11063–11067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonso, T.B.; Costa, M.S.; Rezende de Castro, R.; Freitas, S.; Silva, A.; Schneider, M.P.C.; Martins, R.R.; Leão, P.N. Bartolosides e–k from a marine coccoid cyanobacterium. J. Nat. Prod. 2016, 79, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, J.B.; Molinski, T.F. Caylobolide A, a unique 36-membered macrolactone from a Bahamian Lyngbya majuscula. Org. Lett. 2002, 4, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Paul, V.J.; Luesch, H. Caylobolide B, a macrolactone from symplostatin 1-producing marine cyanobacteria Phormidium spp. from Florida. J. Nat. Prod. 2010, 73, 1606–1609. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Williams, H.; Cagle, D.; Karanovich, K.; Horgen, F.D.; Watanabe, C.M. Macrolactone nuiapolide, isolated from a Hawaiian marine cyanobacterium, exhibits anti-chemotactic activity. Mar. Drugs 2015, 13, 6274–6290. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, P.; Zhang, D.; Glukhov, E.; Gerwick, L.; Zhang, C.; Murray, T.F.; Gerwick, W.H. Samholides, swinholide-related metabolites from a marine cyanobacterium cf. Phormidium sp. J. Org. Chem. 2018, 83, 3034–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmely, S.; Kashman, Y. Structure of swinholide-A, a new macrolide from the marine sponge Theonella swinhoei. Tetrahedron Lett. 1985, 26, 511–514. [Google Scholar] [CrossRef]
- Saito, S.-y.; Watabe, S.; Ozaki, H.; Kobayashi, M.; Suzuki, T.; Kobayashi, H.; Fusetani, N.; Karaki, H. Actin-depolymerizing effect of dimeric macrolides, bistheonellide A and swinholide A. J. Biochem. 1998, 123, 571–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, M.; Ishida, T.; Kobayashi, M.; Kitagawa, I. Molecular conformation of swinholide A, a potent cytotoxic dimeric macrolide from the Okinawan marine sponge Theonella swinhoei: X-ray crystal structure of its diketone derivative. J. Org. Chem. 1991, 56, 3629–3632. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kawazoe, K.; Okamoto, T.; Sasaki, T.; Kitagawa, I. Structure-activity correlation of a potent cytotoxic dimeric macrolide swinholide A, from the Okinawan marine sponge Theonella swinhoei, and its isomers. Chem. Pharm. Bull. 1994, 42, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teruya, T.; Sasaki, H.; Fukazawa, H.; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2009, 11, 5062–5065. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Teruya, T.; Fukazawa, H.; Suenaga, K. Revised structure and structure–activity relationship of bisebromoamide and structure of norbisebromoamide from the marine cyanobacterium Lyngbya sp. Tetrahedron 2011, 67, 990–994. [Google Scholar] [CrossRef]
- Suzuki, K.; Mizuno, R.; Suenaga, K.; Teruya, T.; Tanaka, N.; Kosaka, T.; Oya, M. Bisebromoamide, an extract from Lyngbya species, induces apoptosis through ERK and mTOR inhibitions in renal cancer cells. Cancer Med. 2013, 2, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.J.; Boys, S.K.; Makda, A.; Carragher, N.O.; Hulme, A.N. Naturally inspired peptide leads: Alanine scanning reveals an actin-targeting thiazole analogue of bisebromoamide. ChemBioChem 2016, 17, 1621. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.T.; Phyo, M.Y. Marine cyanobacteria: A source of lead compounds and their clinically-relevant molecular targets. Molecules 2020, 25, 2197. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; Kale, A.J.; Fenley, A.T.; Byrum, T.; Debonsi, H.M.; Gilson, M.K.; Valeriote, F.A.; Moore, B.S.; Gerwick, W.H. The carmaphycins, new proteasome inhibitors exhibiting an α, β-epoxyketone warhead from a marine cyanobacterium. ChemBioChem 2012, 13, 810. [Google Scholar] [CrossRef] [Green Version]
- Almaliti, J.; Miller, B.; Pietraszkiewicz, H.; Glukhov, E.; Naman, C.B.; Kline, T.; Hanson, J.; Li, X.; Zhou, S.; Valeriote, F.A. Exploration of the carmaphycins as payloads in antibody drug conjugate anticancer agents. Eur. J. Med. Chem. 2019, 161, 416–432. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Brumley, D.A.; Gunasekera, S.P.; Sauvage, T.; Dos Santos, L.A.; Chen, Q.-Y.; Paul, V.J.; Luesch, H. Discovery, synthesis, and biological evaluation of anaenamides C and D from a new marine cyanobacterium, Hormoscilla sp. J. Nat. Prod. 2022, 85, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from Guam and Palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin D, a potent cytotoxic cyclodepsipeptide from Papua New Guinea collections of the marine cyanobacteria Lyngbya majuscula and Lyngbya sordida. J Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef]
- Tidgewell, K.; Engene, N.; Byrum, T.; Media, J.; Doi, T.; Valeriote, F.A.; Gerwick, W.H. Evolved diversification of a modular natural product pathway: Apratoxins F and G, two cytotoxic cyclic depsipeptides from a Palmyra collection of Lyngbya bouillonii. ChemBioChem 2010, 11, 1458. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Cowley, E.S.; Sikorska, J.; Shaala, L.A.; Ishmael, J.E.; Youssef, D.T.; McPhail, K.L. Apratoxin H and apratoxin A sulfoxide from the Red Sea cyanobacterium Moorea producens. J. Nat. Prod. 2013, 76, 1781–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthew, S.; Schupp, P.J.; Luesch, H. Apratoxin E, a cytotoxic peptolide from a Guamanian collection of the marine cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2008, 71, 1113–1116. [Google Scholar] [CrossRef]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Belmonte, J.C.I.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef]
- Liu, Y.; Law, B.K.; Luesch, H. Apratoxin A reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol. Pharmacol. 2009, 76, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi, S.; Kawaguchi, S.; Badr, C.E.; Mattos, D.R.; Ruiz-Saenz, A.; Serrill, J.D.; Moasser, M.M.; Dolan, B.P.; Paavilainen, V.O.; Oishi, S. Targeting of HER/ErbB family proteins using broad spectrum Sec61 inhibitors coibamide A and apratoxin A. Biochem. Pharmacol. 2021, 183, 114317. [Google Scholar] [CrossRef] [PubMed]
- Paatero, A.O.; Kellosalo, J.; Dunyak, B.M.; Almaliti, J.; Gestwicki, J.E.; Gerwick, W.H.; Taunton, J.; Paavilainen, V.O. Apratoxin kills cells by direct blockade of the Sec61 protein translocation channel. Cell Chem. Biol. 2016, 23, 561–566. [Google Scholar]
- Huang, K.-C.; Chen, Z.; Jiang, Y.; Akare, S.; Kolber-Simonds, D.; Condon, K.; Agoulnik, S.; Tendyke, K.; Shen, Y.; Wu, K.-M. Apratoxin A shows novel pancreas-targeting activity through the binding of Sec 61. Mol. Cancer Ther. 2016, 15, 1208–1216. [Google Scholar]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-Y.; Liu, Y.; Luesch, H. Systematic chemical mutagenesis identifies a potent novel apratoxin A/E hybrid with improved in vivo antitumor activity. ACS Med. Chem. Lett. 2011, 2, 861–865. [Google Scholar]
- Onda, Y.; Masuda, Y.; Yoshida, M.; Doi, T. Conformation-based design and synthesis of apratoxin A mimetics modified at the α, β-unsaturated thiazoline moiety. J. Med. Chem. 2017, 60, 6751–6765. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.-Y.; Liu, Y.; Cai, W.; Luesch, H. Improved total synthesis and biological evaluation of potent apratoxin S4 based anticancer agents with differential stability and further enhanced activity. J. Med. Chem. 2014, 57, 3011–3029. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Chen, Q.-Y.; Dang, L.H.; Luesch, H. Apratoxin S10, a dual inhibitor of angiogenesis and cancer cell growth to treat highly vascularized tumors. ACS Med. Chem. Lett. 2017, 8, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Ratnayake, R.; Gerber, M.H.; Chen, Q.-Y.; Yu, Y.; Derendorf, H.; Trevino, J.G.; Luesch, H. Development of apratoxin S10 (Apra S10) as an anti-pancreatic cancer agent and its preliminary evaluation in an orthotopic patient-derived xenograft (PDX) model. Investig. New Drugs 2019, 37, 364–374. [Google Scholar] [CrossRef]
- Michon, S.; Cavelier, F.; Salom-Roig, X.J. Synthesis and biological activities of cyclodepsipeptides of Aurilide family from marine origin. Mar. Drugs 2021, 19, 55. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, K.; Mutou, T.; Shibata, T.; Itoh, T.; Kigoshi, H.; Yamada, K. Isolation and stereostructure of aurilide, a novel cyclodepsipeptide from the Japanese sea hare Dolabella auricularia. Tetrahedron Lett. 1996, 37, 6771–6774. [Google Scholar] [CrossRef]
- Han, B.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Quon, M.K.; Moore, R.E.; Paul, V.J. The structure of palau’amide, a potent cytotoxin from a species of the marine cyanobacterium Lyngbya. J. Nat. Prod. 2003, 66, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Kaneda, M.; Kawaguchi, S.; Fujii, N.; Ohno, H.; Oishi, S. Structure–activity relationship study on odoamide: Insights into the bioactivities of Aurilide-family hybrid peptide–polyketides. ACS Med. Chem. Lett. 2018, 9, 365–369. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.-K.; Tan, L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef]
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Putra, M.Y.; Ye, T.; Paul, V.J.; Luesch, H. Isolation, structure elucidation and biological evaluation of lagunamide D: A new cytotoxic macrocyclic depsipeptide from marine cyanobacteria. Mar. Drugs 2019, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.-i.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine natural product aurilide activates the OPA1-mediated apoptosis by binding to prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Takase, S.; Kurokawa, R.; Arai, D.; Kanto, K.K.; Okino, T.; Nakao, Y.; Kushiro, T.; Yoshida, M.; Matsumoto, K. A quantitative shRNA screen identifies ATP1A1 as a gene that regulates cytotoxicity by aurilide B. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasekera, S.P.; Owle, C.S.; Montaser, R.; Luesch, H.; Paul, V.J. Malyngamide 3 and cocosamides A and B from the marine cyanobacterium Lyngbya majuscula from Cocos Lagoon, Guam. J. Nat. Prod. 2011, 74, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Wang, W.; Ao, L.; Cheng, Z.; Wu, C.; Pan, Z.; Liu, K.; Li, H.; Su, W.; Fang, L. Improved total synthesis and biological evaluation of coibamide A analogues. J. Med. Chem. 2018, 61, 8908–8916. [Google Scholar] [CrossRef]
- Nabika, R.; Suyama, T.L.; Hau, A.M.; Misu, R.; Ohno, H.; Ishmael, J.E.; McPhail, K.L.; Oishi, S.; Fujii, N. Synthesis and biological evaluation of the [d-MeAla11]-epimer of coibamide A. Bioorg. Med. Chem. Lett. 2015, 25, 302–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Cheng, Z.; Lu, D.; Liu, K.; Cheng, Y.; Wang, P.; Zhou, Y.; Li, M.; Shao, X.; Li, H. Novel N-methylated cyclodepsipeptide prodrugs for targeted cancer therapy. J. Med. Chem. 2021, 64, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Hau, A.M.; Greenwood, J.A.; Löhr, C.V.; Serrill, J.D.; Proteau, P.J.; Ganley, I.G.; McPhail, K.L.; Ishmael, J.E. Coibamide A induces mTOR-independent autophagy and cell death in human glioblastoma cells. PLoS ONE 2013, 8, e65250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Lu, D.; Wu, C.; Li, M.; Ding, Z.; Li, Y.; Chen, B.; Lin, X.; Su, W.; Shao, X. Coibamide A kills cancer cells through inhibiting autophagy. Biochem. Biophys. Res. Commun. 2021, 547, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Serrill, J.D.; Wan, X.; Hau, A.M.; Jang, H.S.; Coleman, D.J.; Indra, A.K.; Alani, A.W.; McPhail, K.L.; Ishmael, J.E. Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Investig. New Drugs 2016, 34, 24–40. [Google Scholar] [CrossRef]
- Tranter, D.; Paatero, A.O.; Kawaguchi, S.; Kazemi, S.; Serrill, J.D.; Kellosalo, J.; Vogel, W.K.; Richter, U.; Mattos, D.R.; Wan, X. Coibamide A targets Sec61 to prevent biogenesis of secretory and membrane proteins. ACS Chem. Biol. 2020, 15, 2125–2136. [Google Scholar] [CrossRef]
- Engene, N.; Tronholm, A.; Salvador-Reyes, L.A.; Luesch, H.; Paul, V.J. Caldora penicillata gen. nov. comb. nov. (Cyanobacteria), a pantropical marine species with biomedical relevance. J. Phycol. 2015, 51, 670–681. [Google Scholar] [CrossRef] [Green Version]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef] [Green Version]
- Al-Awadhi, F.H.; Salvador-Reyes, L.A.; Elsadek, L.A.; Ratnayake, R.; Chen, Q.-Y.; Luesch, H. Largazole is a brain-penetrant class I HDAC inhibitor with extended applicability to glioblastoma and CNS diseases. ACS Chem. Neurosci. 2020, 11, 1937–1943. [Google Scholar] [CrossRef]
- Clausen, D.J.; Smith, W.B.; Haines, B.E.; Wiest, O.; Bradner, J.E.; Williams, R.M. Modular synthesis and biological activity of pyridyl-based analogs of the potent class I histone deacetylase inhibitor largazole. Bioorg. Med. Chem. 2015, 23, 5061–5074. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Huang, P.-H.; Chen, C.-S.; Forsyth, C.J. Total syntheses of the histone deacetylase inhibitors largazole and 2-epi-largazole: Application of N-heterocyclic carbene mediated acylations in complex molecule synthesis. J. Org. Chem. 2011, 76, 1140–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.-C.; Wen, Z.-S.; Qiu, Y.-T.; Chen, X.-Q.; Chen, H.-B.; Wei, M.-M.; Liu, Z.; Jiang, S.; Zhou, G.-B. Largazole arrests cell cycle at G1 phase and triggers proteasomal degradation of E2F1 in lung cancer cells. ACS Med. Chem. Lett. 2013, 4, 921–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousquet, M.S.; Ma, J.J.; Ratnayake, R.; Havre, P.A.; Yao, J.; Dang, N.H.; Paul, V.J.; Carney, T.J.; Dang, L.H.; Luesch, H. Multidimensional screening platform for simultaneously targeting oncogenic KRAS and hypoxia-inducible factors pathways in colorectal cancer. ACS Chem. Biol. 2016, 11, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Ungermannova, D.; Parker, S.J.; Nasveschuk, C.G.; Wang, W.; Quade, B.; Zhang, G.; Kuchta, R.D.; Phillips, A.J.; Liu, X. Largazole and its derivatives selectively inhibit ubiquitin activating enzyme (E1). PLoS ONE 2012, 7, e29208. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ruan, Z.-W.; Luo, D.; Zhu, Y.; Ding, T.; Sui, Q.; Lei, X. Unexpected enhancement of HDACs inhibition by MeS substitution at C-2 position of fluoro largazole. Mar. Drugs 2020, 18, 344. [Google Scholar]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Pilon, J.L.; Clausen, D.J.; Hansen, R.J.; Lunghofer, P.J.; Charles, B.; Rose, B.J.; Thamm, D.H.; Gustafson, D.L.; Bradner, J.E.; Williams, R.M. Comparative pharmacokinetic properties and antitumor activity of the marine HDACi largazole and largazole peptide isostere. Cancer Chemother. Pharmacol. 2015, 75, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef]
- Zeng, X.; Yin, B.; Hu, Z.; Liao, C.; Liu, J.; Li, S.; Li, Z.; Nicklaus, M.C.; Zhou, G.; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef]
- Law, M.; Corsino, P.; Jahn, S.; Davis, B.; Chen, S.; Patel, B.; Pham, K.; Lu, J.; Sheppard, B.; Nørgaard, P. Glucocorticoids and histone deacetylase inhibitors cooperate to block the invasiveness of basal-like breast cancer cells through novel mechanisms. Oncogene 2013, 32, 1316–1329. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Poli, G.; Di Fabio, R.; Ferrante, L.; Summa, V.; Botta, M. Largazole analogues as histone deacetylase inhibitors and anticancer agents: An overview of structure–activity relationships. ChemMedChem 2017, 12, 1917–1926. [Google Scholar] [CrossRef]
- Kagihara, J.A.; Corr, B.; Pacheco, J.M.; Davis, S.L.; Lieu, C.H.; Kim, S.S.; Jimeno, A.; Heim, A.M.; DeMattei, J.A.; Gordon, G. Phase 1 study of OKI-179, an oral class 1-selective depsipeptide HDAC inhibitor, in patients with advanced solid tumors: Final results. J. Clin. Oncol. 2021, 39, 3075. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Lefranc, F.; Carbone, M.; Mollo, E.; Gavagnin, M.; Betancourt, T.; Dasari, R.; Kornienko, A.; Kiss, R. Marine mollusk-derived agents with antiproliferative activity as promising anticancer agents to overcome chemotherapy resistance. Med. Res. Rev. 2017, 37, 702–801. [Google Scholar] [CrossRef]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef]
- Catassi, A.; Cesario, A.; Arzani, D.; Menichini, P.; Alama, A.; Bruzzo, C.; Imperatori, A.; Rotolo, N.; Granone, P.; Russo, P. Characterization of apoptosis induced by marine natural products in non small cell lung cancer A549 cells. Cell. Mol. Life Sci. 2006, 63, 2377–2386. [Google Scholar] [CrossRef]
- Turner, T.; Jackson, W.H.; Pettit, G.R.; Wells, A.; Kraft, A.S. Treatment of human prostate cancer cells with dolastatin 10, a peptide isolated from a marine shell-less mollusc. Prostate 1998, 34, 175–181. [Google Scholar] [CrossRef]
- Beckwith, M.; Urba, W.J.; Longo, D.L. Growth inhibition of human lymphoma cell lines by the marine products, dolastatins 10 and 15. J. Natl. Cancer Inst. 1993, 85, 483–488. [Google Scholar] [CrossRef]
- Kalemkerian, G.P.; Ou, X.; Adil, M.R.; Rosati, R.; Khoulani, M.M.; Madan, S.K.; Pettit, G.R. Activity of dolastatin 10 against small-cell lung cancer in vitro and in vivo: Induction of apoptosis and bcl-2 modification. Cancer Chemother. Pharmacol. 1999, 43, 507–515. [Google Scholar] [CrossRef]
- Aherne, G.W.; Hardcastle, A.; Valenti, M.; Bryant, A.; Rogers, P.; Pettit, G.R.; Srirangam, J.K.; Kelland, L.R. Antitumour evaluation of dolastatins 10 and 15 and their measurement in plasma by radioimmunoassay. Cancer Chemother. Pharmacol. 1996, 38, 225–232. [Google Scholar] [CrossRef]
- Ratnayake, R.; Gunasekera, S.P.; Ma, J.J.; Dang, L.H.; Carney, T.J.; Paul, V.J.; Luesch, H. Dolastatin 15 from a marine cyanobacterium suppresses HIF-1α mediated cancer cell viability and vascularization. ChemBioChem 2020, 21, 2356. [Google Scholar] [CrossRef]
- Mooberry, S.L.; Leal, R.M.; Tinley, T.L.; Luesch, H.; Moore, R.E.; Corbett, T.H. The molecular pharmacology of symplostatin 1: A new antimitotic dolastatin 10 analog. Int. J. Cancer 2003, 104, 512–521. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Symplostatin 3, a new dolastatin 10 analogue from the marine cyanobacterium Symploca sp. VP452. J. Nat. Prod. 2002, 65, 16–20. [Google Scholar] [CrossRef]
- Pitot, H.C.; McElroy, E.A.; Reid, J.M.; Windebank, A.J.; Sloan, J.A.; Erlichman, C.; Bagniewski, P.G.; Walker, D.L.; Rubin, J.; Goldberg, R.M. Phase I trial of dolastatin-10 (NSC 376128) in patients with advanced solid tumors. Clin. Cancer Res. 1999, 5, 525–531. [Google Scholar] [CrossRef]
- Madden, T.; Tran, H.T.; Beck, D.; Huie, R.; Newman, R.A.; Pusztai, L.; Wright, J.J.; Abbruzzese, J.L. Novel marine-derived anticancer agents: A phase I clinical, pharmacological, and pharmacodynamic study of dolastatin 10 (NSC 376128) in patients with advanced solid tumors. Clin. Cancer Res. 2000, 6, 1293–1301. [Google Scholar]
- Deng, C.; Pan, B.; O’Connor, O. Brentuximab vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Minich, S.S. Brentuximab vedotin: A new age in the treatment of Hodgkin lymphoma and anaplastic large cell lymphoma. Ann. Pharmacother. 2012, 46, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.; Masri, N.; Abou Dalle, I.; El-Cheikh, J.; Ghanem, H.; Bazarbachi, A. Polatuzumab vedotin: Current role and future applications in the treatment of patients with diffuse large B-cell lymphoma. Clin. Hematol. Int. 2021, 3, 21–26. [Google Scholar] [CrossRef]
- Maderna, A.; Leverett, C.A. Recent advances in the development of new auristatins: Structural modifications and application in antibody drug conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Y.; Hua, H.; Li, D.; Tang, C. Marine antitumor peptide dolastatin 10: Biological activity, structural modification and synthetic chemistry. Mar. Drugs 2021, 19, 333. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar]
- Hammond, L.; Ruvuna, F.; Cunningham, C.; Ebbinghaus, S.; Rubin, E.; Mita, A.; Hersh, E.; Eder, J.; Weiss, J.; Rowinsky, E. Phase (Ph) I evaluation of the dolastatin analogue synthadotin (SYN-D; ILX651): Pooled data analysis of three alternate schedules in patients (pts) with advanced solid tumors. Am. J. Clin. Oncol. 2004, 22 (Suppl. 14), 3068. [Google Scholar] [CrossRef]
- Ebbinghaus, S.; Hersh, E.; Cunningham, C.; O’Day, S.; McDermott, D.; Stephenson, J.; Richards, D.; Eckardt, J.; Haider, O.; Hammond, L. Phase II study of synthadotin (SYN-D; ILX651) administered daily for 5 consecutive days once every 3 weeks (qdx5q3w) in patients (Pts) with inoperable locally advanced or metastatic melanoma. Am. J. Clin. Oncol. 2004, 22 (Suppl. 14), 7530. [Google Scholar] [CrossRef]
- Pettit, G.R.; Xu, J.-p.; Hogan, F.; Williams, M.D.; Doubek, D.L.; Schmidt, J.M.; Cerny, R.L.; Boyd, M.R. Isolation and structure of the human cancer cell growth inhibitory cyclodepsipeptide dolastatin 16, 1. J. Nat. Prod. 1997, 60, 752–754. [Google Scholar] [CrossRef]
- Quintana, J.; Bayona, L.M.; Castellanos, L.; Puyana, M.; Camargo, P.; Aristizábal, F.; Edwards, C.; Tabudravu, J.N.; Jaspars, M.; Ramos, F.A. Almiramide D, cytotoxic peptide from the marine cyanobacterium Oscillatoria nigroviridis. Bioorg. Med. Chem. 2014, 22, 6789–6795. [Google Scholar] [CrossRef]
- Yu, H.-B.; Glukhov, E.; Li, Y.; Iwasaki, A.; Gerwick, L.; Dorrestein, P.C.; Jiao, B.-H.; Gerwick, W.H. Cytotoxic microcolin lipopeptides from the marine cyanobacterium Moorea producens. J. Nat. Prod. 2019, 82, 2608–2619. [Google Scholar] [CrossRef]
- Meickle, T.; Matthew, S.; Ross, C.; Luesch, H.; Paul, V. Bioassay-guided isolation and identification of desacetylmicrocolin B from Lyngbya cf. polychroa. Planta Med. 2009, 75, 1427–1430. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, S.; Kobayashi, M.; Sato, R.; Shinomiya, S.; Iwasaki, A.; Suda, S.; Teruya, T.; Inuzuka, T.; Ohno, O.; Suenaga, K. Minnamide A, a linear lipopeptide from the marine cyanobacterium Okeania hirsuta. Org. Lett. 2019, 21, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Marquez, B.L.; Watts, K.S.; Yokochi, A.; Roberts, M.A.; Verdier-Pinard, P.; Jimenez, J.I.; Hamel, E.; Scheuer, P.J.; Gerwick, W.H. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J. Nat. Prod. 2002, 65, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Suntornchashwej, S.; Chaichit, N.; Isobe, M.; Suwanborirux, K. Hectochlorin and morpholine derivatives from the Thai sea hare, Bursatella leachii. J. Nat. Prod. 2005, 68, 951–955. [Google Scholar] [CrossRef]
- Boudreau, P.D.; Monroe, E.A.; Mehrotra, S.; Desfor, S.; Korobeynikov, A.; Sherman, D.H.; Murray, T.F.; Gerwick, L.; Dorrestein, P.C.; Gerwick, W.H. Expanding the described metabolome of the marine cyanobacterium Moorea producens JHB through orthogonal natural products workflows. PLoS ONE 2015, 10, e0133297. [Google Scholar] [CrossRef]
- Choi, H.; Mevers, E.; Byrum, T.; Valeriote, F.A.; Gerwick, W.H. Lyngbyabellins KN from two Palmyra atoll collections of the marine cyanobacterium Moorea bouillonii. Eur. J. Org. Chem. 2012, 2012, 5141. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef]
- Han, B.; McPhail, K.L.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Isolation and structure of five lyngbyabellin derivatives from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. Tetrahedron 2005, 61, 11723–11729. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and structure of the cytotoxin lyngbyabellin B and absolute configuration of lyngbyapeptin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Continuing studies on the cyanobacterium Lyngbya sp.: Isolation and structure determination of 15-norlyngbyapeptin A and lyngbyabellin D. J. Nat. Prod. 2003, 66, 595–598. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Structurally diverse new alkaloids from Palauan collections of the apratoxin-producing marine cyanobacterium Lyngbya sp. Tetrahedron 2002, 58, 7959–7966. [Google Scholar] [CrossRef]
- Matthew, S.; Salvador, L.A.; Schupp, P.J.; Paul, V.J.; Luesch, H. Cytotoxic halogenated macrolides and modified peptides from the apratoxin-producing marine cyanobacterium Lyngbya bouillonii from Guam. J. Nat. Prod. 2010, 73, 1544–1552. [Google Scholar] [CrossRef] [Green Version]
- Petitbois, J.G.; Casalme, L.O.; Lopez, J.A.V.; Alarif, W.M.; Abdel-Lateff, A.; Al-Lihaibi, S.S.; Yoshimura, E.; Nogata, Y.; Umezawa, T.; Matsuda, F. Serinolamides and lyngbyabellins from an Okeania sp. cyanobacterium collected from the Red Sea. J. Nat. Prod. 2017, 80, 2708–2715. [Google Scholar] [CrossRef]
- Fathoni, I.; Petitbois, J.G.; Alarif, W.M.; Abdel-Lateff, A.; Al-Lihaibi, S.S.; Yoshimura, E.; Nogata, Y.; Vairappan, C.S.; Sholikhah, E.N.; Okino, T. Bioactivities of lyngbyabellins from cyanobacteria of Moorea and Okeania genera. Molecules 2020, 25, 3986. [Google Scholar] [CrossRef]
- Caro-Diaz, E.J.; Valeriote, F.A.; Gerwick, W.H. Highly convergent total synthesis and assignment of absolute configuration of majusculamide D, a potent and selective cytotoxic metabolite from Moorea sp. Org. Lett. 2019, 21, 793–796. [Google Scholar] [CrossRef]
- Ali, M.A.; Bates, R.B.; Crane, Z.D.; Dicus, C.W.; Gramme, M.R.; Hamel, E.; Marcischak, J.; Martinez, D.S.; McClure, K.J.; Nakkiew, P. Dolastatin 11 conformations, analogues and pharmacophore. Bioorg. Med. Chem. 2005, 13, 4138–4152. [Google Scholar] [CrossRef]
- Simmons, T.L.; Nogle, L.M.; Media, J.; Valeriote, F.A.; Mooberry, S.L.; Gerwick, W.H. Desmethoxymajusculamide C, a cyanobacterial depsipeptide with potent cytotoxicity in both cyclic and ring-opened forms. J. Nat. Prod. 2009, 72, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivonen, K.; Leikoski, N.; Fewer, D.P.; Jokela, J. Cyanobactins—Ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 2010, 86, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Ireland, C.M.; Durso Jr, A.R.; Newman, R.A.; Hacker, M.P. Antineoplastic cyclic peptides from the marine tunicate Lissoclinum patella. J. Org. Chem. 1982, 47, 1807–1811. [Google Scholar] [CrossRef]
- Fu, X.; Do, T.; Schmitz, F.J.; Andrusevich, V.; Engel, M.H. New cyclic peptides from the ascidian Lissoclinum patella. J. Nat. Prod. 1998, 61, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Fujiki, H.; Tanaka, Y.; Miyake, R.; Kikkawa, U.; Nishizuka, Y.; Sugimura, T. Activation of calcium-activated, phospholipid-dependent protein kinase (protein kinase C) by new classes of tumor promoters: Teleocidin and debromoaplysiatoxin. Biochem. Biophys. Res. Commun. 1984, 120, 339–343. [Google Scholar] [CrossRef]
- Nakamura, H.; Kishi, Y.; Pajares, M.A.; Rando, R.R. Structural basis of protein kinase C activation by tumor promoters. Proc. Natl. Acad. Sci. USA 1989, 86, 9672–9676. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Satake, M.; Zhang, B.-T.; Xiao, Y.-Y.; Fukuoka, M.; Uchida, H.; Nagai, H. Neo-aplysiatoxin A isolated from Okinawan cyanobacterium Moorea producens. Molecules 2020, 25, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, H.; Watanabe, M.; Sato, S.; Kawaguchi, M.; Xiao, Y.-Y.; Hayashi, K.; Watanabe, R.; Uchida, H.; Satake, M. New aplysiatoxin derivatives from the Okinawan cyanobacterium Moorea producens. Tetrahedron 2019, 75, 2486–2494. [Google Scholar] [CrossRef]
- Tang, Y.-H.; Liang, T.-T.; Fan, T.-T.; Keen, L.J.; Zhang, X.-D.; Xu, L.; Zhao, Q.; Zeng, R.; Han, B.-N. Neo-debromoaplysiatoxin C, with new structural rearrangement, derived from debromoaplysiatoxin. Nat. Prod. Res. 2020, 34, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Sato, S.; Iida, K.; Hayashi, K.; Kawaguchi, M.; Uchida, H.; Satake, M. Oscillatoxin I: A new aplysiatoxin derivative, from a marine cyanobacterium. Toxins 2019, 11, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, O.; Iwasaki, A.; Same, K.; Kudo, C.; Aida, E.; Sugiura, K.; Sumimoto, S.; Teruya, T.; Tashiro, E.; Simizu, S. Isolation of caldorazole, a thiazole-containing polyketide with selective cytotoxicity under glucose-restricted conditions. Org. Lett. 2022, 24, 4547–4551. [Google Scholar] [CrossRef]
- Kurisawa, N.; Iwasaki, A.; Teranuma, K.; Dan, S.; Toyoshima, C.; Hashimoto, M.; Suenaga, K. Structural determination, total synthesis, and biological activity of iezoside, a highly potent Ca2+-ATPase inhibitor from the marine cyanobacterium Leptochromothrix valpauliae. J. Am. Chem. Soc. 2022, 144, 11019–11032. [Google Scholar] [CrossRef]
- Gromek, S.M.; deMayo, J.A.; Maxwell, A.T.; West, A.M.; Pavlik, C.M.; Zhao, Z.; Li, J.; Wiemer, A.J.; Zweifach, A.; Balunas, M.J. Synthesis and biological evaluation of santacruzamate A analogues for anti-proliferative and immunomodulatory activity. Bioorg. Med. Chem. 2016, 24, 5183–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlik, C.M.; Wong, C.Y.; Ononye, S.; Lopez, D.D.; Engene, N.; McPhail, K.L.; Gerwick, W.H.; Balunas, M.J. Santacruzamate A, a potent and selective histone deacetylase inhibitor from the Panamanian marine cyanobacterium cf. Symploca sp. J. Nat. Prod. 2013, 76, 2026–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A. Pharmacological or transcriptional inhibition of both HDAC 1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018, 51, e12447. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wei, D.; Lu, T.; Ma, D.; Yu, K.; Fang, Q.; Zhang, Z.; Wang, W.; Wang, J. CAY10683 and imatinib have synergistic effects in overcoming imatinib resistance via HDAC2 inhibition in chronic myeloid leukemia. RSC Adv. 2020, 10, 828–844. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; He, D.; Xiao, M.; Zhu, Y.; Zhou, J.; Cao, K. Long noncoding RNA LINC00518 induces radioresistance by regulating glycolysis through an miR-33a-3p/HIF-1α negative feedback loop in melanoma. Cell Death Dis. 2021, 12, 245. [Google Scholar] [CrossRef]
- Randino, R.; Gazzerro, P.; Mazitschek, R.; Rodriquez, M. Synthesis and biological evaluation of santacruzamate-A based analogues. Bioorg. Med. Chem. 2017, 25, 6486–6491. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Luo, D.; Luesch, H. Advances in exploring the therapeutic potential of marine natural products. Pharmacol. Res. 2019, 147, 104373. [Google Scholar] [CrossRef] [PubMed]
- Qamar, H.; Hussain, K.; Soni, A.; Khan, A.; Hussain, T.; Chénais, B. Cyanobacteria as natural therapeutics and pharmaceutical potential: Role in antitumor activity and as nanovectors. Molecules 2021, 26, 247. [Google Scholar] [CrossRef]
- Nakao, Y.; Yoshida, W.Y.; Takada, Y.; Kimura, J.; Yang, L.; Mooberry, S.L.; Scheuer, P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004, 67, 1332–1340. [Google Scholar]
- Marino, S.D.; Festa, C.; D’Auria, M.V.; Cresteil, T.; Debitus, C.; Zampella, A. Swinholide J, a potent cytotoxin from the marine sponge Theonella swinhoei. Mar. Drugs 2011, 9, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Humisto, A.; Jokela, J.; Liu, L.; Wahlsten, M.; Wang, H.; Permi, P.; Machado, J.P.; Antunes, A.; Fewer, D.P.; Sivonen, K. The swinholide biosynthesis gene cluster from a terrestrial cyanobacterium, Nostoc sp. strain UHCC 0450. Appl. Environ. Microbiol. 2018, 84, e02321-17. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.S.; Hathaway, B.J.; Sudek, S.; Haygood, M.G.; Rosovitz, M.; Ravel, J.; Schmidt, E.W. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat. Chem. Biol. 2006, 2, 729–735. [Google Scholar] [CrossRef]
- Houssen, W.E.; Jaspars, M. Azole-based cyclic peptides from the sea squirt Lissoclinum patella: Old scaffolds, new avenues. ChemBioChem 2010, 11, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chung-Davidson, Y.-W.; Bussy, U.; Li, W. Recent advances and applications of experimental technologies in marine natural product research. Mar. Drugs 2015, 13, 2694–2713. [Google Scholar] [CrossRef] [Green Version]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; De Marsac, N.T.; Rippka, R. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naman, C.B.; Rattan, R.; Nikoulina, S.E.; Lee, J.; Miller, B.W.; Moss, N.A.; Armstrong, L.; Boudreau, P.D.; Debonsi, H.M.; Valeriote, F.A. Integrating molecular networking and biological assays to target the isolation of a cytotoxic cyclic octapeptide, samoamide A, from an American Samoan marine cyanobacterium. J. Nat. Prod. 2017, 80, 625–633. [Google Scholar] [CrossRef]
- Costa, M.; Costa-Rodrigues, J.; Fernandes, M.H.; Barros, P.; Vasconcelos, V.; Martins, R. Marine cyanobacteria compounds with anticancer properties: A review on the implication of apoptosis. Mar. Drugs 2012, 10, 2181–2207. [Google Scholar] [CrossRef] [Green Version]
- Salvador-Reyes, L.A.; Luesch, H. Biological targets and mechanisms of action of natural products from marine cyanobacteria. Nat. Prod. Rep. 2015, 32, 478–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel-López, E.; Robles-Bañuelos, B.; Guadiana-Ramírez, N.; Alvarez-Garduño, V.; Galván-Arzate, S.; Zazueta, C.; Karasu, C.; Túnez, I.; Tinkov, A.; Aschner, M. Thallium induces antiproliferative and cytotoxic activity in glioblastoma C6 and U373 cell cultures via apoptosis and changes in cell cycle. Neurotox Res. 2022, 40, 814–824. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 2021, 14, 8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Genera | Metabolites | Selected References |
|---|---|---|---|---|
| Oscillatoriales | Oscillatoriaceae | Lyngbya, Oscillatoria, Moorea, Okeania, Phormidium Neolyngbya | Almiramides, anaenamides, aplysiatoxins, apratoxins, aurilides, bisebromoamides, carmaphycins, caldorazole, caylobolides, cocosamides, dolastatins, hectochlorins, iezoside, largazole, lyngbyabellins, majusculamides, microcolins, santacruzamate A, swinholide-type, wenchangamides | [2,12,13,14,15] |
| Coleofasciculaceae | Geitlerinema | |||
| Gomontiellaceae | Hormoscilla | |||
| Microcoleaceae | Trichodesmium, Symploca, Caldora | |||
| Vermifilaceae | Leptochromothrix | |||
| Synechococcales | Procholortrichaceae | Nodosilinea | Aplysiatoxins, bartolosides, coibamide A, dolastatins, patellamides | [10,16,17,18] |
| Leptolyngbyeaceae | Leptolingbya | |||
| Prochloraceae | Prochloron | |||
| Merismopediaceae | Synechocystis | |||
| Schizotrichaceae | Schizothrix |
| Chemical Family | Metabolites | |
|---|---|---|
| Glycolipids | Bartolosides | |
| Macrolides | Caylobolides Swinholide-type compounds | |
| Peptides | Bisebromoamides Carmaphycins Dolastatin 10 * | |
| Subfamily: Depsipeptides Linear depsipeptides Cyclic depsipeptides | Anaenamides Apratoxins Aurilides Cocosamides Coibamide A Largazole Dolastatins 15 and 16 * | |
| Subfamily: Lipopeptides Linear lipopeptides Cyclic lipopeptides | Almiramides Microcolins Wenchangamides Hectochlorins | |
| Peptolide † | Lyngbyabellins Majusculamides Patellamides # | |
| Polyketides | Aplysiatoxins Caldorazole | |
| Metabolites from other chemical families | Iezoside Santacruzamate A | |
| Cell Line | Molecule | References | ||
|---|---|---|---|---|
| Swinholide A | Ankaraholide A | Samholide A–I | ||
| L1210 | 0.03 | [13,23,26,27] | ||
| KB | 0.04 | |||
| SW-480 | 0.07 | |||
| KATO-III | 0.05 | |||
| HT-1080 | 0.017 | |||
| T-24 | 0.046 | |||
| PC-3 | 6.0 | |||
| PC-8 | 0.12 | |||
| PC-9 | 0.13 | |||
| PC-10 | 0.11 | |||
| PC-13 | 0.10 | |||
| QG-56 | 0.04 | |||
| Daudi | 0.036 | |||
| NCI-H460 | 119 * | 170–910 * | ||
| Neuro-2a | 262 * | |||
| MDA-MB-435 | 8.9 * | |||
| Apratoxin Analog | Cell Line (IC50/* GI50 Values in nM) | Selected References | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| KB | LoVo | NCI-H460 | HT-29 | HeLa | U20S | HCT-116 | NCI-60 1 | Neuro-2a | Panel HCCL 2 | ||
| A | 0.52 | 0.36 | 2.5 | 1.4 | 10 | 10 | 1.21 | 1–3 * | 1000 | 4.9–41 * | [37,38,39,40,41,42] |
| A SO * | 89.9 | ||||||||||
| B | 21.3 | 10.8 | |||||||||
| C | 1 | 0.73 | |||||||||
| D | 2.6 | ||||||||||
| E | 21 | 72 | 59 | ||||||||
| F | 2 | 36.7 | |||||||||
| G | 14 | ||||||||||
| H | 3.4 | ||||||||||
| Molecule | Cell Line (IC50 / LC50 Values in nM) | References | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HeLa S3 | P388 | BJ | BJ Shp 53 | PC3 | SK-OV-3 | HCT8 | Neuro-2a | NCI-H460 | KB | A549 | ||
| Aur B | 10 * | 40 * | [55,56,57,58,59,60,61] | |||||||||
| Aur C | 50 * | 130 * | ||||||||||
| Palau | 13 | |||||||||||
| Odo | 26.3 | 4.2 | ||||||||||
| Lag A | 6.4 | 20.2 | 58.8 | 2.5 | 3.8 | 1.6 | 2.9 | |||||
| Lag B | 20.5 | 5.2 | ||||||||||
| Lag C | 24.4 | 2.6 | 4.5 | 2.1 | 2.4 | |||||||
| Lag D | 7.1 | |||||||||||
| Lag ’ | 68.2 | |||||||||||
| Tested Cell Lines | LC50/EC50/GI50 Values (nM) | Cellular Effects | References |
|---|---|---|---|
| NCI-H460 and | LC50 ≤ 23 | Arrest in the G1 phase of the cell cycle; mTOR-independent autophagy; apoptosis; decreased protein expression due to the inhibition of Sec9 | [17,44,67,68] |
| Neuro-2a | |||
| MDA-MB-231 | GI50 = 1–7.4 | ||
| LOX IMVI | GI50 = 7.4 | ||
| HL-60 | GI50 = 7.4 | ||
| SNB-75 | GI50 = 7.6 | ||
| U87-MG | EC50 = 28.8 | ||
| SF-295 | EC50 = 96.2 | ||
| A549 | GI50 = 5.4 | ||
| PANC-1 | GI50 = 3.1 | ||
| MDA-MB-436 | EC50 = 0.9 | ||
| MDA-MB-468 | EC50 = 0.4 | ||
| HS578T | EC50 = 4.0 | ||
| BT474 | EC50 = 0.8 |
| Tested Cell Lines | GI50 or IC50 Values (nM) | Cellular Effects | References |
|---|---|---|---|
| MDA-MB-231 | GI50 = 7.7 | Histone deacetylases inhibitor; apoptosis; modulation of the levels of cell cycle regulators; antagonism of the AKT, KRAS, and HIF pathways; reduction of the epidermal growth factor receptors levels; inhibition of ubiquitin-activating enzyme (E1); proteasomal degradation of E2F1; antiangiogenic activity; upregulation of the Pax6 gene | [74,75,76,77,78,79,80,81,82,83,84,85] |
| U2OS | GI50 = 55 | ||
| HT-29 | GI50 = 12 | ||
| IMR-32 | GI50 = 16 | ||
| HCT-116 | GI50 = 80 | ||
| A549 | GI50 = 320 | ||
| SK-OV-3 | IC50 = 250 | ||
| HeLa | IC50 = 170 | ||
| Eca-109 | IC50 = 100 | ||
| Bel 7402 | IC50 = 170 | ||
| U937 | IC50 = 20 | ||
| 797 | IC50 = 24 | ||
| 10326 | IC50 = 25 | ||
| PC3 | IC50 ≤ 500 | ||
| LNCap | IC50 ≤ 500 | ||
| Panel of melanoma cell lines | IC50 = 45–315 | ||
| NCI-H1975 | IC50 = 83 | ||
| NCI-H460 | IC50 = 120 | ||
| GLC-82 | IC50 = 190 | ||
| L78 | IC50 = 570 | ||
| SPC-A1 | IC50= 140 | ||
| 95D | IC50 = 420 | ||
| NCI-H466 | IC50 = 520 | ||
| SW620 | IC50 = 26.5 | ||
| MiaPaCa | IC50 = 206.4 | ||
| SH-SY5Y | IC50 = 102 | ||
| SF-268 | IC50 = 62 | ||
| SF-295 | IC50 = 68 |
| Cell Line (IC50 Values in nM) | Dolastatin (D) or Symplostatin (Sy) | References | |||
|---|---|---|---|---|---|
| D10 | D15 | Sy1 | Sy3 | ||
| KB | 0.052 | 0.15–0.20 | 3.9 | [91,92,93,94,95,96,97,98,99] | |
| LoVo | 0.076 | 0.34–0.50 | 10.3 | ||
| A549 | 0.97 | 0.74 | |||
| MDA-MB-435 | 0.15 | ||||
| SK-OV-3 | 0.09 | ||||
| NCI/ADR | 2.9 | ||||
| DB, HT, RL, SR | 0.00013–0.13 | 0.0013 to 0.13 | |||
| H82, H446, H69, H510 | 0.032–0.184 | ||||
| BE, HT-29, MAWI, SW480, SW620 | 0.018–0.16 | 0.12–2.24 | |||
| A2780, CHI, 41M, HX/62 | 0.046–1.8 | 0.061–10 | |||
| L1210 | 0.4 | 3 | |||
| DU-145 | 0.5 | ||||
| HCT-116 | 2.2 | ||||
| Lyngbyabellin | Cell Line | References | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HT-29 | HeLa | KB | LoVo | NCI-H460 | Neuro-2a | HCT-116 | MCF7 | CA46 | ||
| A | 0.047 * | 0.022 * | 0.03 ** | 0.5 ** | [115,118,119,120,122,123,124,125,126] | |||||
| 27-Deoxy A | 0.012 * | 0.0073 * | 0.31 * | |||||||
| B | 1.1 * | 0.71 * | 0.1 ** | 0.83 ** | 0.1 * | |||||
| C | 2.1 | 5.3 | ||||||||
| D | 0.1 | |||||||||
| E | 0.4 ‡ | 1.2 ‡ | ||||||||
| F | 1 ‡ | 1.8 ‡ | ||||||||
| G | 2.2 ‡ | 4.8 ‡ | ||||||||
| H | 0.2 ‡ | 1.4 ‡ | 0.07 * | |||||||
| I | 1 ‡ | 0.7 ‡ | ||||||||
| J | 0.054 * | 0.041 * | ||||||||
| N | 0.0048–1.8 * | 0.0409 * | ||||||||
| P | 9 * | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robles-Bañuelos, B.; Durán-Riveroll, L.M.; Rangel-López, E.; Pérez-López, H.I.; González-Maya, L. Marine Cyanobacteria as Sources of Lead Anticancer Compounds: A Review of Families of Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects. Molecules 2022, 27, 4814. https://doi.org/10.3390/molecules27154814
Robles-Bañuelos B, Durán-Riveroll LM, Rangel-López E, Pérez-López HI, González-Maya L. Marine Cyanobacteria as Sources of Lead Anticancer Compounds: A Review of Families of Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects. Molecules. 2022; 27(15):4814. https://doi.org/10.3390/molecules27154814
Chicago/Turabian StyleRobles-Bañuelos, Benjamín, Lorena María Durán-Riveroll, Edgar Rangel-López, Hugo Isidro Pérez-López, and Leticia González-Maya. 2022. "Marine Cyanobacteria as Sources of Lead Anticancer Compounds: A Review of Families of Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects" Molecules 27, no. 15: 4814. https://doi.org/10.3390/molecules27154814
APA StyleRobles-Bañuelos, B., Durán-Riveroll, L. M., Rangel-López, E., Pérez-López, H. I., & González-Maya, L. (2022). Marine Cyanobacteria as Sources of Lead Anticancer Compounds: A Review of Families of Metabolites with Cytotoxic, Antiproliferative, and Antineoplastic Effects. Molecules, 27(15), 4814. https://doi.org/10.3390/molecules27154814