
First Example of Fluorinated Phenanthroline Diamides: Synthesis, Structural Study, and Complexation with Lanthanoids

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results and Discussions
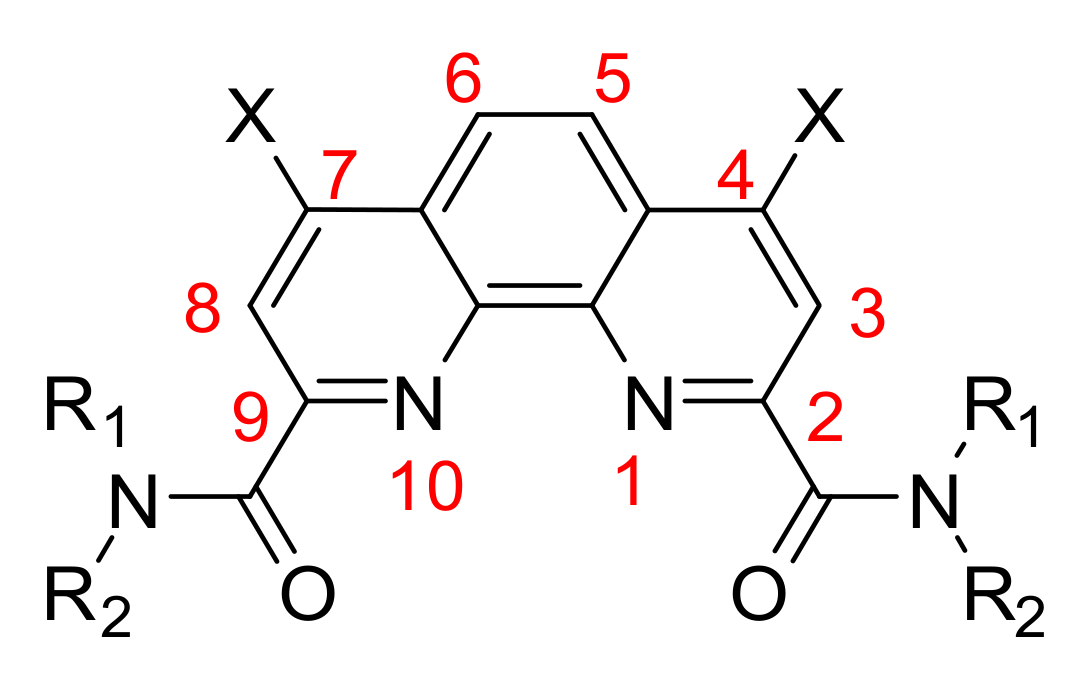
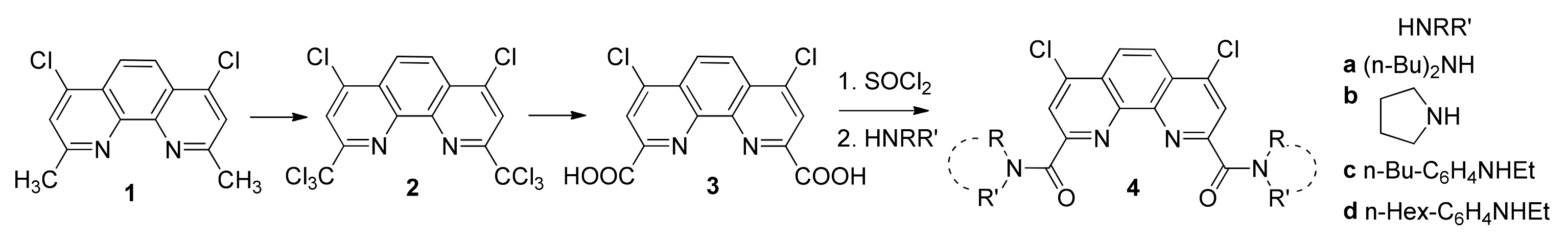
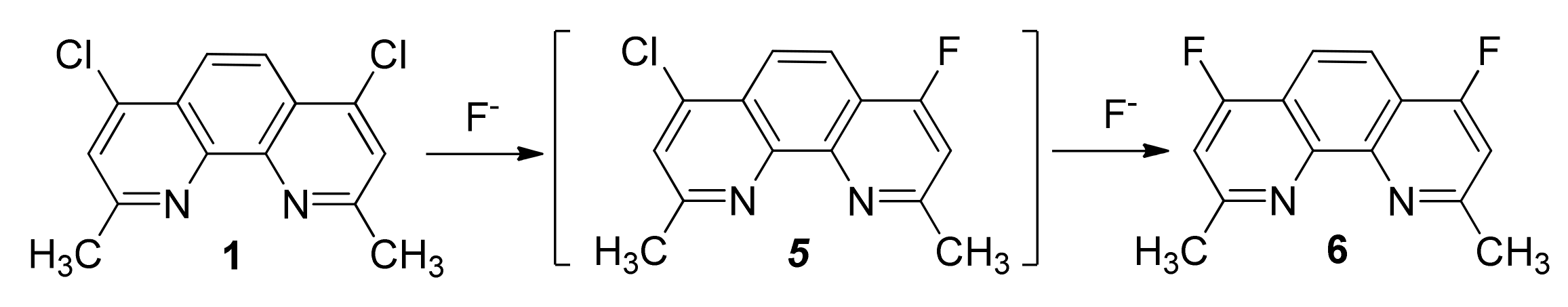
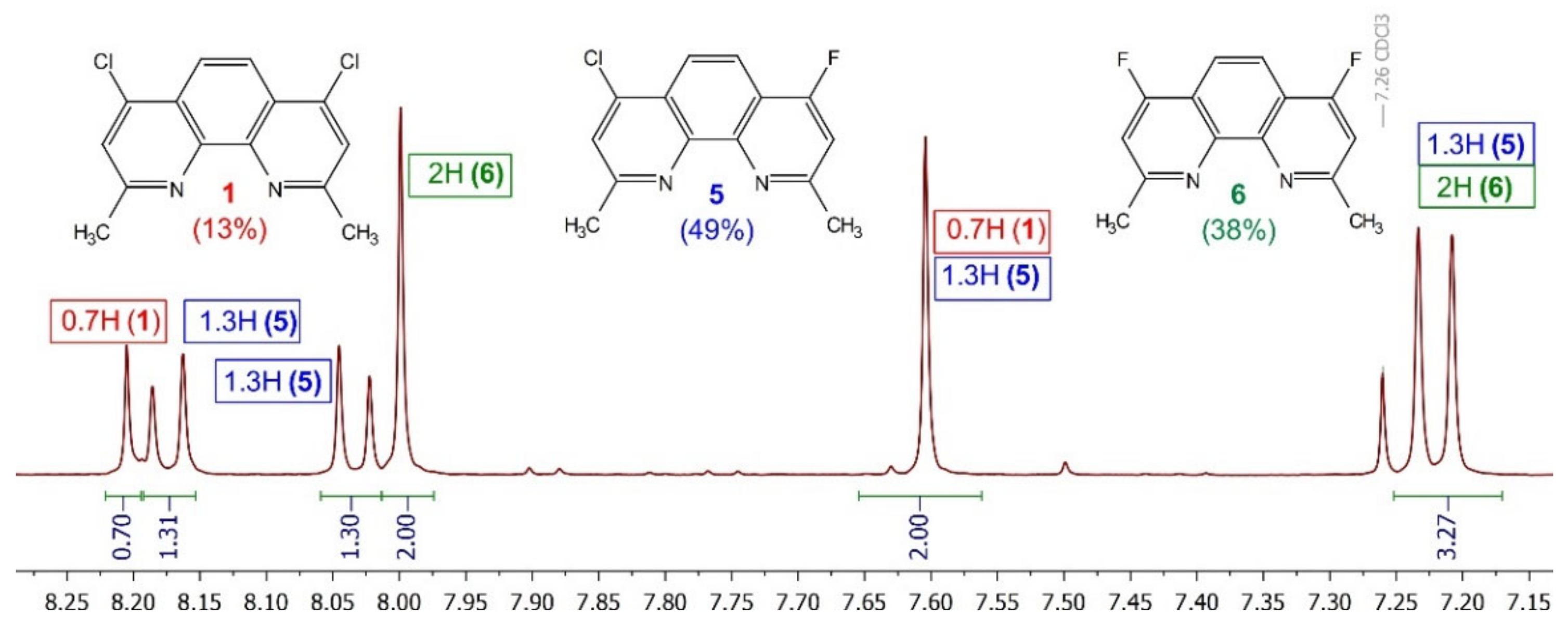
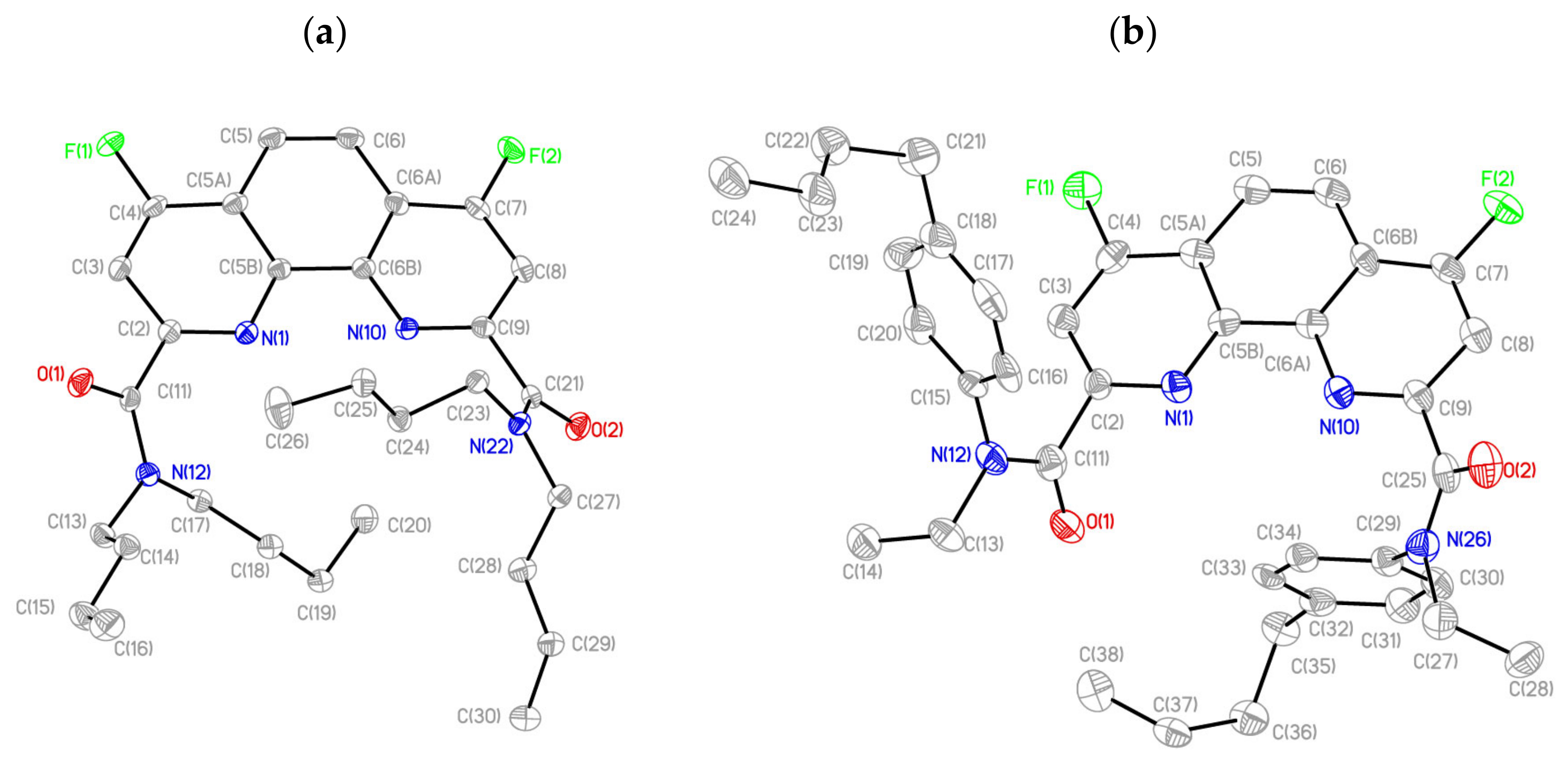
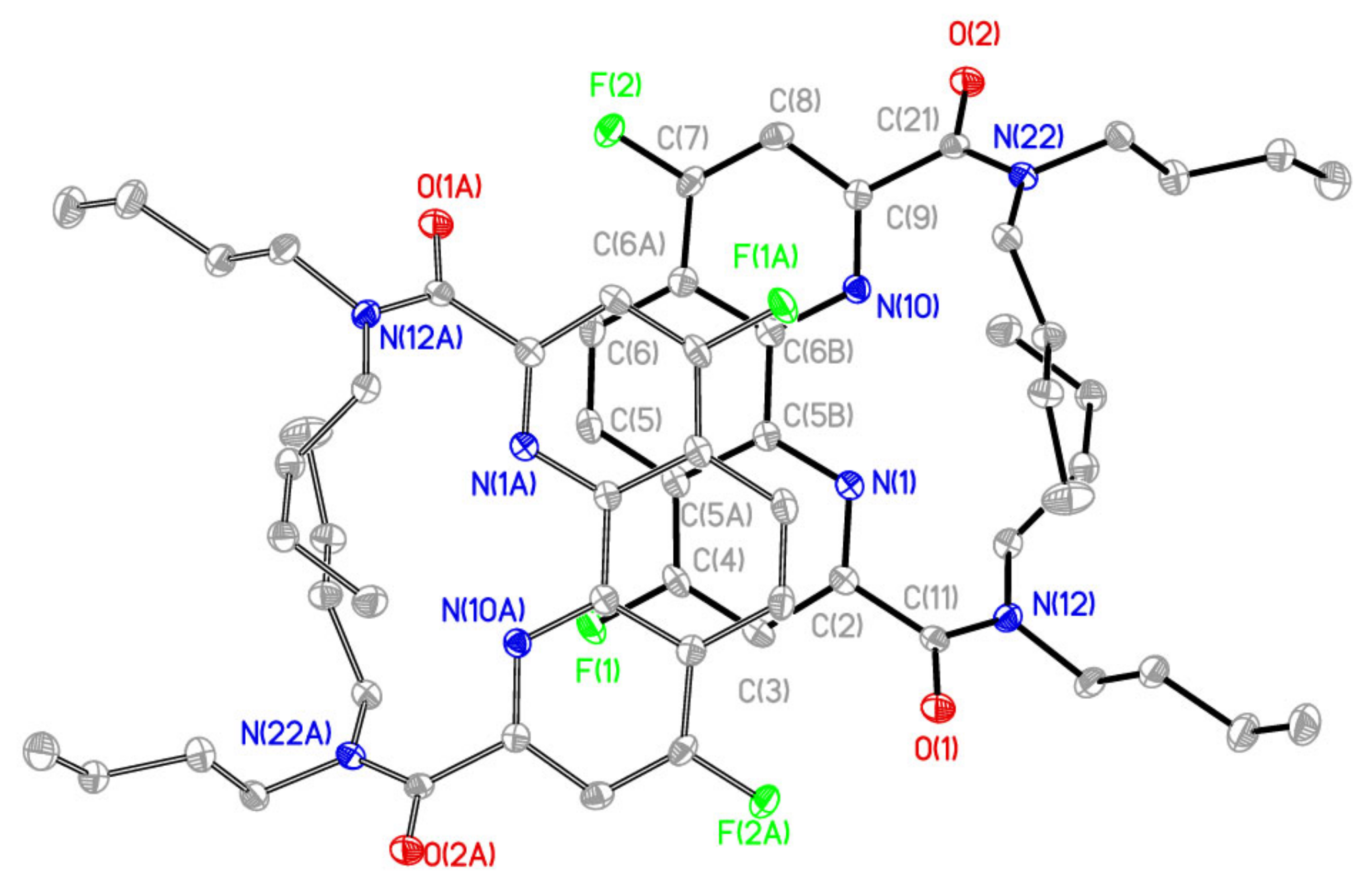
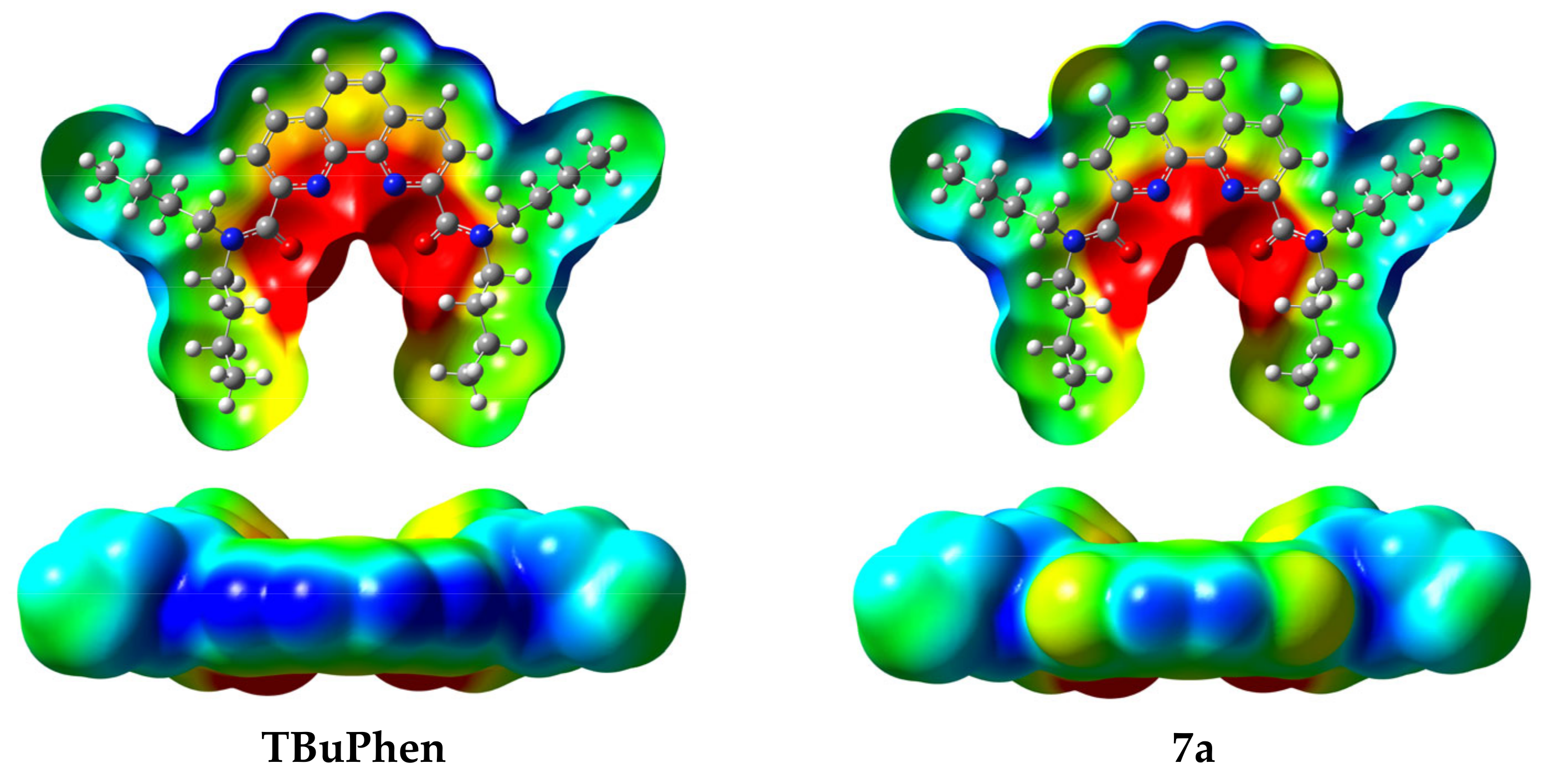
2.1. Synthesis and Structure of Fluorinated Phenanthrolinediamide
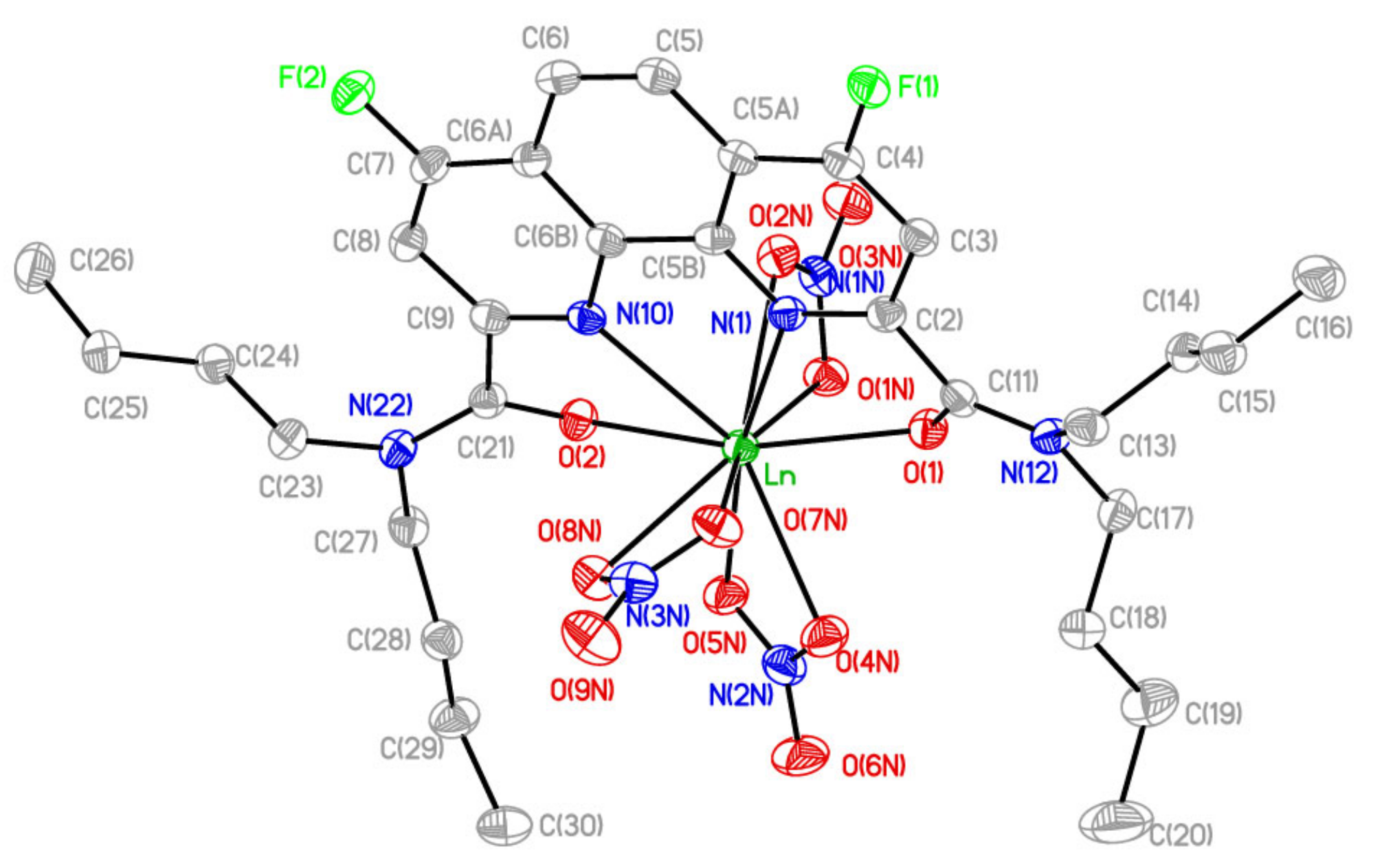
2.2. Preparation of Complexes with Nitrates of REE
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Synthesis and Analytical Data
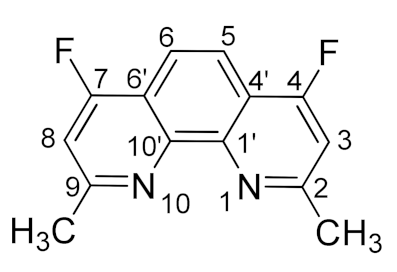
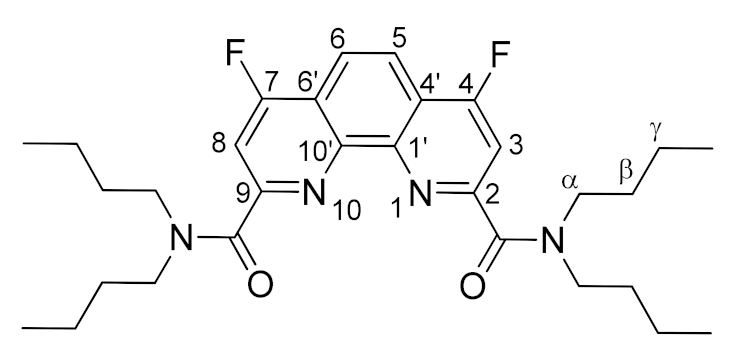
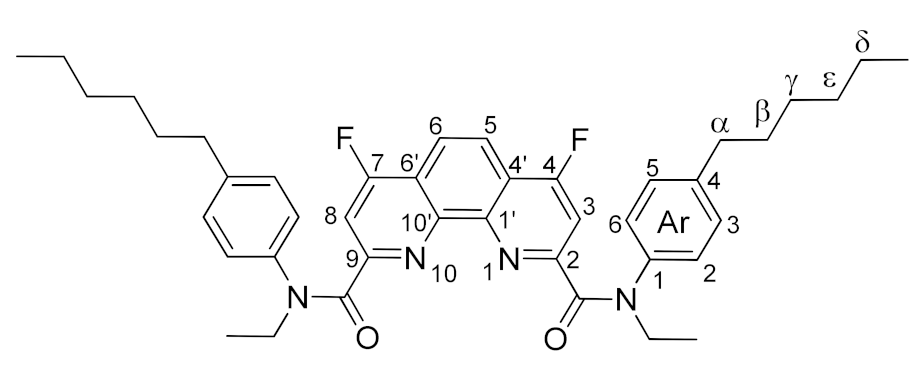
3.3.1. General Method for Synthesis of 4,7-Difluorosubstituted 1,10-Phenanthroline-2,9-dicarboxamides


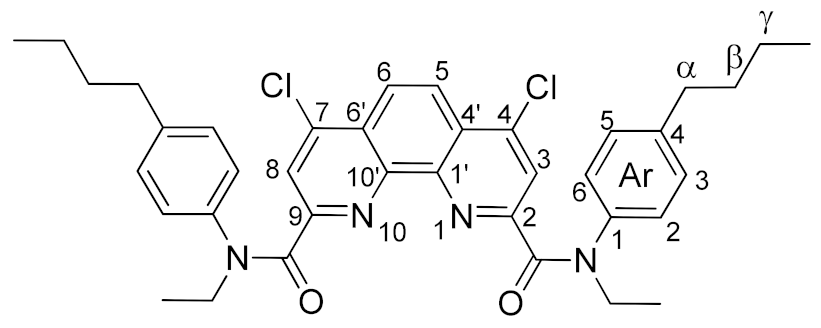
3.3.2. General Method for Synthesis of Complexes 7a•Ln(NO3)3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Poinssot, C.; Bourg, S.; Ouvrier, N.; Combernoux, N.; Rostaing, C.; Vargas-Gonzalez, M.; Bruno, J. Assessment of the Environmental Footprint of Nuclear Energy Systems. Comparison between Closed and Open Fuel Cycles. Energy 2014, 69, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Leoncini, A.; Huskens, J.; Verboom, W. Ligands for f-element extraction used in the nuclear fuel cycle. Chem. Soc. Rev. 2017, 46, 7229–7273. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, D.N.; Bushkov, N.S.; Lavrov, H.V.; Dolgushin, F.M.; Ustynyuk, N.A.; Ustynyuk, Y.A. 4,7-Di-n-butoxy-1,10-phenanthroline-2,9-dicarboxamide: A Tetradentate Ligand Featuring Excellent Solubility in Nonpolar Media. INEOS OPEN 2019, 2, 130–133. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, X.; Yuan, L.; Chai, Z.; Shi, W. Coordination of Eu(III) with 1,10-Phenanthroline-2,9-dicarboxamide Derivatives: A Combined Study by MS, TRLIF, and DFT. Inorg. Chem. 2019, 58, 10239–10247. [Google Scholar] [CrossRef]
- Distler, P.; Mindov’a, M.; John, J.; Babain, V.A.; Alyapyshev, M.Y.; Tkachenko, L.I.; Kenf, E.V.; Harwood, L.M.; Afsar, A. Fluorinated Carbonates as New Diluents for Extraction and Separation of f-Block Elements. Solvent Extr. Ion Exch. 2020, 38, 180–193. [Google Scholar] [CrossRef]
- Gutorova, S.V.; Matveev, P.I.; Lemport, P.S.; Trigub, A.L.; Pozdeev, A.S.; Yatsenko, A.V.; Tarasevich, B.N.; Konopkina, E.A.; Khult, E.K.; Roznyatovsky, V.A.; et al. Structural Insight into Complexation Ability and Coordination of Uranyl Nitrate by 1,10-Phenanthroline-2,9-diamides. Inorg. Chem. 2022, 61, 384–398. [Google Scholar] [CrossRef]
- Lemport, P.S.; Matveev, P.I.; Yatsenko, A.V.; Evsiunina, M.V.; Petrov, V.S.; Tarasevich, B.N.; Roznyatovsky, V.A.; Dorovatovskii, P.V.; Khrustalev, V.N.; Zhokhov, S.S.; et al. The impact of alicyclic substituents on the extraction ability of new family of 1,10-phenanthroline-2,9-diamides. RSC Adv. 2020, 10, 26022–26033. [Google Scholar] [CrossRef]
- Ustynyuk, Y.A.; Borisova, N.E.; Babain, V.A.; Gloriozov, I.P.; Manuilov, A.Y.; Kalmykov, S.N.; Alyapyshev, M.Y.; Tkachenko, L.I.; Kenf, E.V.; Ustynyuk, N.A. N,N′-Dialkyl-N,N′-diaryl-1,10-phenanthroline-2,9-dicarboxamides as donor ligands for separation of rare earth elements with a high and unusual selectivity. DFT computational and experimental studies. Chem. Commun. 2015, 51, 7466–7469. [Google Scholar] [CrossRef]
- Ustynyuk, Y.A.; Lemport, P.S.; Roznyatovsky, V.A.; Lyssenko, K.A.; Gudovannyy, A.O.; Matveev, P.I.; Khult, E.K.; Evsiunina, M.V.; Petrov, V.G.; Gloriozov, I.P.; et al. First Trifluoromethylated Phenanthrolinediamides: Synthesis, Structure, Stereodynamics and Complexation with Ln(III). Molecules 2022, 27, 3114. [Google Scholar] [CrossRef]
- Borisova, N.E.; Kostin, A.A.; Reshetova, M.D.; Lyssenko, K.A.; Belova, E.V.; Myasoedov, B.F. The structurally rigid tetradentate N,N0,O,O0-ligands based on phenanthroline for binding of f-elements: The substituents vs. Structures of the complexes. Inorg. Chim. Acta 2018, 478, 148–154. [Google Scholar] [CrossRef]
- Morss, L.R.; Edelstein, N.M.; Fuger, J. The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, The Netherlands, 2010; ISBN 978-94-007-0210-3/978-94-007-0211-0. [Google Scholar]
- Matveev, P.I.; Mitrofanov, A.A.; Petrov, V.G.; Zhokhov, S.S.; Smirnova, A.A.; Ustynyuk, Y.A.; Kalmykov, S.N. Testing a simple approach for theoretical evaluation of radiolysis products in extraction systems. A case of N,O-donor ligands for Am/Eu separation. RSC Adv. 2017, 7, 55441–55449. [Google Scholar] [CrossRef] [Green Version]
- Gouverneur, V.; Seppelt, K. Special Issue Fluorine Chemistry. Chem. Rev. 2015, 115, 563–565. [Google Scholar] [CrossRef]
- Nenajdenko, V.G. Fluorine in Heterocyclic Chemistry; Springer International Publishing: Cham, Switzerland, 2014; ISBN 978-3-319-04346-3. [Google Scholar] [CrossRef]
- Petrov, V.A. Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Gakh, A.A.; Kirk, K.L. Fluorinated Heterocycles; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2009. [Google Scholar]
- Meyer, F. Trifluoromethyl nitrogen heterocycles: Synthetic aspects and potential biological targets. Chemical communications. Chem. Commun. 2016, 52, 3077–3094. [Google Scholar] [CrossRef]
- Hiyama, T. Organofluorine Compounds. Chemistry and Applications; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Chambers, R.D. Fluorine in Organic Chemistry; Blackwell Publishing Ltd.: Oxford, UK, 2004. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Uneyama, K. Organofluorine Chemistry; Blackwell Publishing: Oxford, UK, 2006. [Google Scholar]
- Theodoridis, G. Fluorine-containing agrochemicals: An overview of recent developments. In Advances in Fluorine Science; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 121–175. [Google Scholar] [CrossRef]
- Bégué, J.P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Tressaud, A.; Haufe, G. (Eds.) Fluorine and Health. Molecular Imaging, Biomedical Materials and Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2008; pp. 553–778. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Mikami, K.; Yamazaki, T.; Welch, J.T.; Honek, J.F. (Eds.) Current Fluoroorganic Chemistry. New Synthetic Directions, Technologies, Materials, and Biological Applications; ACS Symposium Series 949; American Chemical Society: Washington, DC, USA, 2006. [Google Scholar]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Lüdtke, C.; Haupt, A.; Wozniak, M.; Kulak, N. Synthesis of fluorine-containing 1,10-phenanthrolines using mild versions of Skraup and Doebner-von Miller reactions. J. Fluor. Chem. 2017, 193, 98–105. [Google Scholar] [CrossRef]
- Lűdtke, C.; Sobottka, S.; Heinrich, J.; Liebing, P.; Wedepohl, S.; Sarkar, B.; Kulak, N. Forty Years after the Discovery of Its Nucleolytic Activity: [Cu(phen)2]2+ Shows Unattended DNA Cleavage Activity upon Fluorination. Chem. Eur. J. 2021, 27, 3273–3277. [Google Scholar] [CrossRef]
- Meurs, J.H.H.; Eilenberg, W. Oxidative fluorination in amine-hf mixturess. Tetrahedron 1991, 47, 705–714. [Google Scholar] [CrossRef]
- Klenner, M.A.; Pascali, G.; Zhang, B.; Sia, T.R.; Spare, L.K.; Krause-Heuer, A.M.; Aldrich-Wright, J.R.; Greguric, I.; Guastella, A.J.; Massi, M.; et al. A Fluorine-18 Radiolabeling Method Enabled by Rhenium(I) Complexation Circumvents the Requirement of Anhydrous Conditions. Chem. Eur. J. 2017, 23, 6499–6503. [Google Scholar] [CrossRef] [Green Version]
- Klenner, M.A.; Zhang, B.; Ciancaleoni, G.; Howard, J.K.; Maynard-Casely, H.E.; Clegg, J.K.; Massi, M.; Fraser, B.H.; Pascali, G. Rhenium(I) complexation–dissociation strategy for synthesising fluorine-18 labelled pyridine bidentate radiotracers. RSC Adv. 2020, 10, 8853–8865. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, H.; Xu, J.; Chen, C.; Pan, Y.; Luo, Z.; Zhang, Z.; Li, H.; Xu, L. Rhodium(III)-Catalyzed Oxidative Annulation of 2,2′-Bipyridine N-Oxides with Alkynes via Dual C–H Bond Activation. Org. Lett. 2018, 20, 3843–3847. [Google Scholar] [CrossRef]
- Maron, A.; Nycz, J.E.; Machura, B.; Małecki, J.G. Luminescence properties of palladium(II) phenanthroline derivative coordination compounds. ChemistrySelect 2016, 4, 798–804. [Google Scholar] [CrossRef]
- Nycz, J.E.; Wantulok, J.; Sokolova, R.; Pajchel, L.; Stankevič, M.; Szala, M.; Malecki, J.G.; Swoboda, D. Synthesis and Electrochemical and Spectroscopic Characterization of 4,7-diamino-1,10-phenanthrolines and Their Precursors. Molecules 2019, 24, 4102. [Google Scholar] [CrossRef] [Green Version]
- Finger, G.C.; Starr, L.D.; Dickerson, D.R.; Gutowsky, H.S.; Hamer, J. Aromatic Fluorine Compounds. XI. Replacement of Chlorine by Fluorine in Halopyridines. J. Org. Chem. 1963, 28, 1666–1668. [Google Scholar] [CrossRef]
- Hong, C.M.; Whittaker, A.M.; Schultz, D.M. Nucleophilic Fluorination of Heteroaryl Chlorides and Aryl Triflates Enabled by Cooperative Catalysis. J. Org. Chem. 2021, 86, 3999–4006. [Google Scholar] [CrossRef]
- Stewart, W.E.; Siddall, T.H. Nuclear magnetic resonance studies of amides. Chem. Rev. 1970, 70, 517–551. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density Functional Theory Is Straying from the Path toward the Exact Functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.A. Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemport, P.S.; Evsiunina, M.V.; Nelyubina, Y.V.; Isakovskaya, K.L.; Khrustalev, V.N.; Petrov, V.S.; Pozdeev, A.S.; Matveev, P.I.; Ustynyuk, Y.A.; Nenajdenko, V.G. Significant impact of lanthanide contraction on the structure of the phenanthroline complexes. Mendeleev Commun. 2021, 31, 853–855. [Google Scholar] [CrossRef]
- Alvarez, S.; Llunell, M. Continuous symmetry measures of penta-coordinate molecules: Berry and non-Berry distortions of the trigonal bipyramid. Dalton Trans. 2000, 19, 3288. [Google Scholar] [CrossRef]
- Casanova, D.; Llunell, M.; Alemany, P.; Alvarez, S. The Rich Stereochemistry of Eight-Vertex Polyhedra: A Continuous Shape Measures Study. Chem. Eur. J. 2005, 11, 1479. [Google Scholar] [CrossRef]
- Gao, Y.; Twamley, B.; Shreeve, J.M. Coordination Networks with Fluorinated Backbones. Inorg. Chem. 2006, 45, 1150–1155. [Google Scholar] [CrossRef]


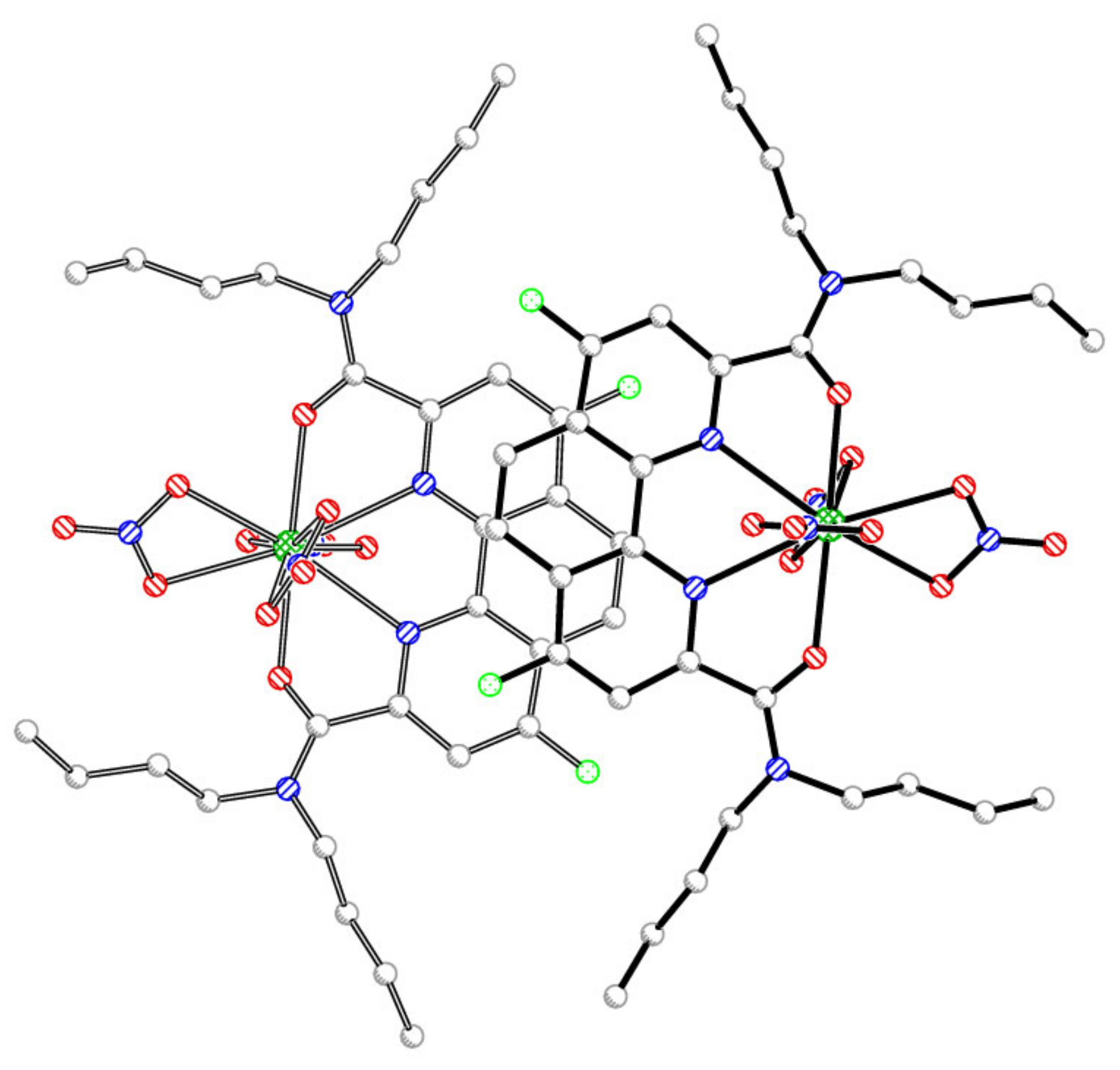
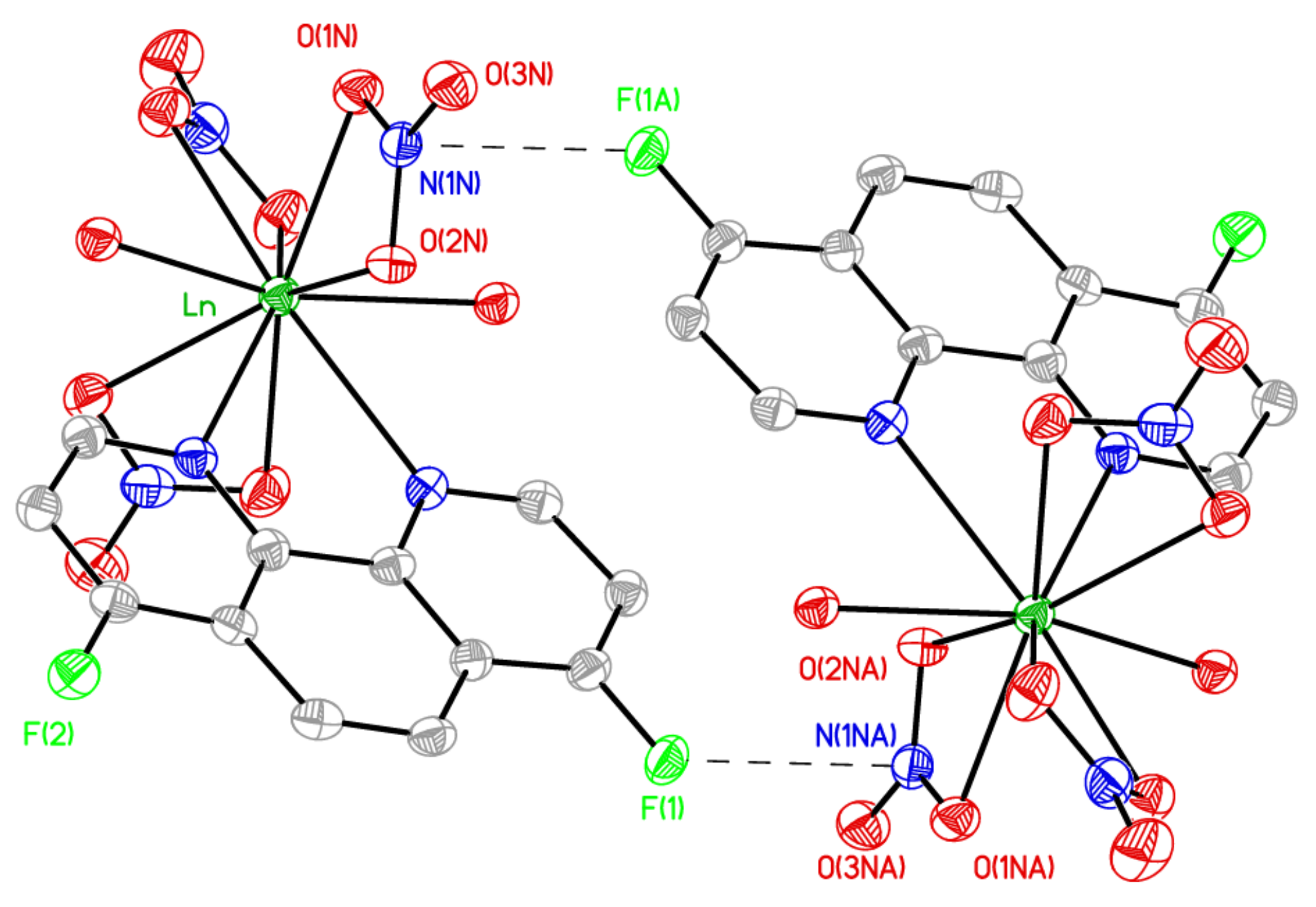
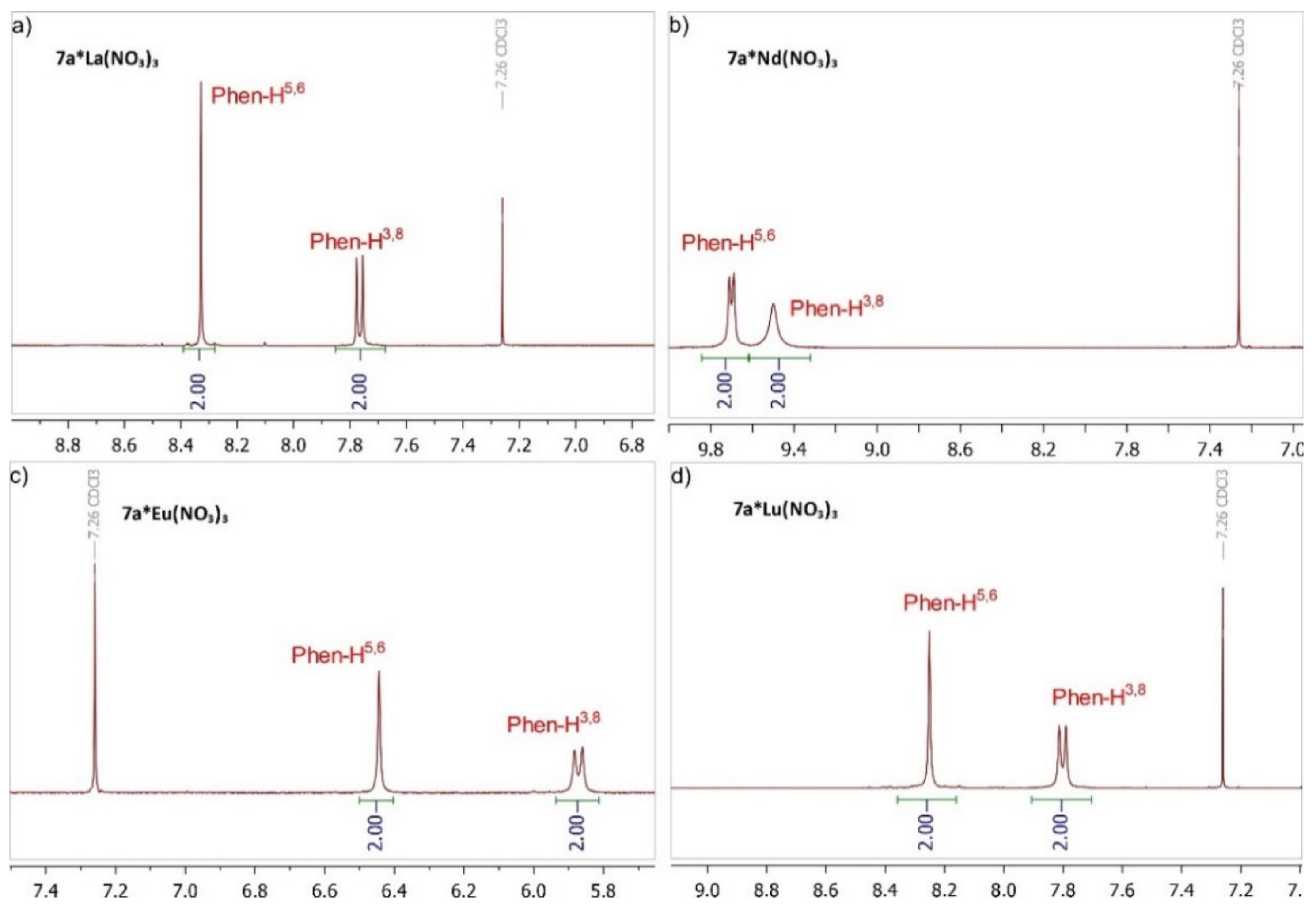

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
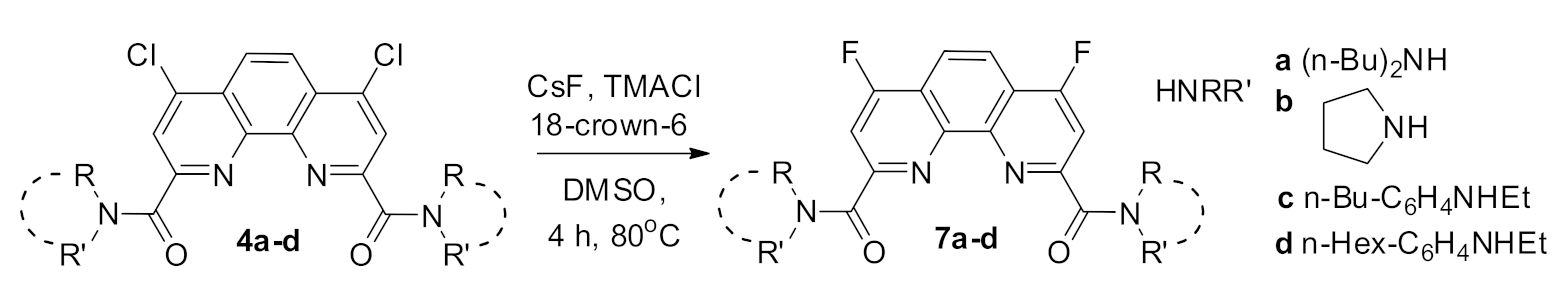
| Code | Yield, % | Tm.p. °C | ν (CO), cm−1 | NMR 19F, δ, ppm | Log p * |
|---|---|---|---|---|---|
| 7a | 88 | 101–103 | 1634 | −110.0 | 5.72 |
| 7b | 80 | 265–267 | 1628, 1618 | −109.3 | 0.22 |
| 7c | 74 | 103–105 | 1655, 1643 | −112.8 | 7.83 |
| 7d | 78 | 108–110 | 1663, 1640 | −112.7 | 10.49 |
| Ligand | Nphen | Oamide |
|---|---|---|
| TBuPhen | −0.301/−0.305 | −0.586/−0.581 |
| 7a | −0.310/−0.313 | −0.582/−0.577 |
| 7a | 7a•La(NO3)3 | 7a•Nd(NO3)3 | 7a•Eu(NO3)3 | 7a•Lu(NO3)3 | |
|---|---|---|---|---|---|
| ν(CO), cm−1 | 1634 | 1603 | 1604 | 1606 | 1609 |
| Δν(CO) | - | 31 | 30 | 28 | 25 |
| 7a•Nd(NO3)3 | TBuPhen•Nd(NO3)3 | 7a | |
|---|---|---|---|
| Nd(1)–N(1) | 2.672(3) | 2.643(2) | |
| Nd(1)–N(10) | 2.631(3) | 2.652(2) | |
| Nd(1)–O(1) | 2.428(2) | 2.434(1) | |
| Nd(1)–O(2) | 2.415(2) | 2.438(1) | |
| C(9)–C(21) | 1.526(5) | 1.519(4) | 1.515(2) |
| C(2)–C(11) | 1.514(4) | 1.516(4) | 1.511(2) |
| N(1)–C(2) | 1.332(4) | 1.335(2) | 1.324(2) |
| C(2)–C(3) | 1.411(4) | 1.403(2) | 1.411(2) |
| C(3)–C(4) | 1.354(4) | 1.376(2) | 1.354(2) |
| C(4)–C(5A) | 1.397(4) | 1.403(2) | 1.397(2) |
| tN(1)C(1)C(11)O(1) | 18.9(1) | 25.07(17) | 131.8(1) |
| N(10)C(9)C(21)O(2) | 28.0(1) | 22.10(18) | 136.3(1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avagyan, N.A.; Lemport, P.S.; Lysenko, K.A.; Gudovannyy, A.O.; Roznyatovsky, V.A.; Petrov, V.S.; Vokuev, M.F.; Ustynyuk, Y.A.; Nenajdenko, V.G. First Example of Fluorinated Phenanthroline Diamides: Synthesis, Structural Study, and Complexation with Lanthanoids. Molecules 2022, 27, 4705. https://doi.org/10.3390/molecules27154705
Avagyan NA, Lemport PS, Lysenko KA, Gudovannyy AO, Roznyatovsky VA, Petrov VS, Vokuev MF, Ustynyuk YA, Nenajdenko VG. First Example of Fluorinated Phenanthroline Diamides: Synthesis, Structural Study, and Complexation with Lanthanoids. Molecules. 2022; 27(15):4705. https://doi.org/10.3390/molecules27154705
Chicago/Turabian StyleAvagyan, Nane A., Pavel S. Lemport, Konstantin A. Lysenko, Alexey O. Gudovannyy, Vitaly A. Roznyatovsky, Valentine S. Petrov, Mikhail F. Vokuev, Yuri A. Ustynyuk, and Valentine G. Nenajdenko. 2022. "First Example of Fluorinated Phenanthroline Diamides: Synthesis, Structural Study, and Complexation with Lanthanoids" Molecules 27, no. 15: 4705. https://doi.org/10.3390/molecules27154705
APA StyleAvagyan, N. A., Lemport, P. S., Lysenko, K. A., Gudovannyy, A. O., Roznyatovsky, V. A., Petrov, V. S., Vokuev, M. F., Ustynyuk, Y. A., & Nenajdenko, V. G. (2022). First Example of Fluorinated Phenanthroline Diamides: Synthesis, Structural Study, and Complexation with Lanthanoids. Molecules, 27(15), 4705. https://doi.org/10.3390/molecules27154705