
Insights into Triazolylidene Ligands Behaviour at a Di-Iron Site Related to [FeFe]-Hydrogenases


 and
and 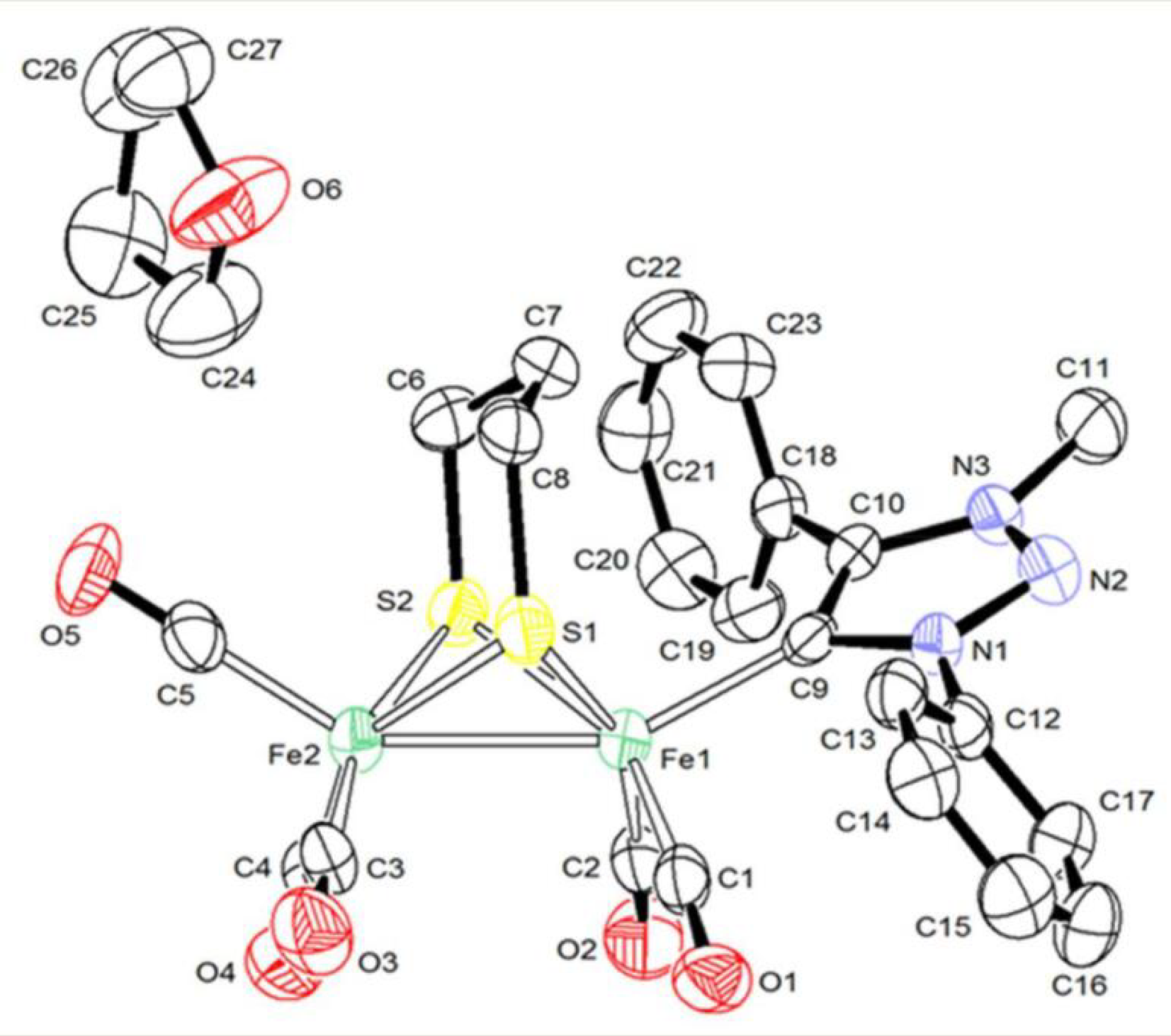{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
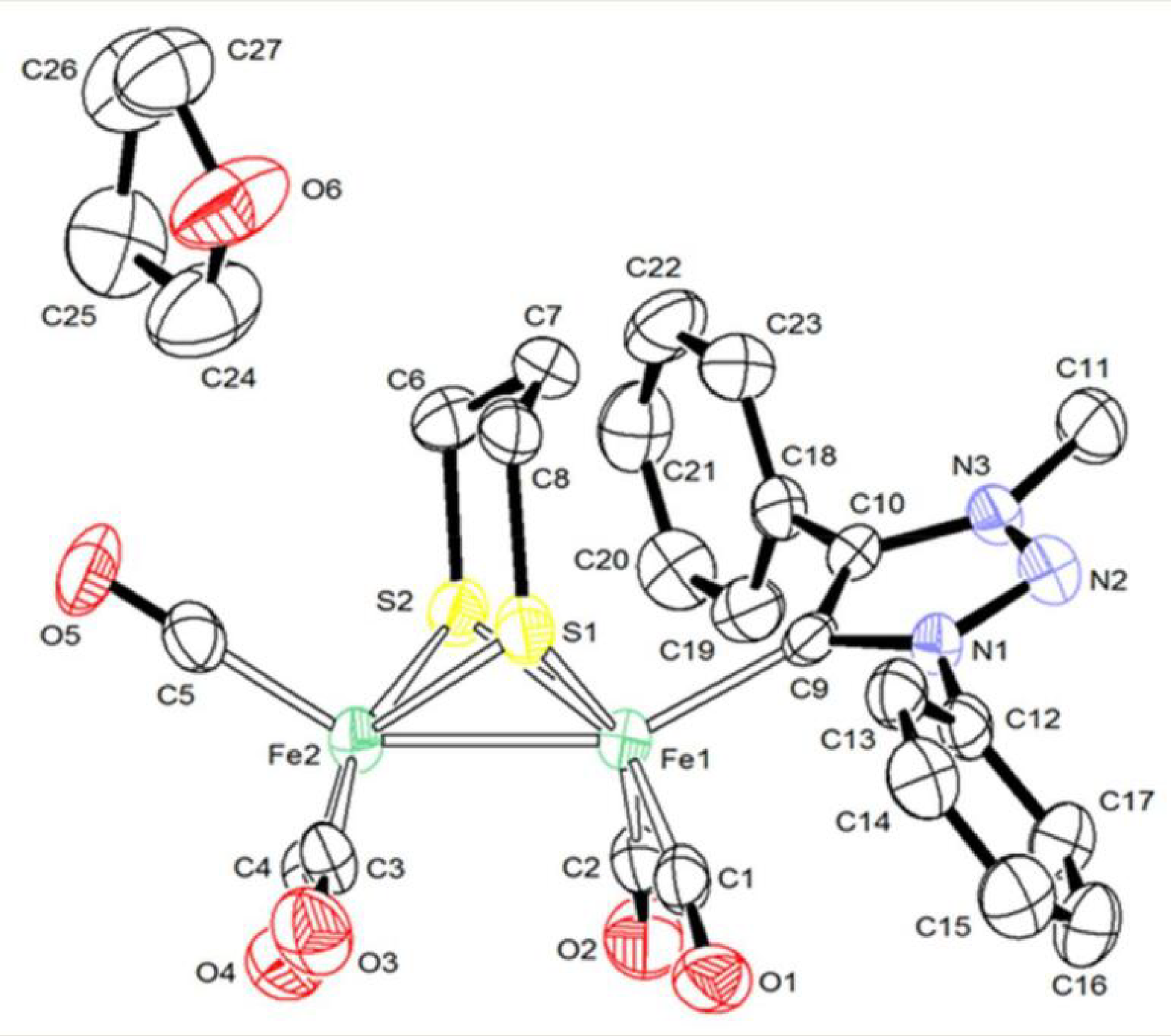
2.1. Synthesis and Characterization
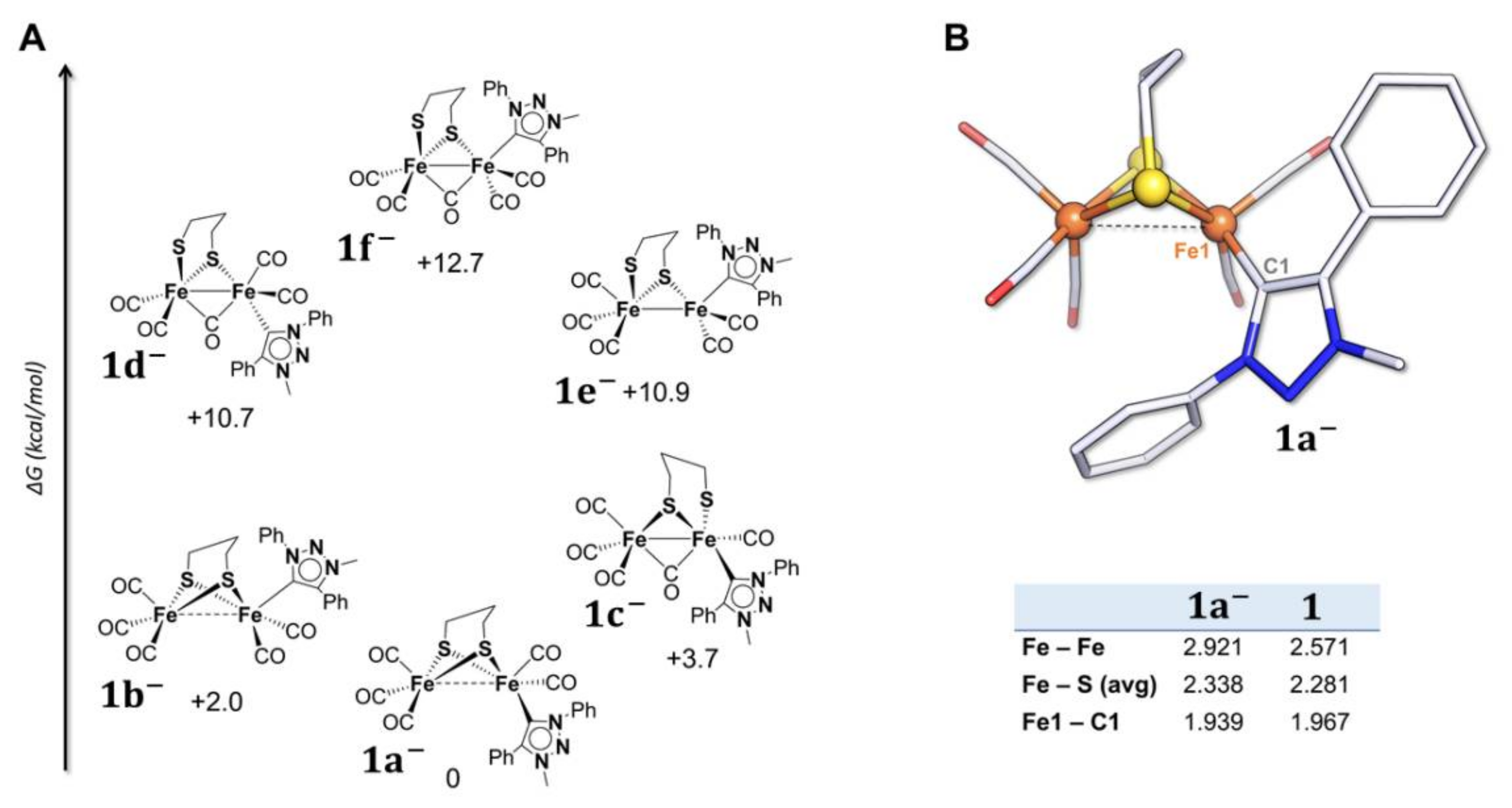
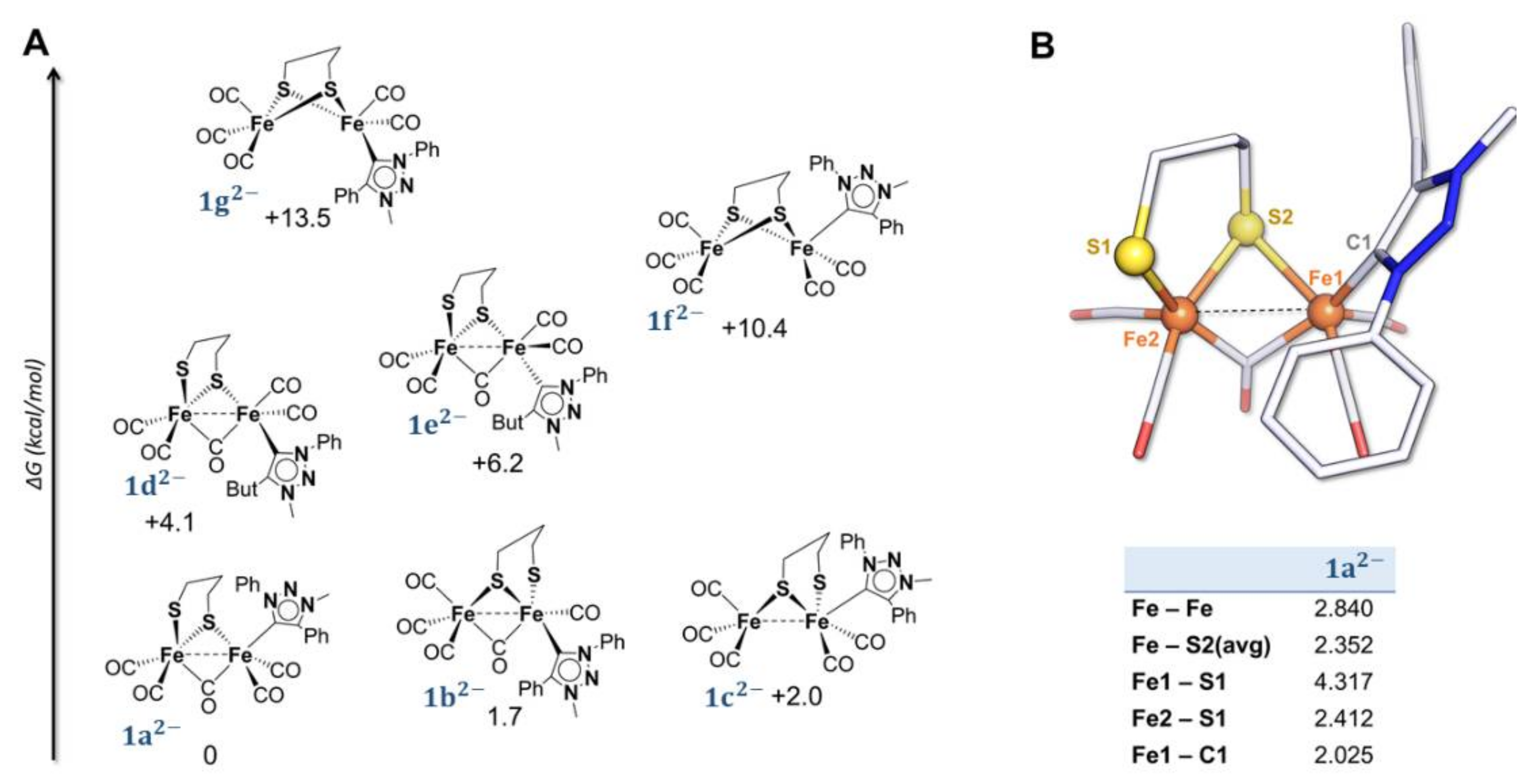
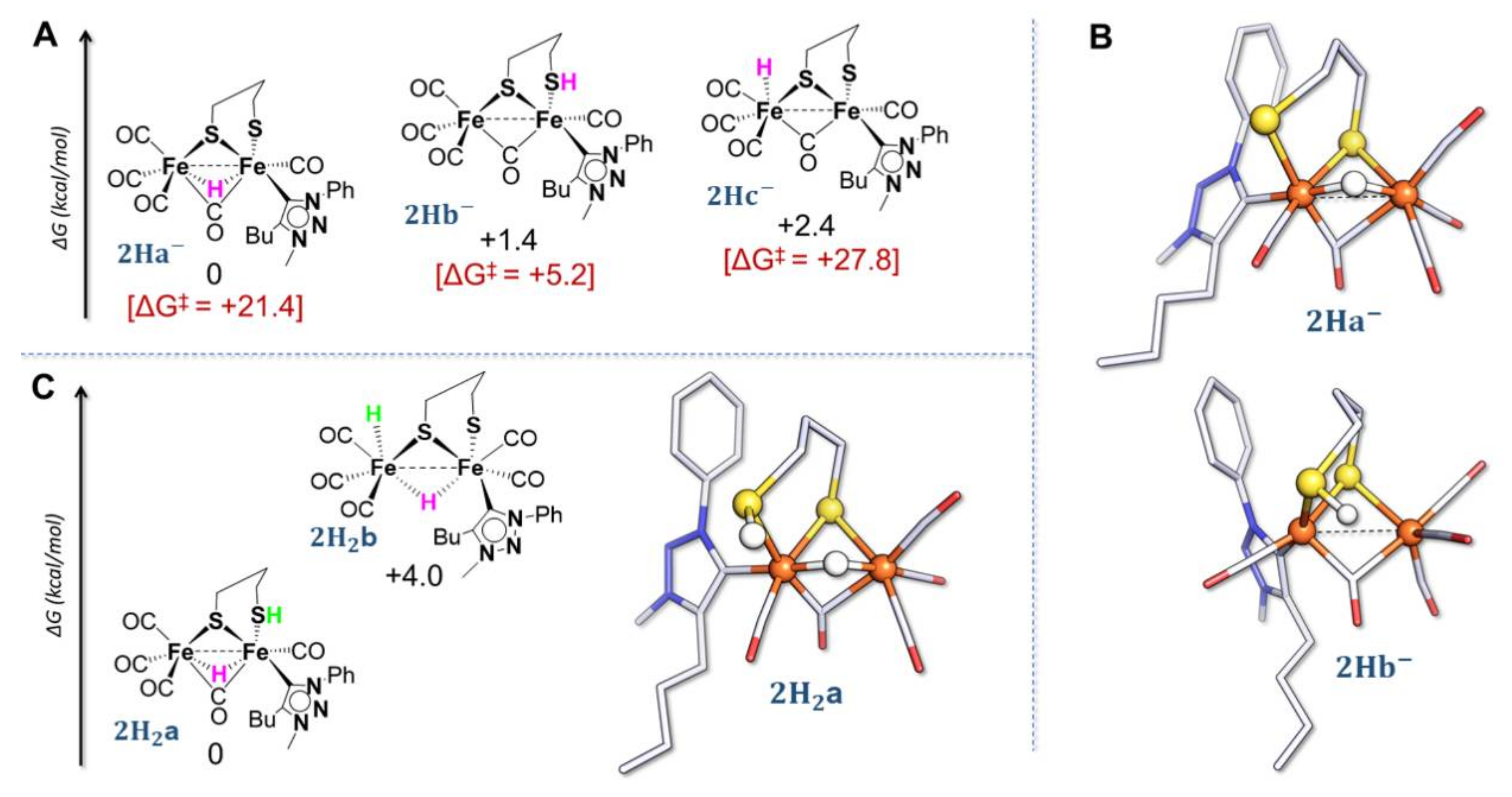
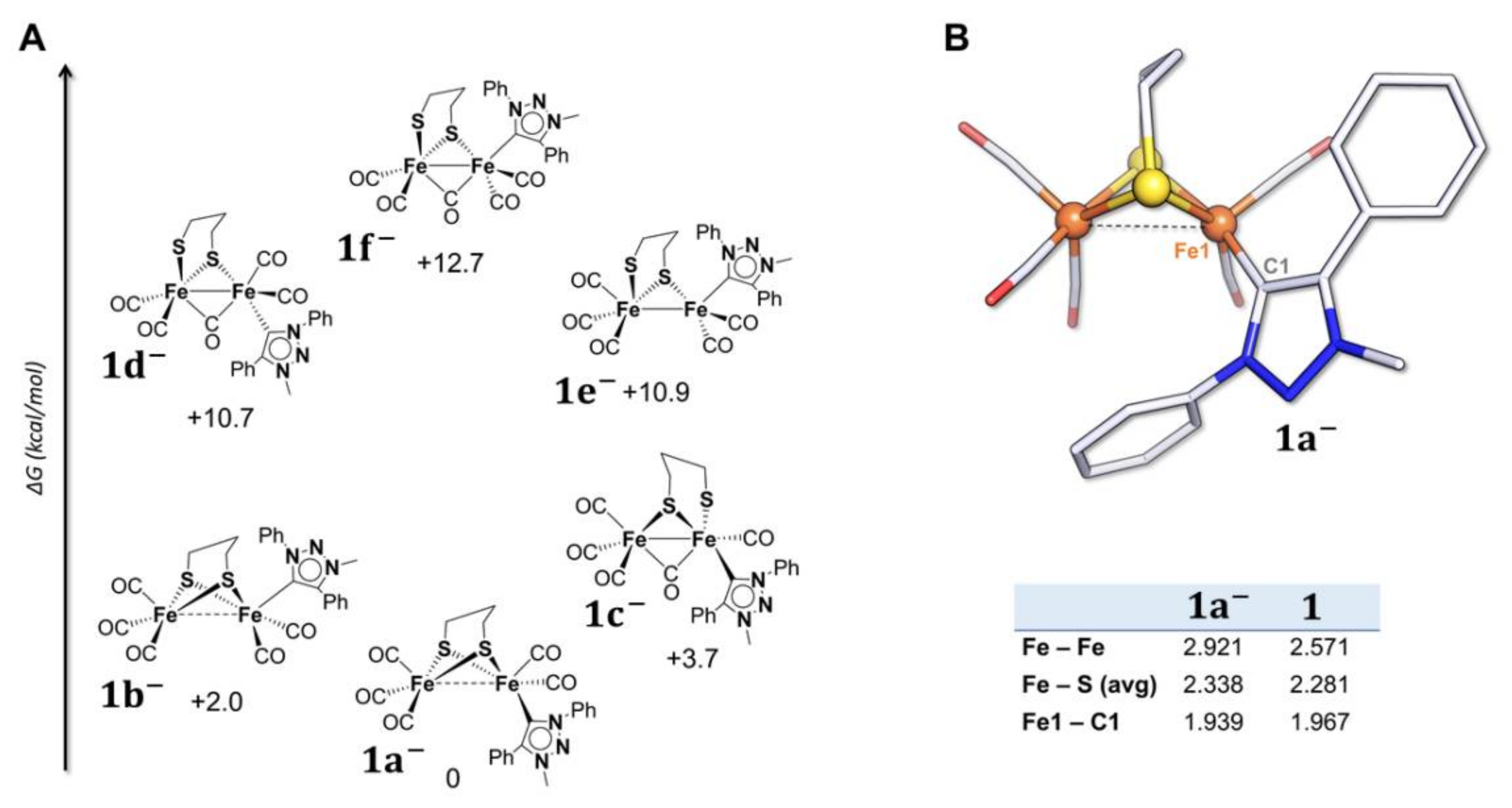
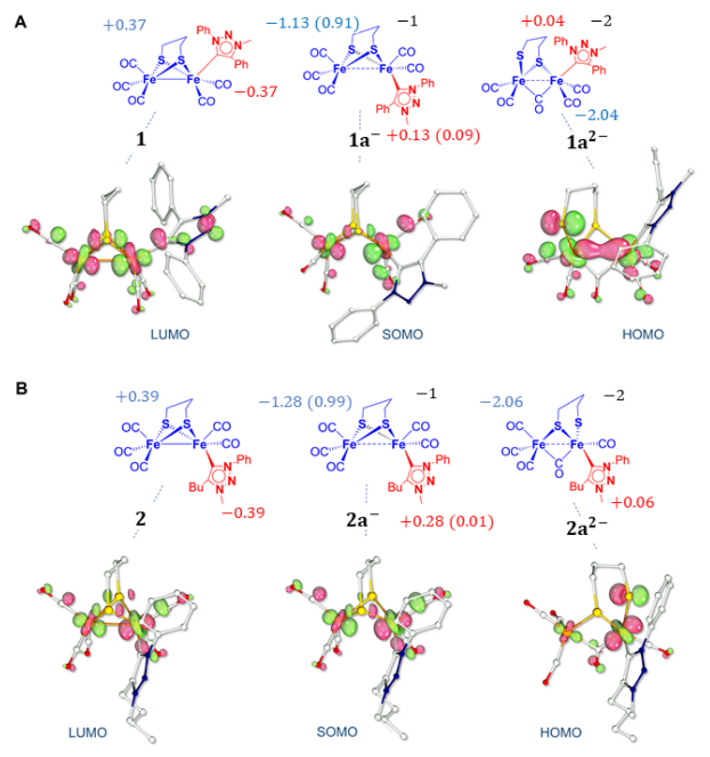
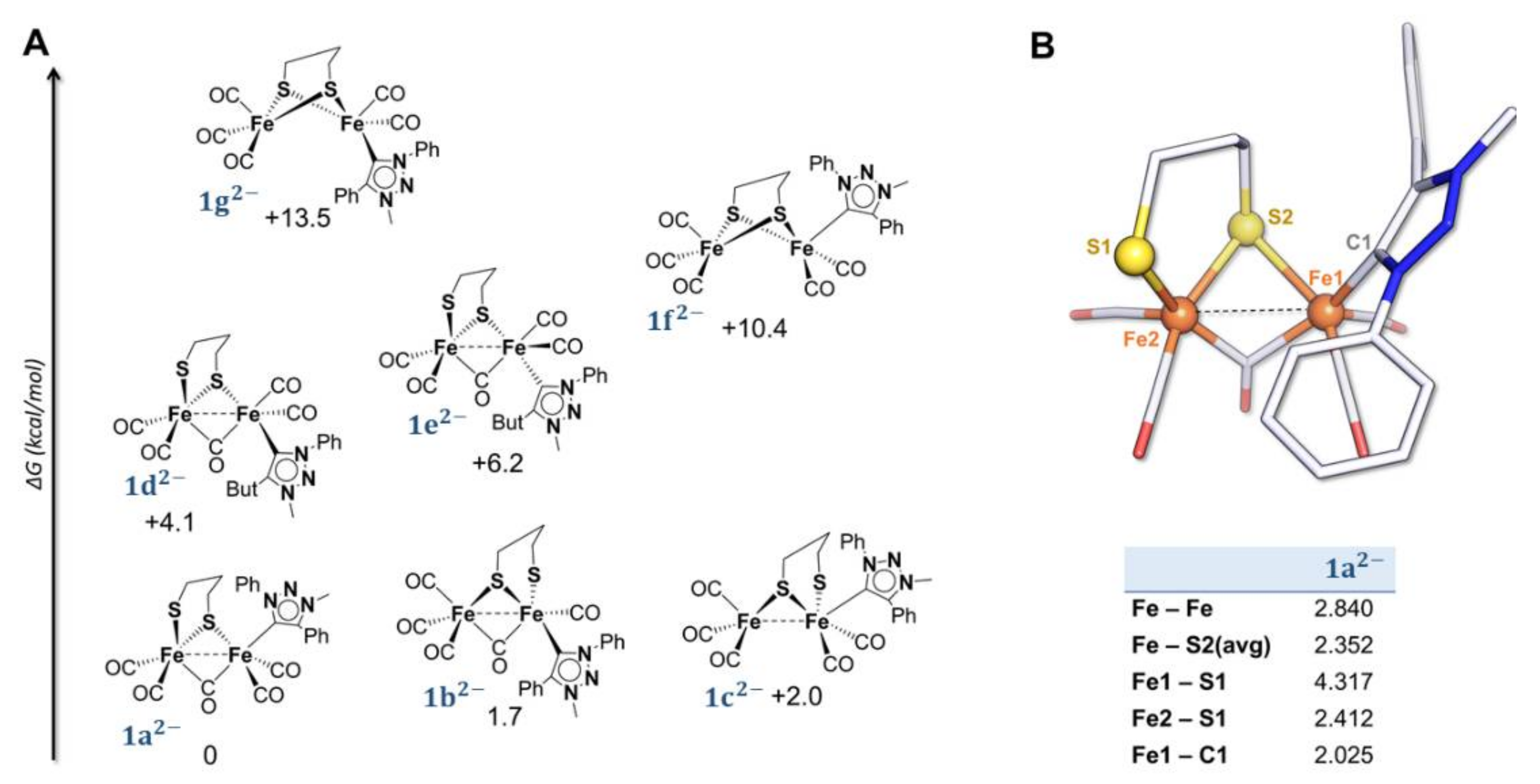
2.2. DFT Investigations of the Geometries of the Mono-Anion and Di-Anion
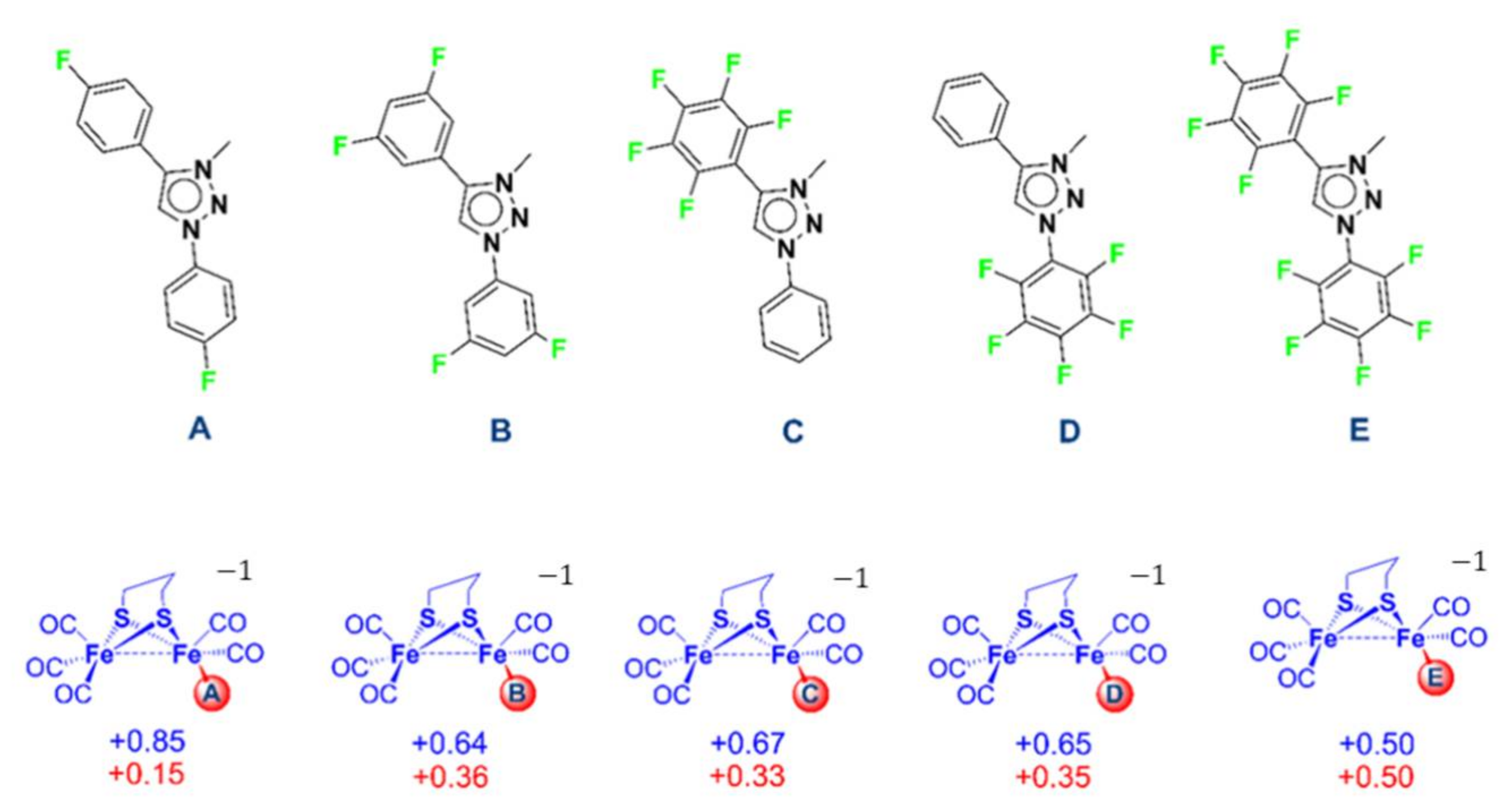
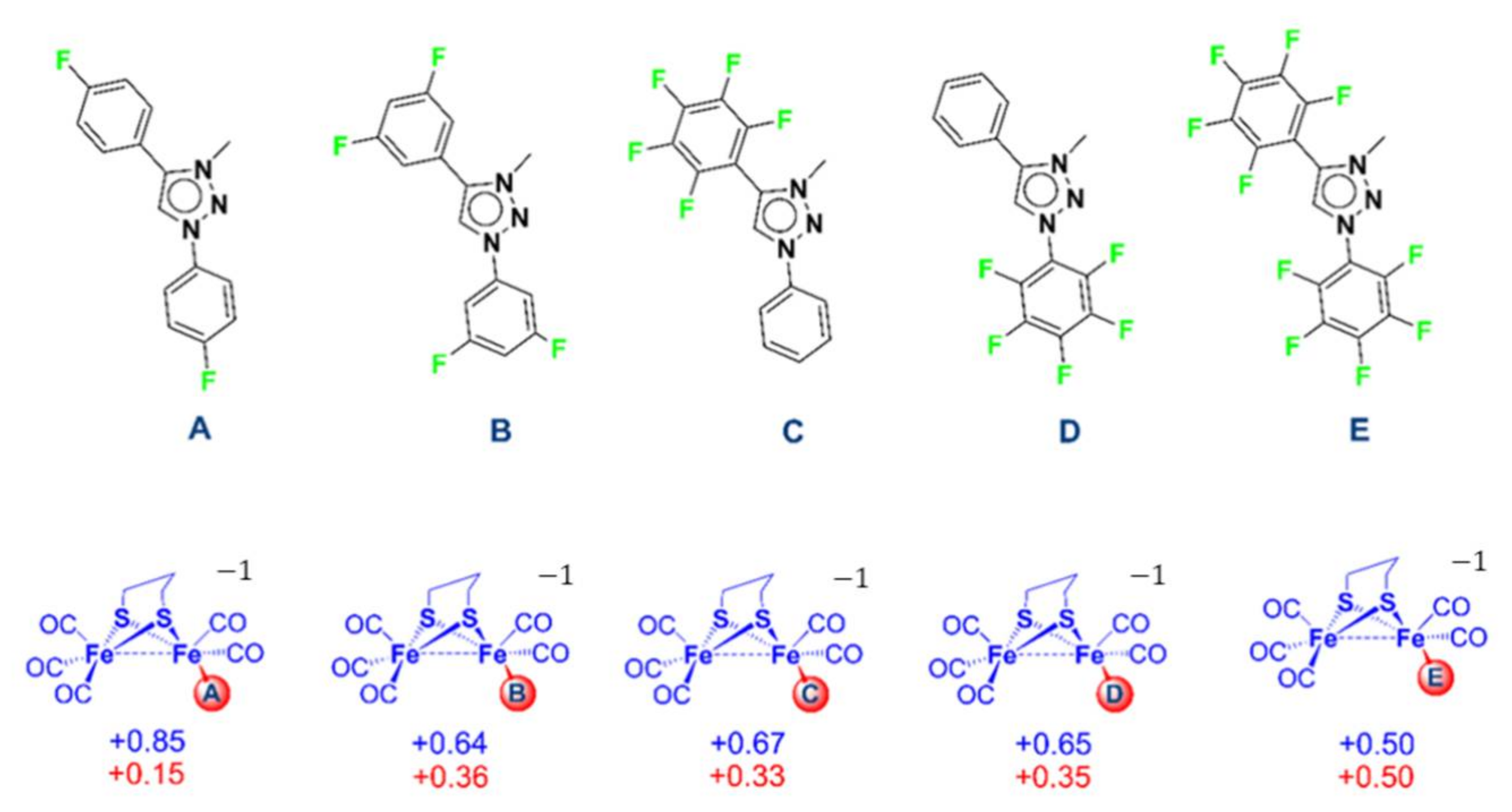
2.3. In Silico Modification of the MIC Ligand
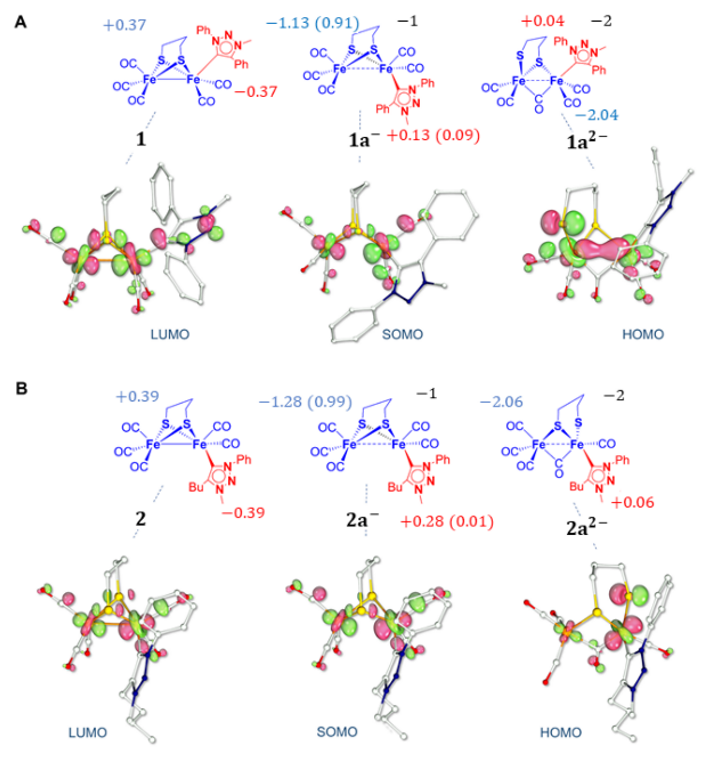
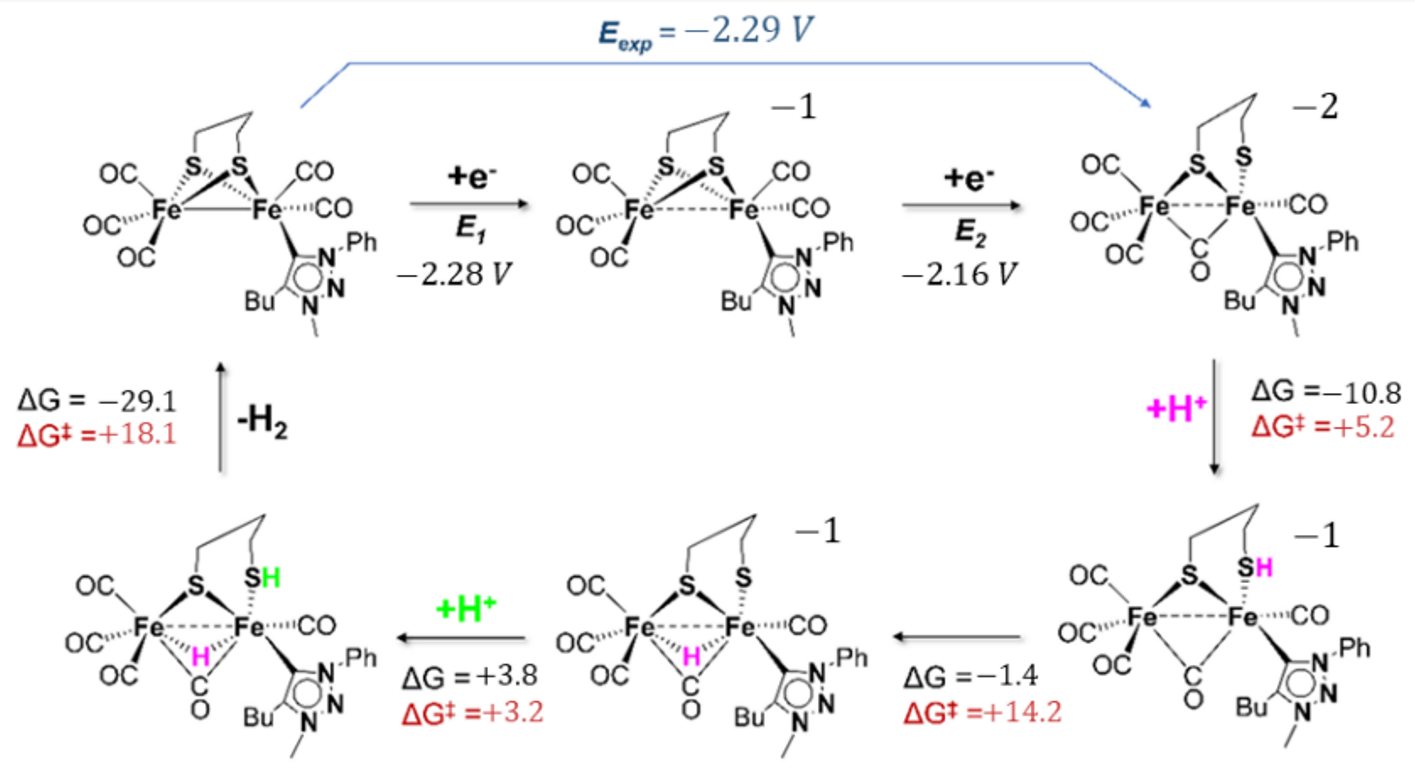
2.4. DFT Investigations of Protonation and Electron Steps in the Proposed Electrocatalytic Pathway
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A
References
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray Crystal Structure of the Fe-Only Hydrogenase (Cpl) from Clostridium pasteurianum to 1.8 Angstrom Resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C.E.; Fontecilla-Camps, J.C. Desulfovibrio desulfuricans iron hydrogenase: The structure shows unusual coordination to an active site Fe binuclear center. Structure 1999, 7, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T.R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J.M.; Reijerse, E.; Lubitz, W.; et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 4, 66–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.-P. [FeFe]-Hydrogenases: Recent developments and future perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef] [Green Version]
- Laun, K.; Baranova, I.; Duan, J.; Kertess, L.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Senger, M.; Stripp, S.T. Site-selective protonation of the one-electron reduced cofactor in [FeFe]-hydrogenase. Dalton Trans. 2021, 50, 3641–3650. [Google Scholar] [CrossRef]
- Birrell, J.A.; Rodríguez-Maciá, P.; Reijerse, E.J.; Martini, M.A.; Lubitz, W. The catalytic cycle of [FeFe] hydrogenase: A tale of two sites. Coord. Chem. Rev. 2021, 449, 214191–214213. [Google Scholar] [CrossRef]
- Caserta, G.; Zuccarello, L.; Barbosa, C.; Silveira, C.M.; Moe, E.; Katz, S.; Hildebrandt, P.; Zebger, I.; Todorovic, S. Unusual structures and unknown roles of FeS clusters in metalloenzymes seen from a resonance Raman spectroscopic perspective. Coord. Chem. Rev. 2022, 452, 214287–214299. [Google Scholar] [CrossRef]
- Apfel, U.-P.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Weigand, W. [FeFe] hydrogenase models: An overview. In Bioinspired Catalysis: Metal-Sulfur Complexes; Weigand, W., Schollhammer, P., Eds.; Wiley-VCH-Verl: Weinheim, Germany, 2015; pp. 79–103. ISBN 978-3-527-33308-0. [Google Scholar]
- Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. [FeFe]-hydrogenases models. In Advances in Bioorganometallic Chemistry: [FeFe]-Hydrogenases Models; Hirao, T., Moriuchi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 17; pp. 347–359. ISBN 9780128141984. [Google Scholar]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase enzymes and their synthetic models: The role of metal hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [Green Version]
- Simmons, T.R.; Berggren, G.; Bacchi, M.; Fontecave, M.; Artero, V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coord. Chem. Rev. 2014, 270–271, 127–150. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Cao, W.; Lv, D. Di-iron analogue of [FeFe]-hydrogenase active site as a molecular electro-catalyst for proton reduction. Catal. Lett. 2020, 150, 3409–3414. [Google Scholar] [CrossRef]
- Van Beek, C.B.; van Leest, N.P.; Lutz, M.; de Vos, S.D.; Klein Gebbink, R.J.M.; de Bruin, B.; Broere, D.L.J. Combining metal–metal cooperativity, metal–ligand cooperativity and chemical non-innocence in diiron carbonyl complexes. Chem. Sci. 2022, 13, 2094–2104. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Organometallic diiron complex chemistry related to the [2Fe]H subsite of [FeFe]H2ase. Eur. J. Inorg. Chem. 2008, 2008, 4671–4681. [Google Scholar] [CrossRef]
- Chouffai, D.; Zampella, G.; Capon, J.-F.; De Gioia, L.; Le Goff, A.; Peétillon, F.Y.; Schollhammer, P.; Talarmin, J. Electrochemical and theoretical studies of the impact of the chelating ligand on the reactivity of [Fe2(CO)4(κ2-LL)(μ-pdt)]+ complexes with different substrates (LL = IMe-CH2-IMe, dppe; IMe = 1-Methylimidazol-2-ylidene). Organometallics 2012, 31, 1082–1091. [Google Scholar] [CrossRef]
- Chouffai, D.; Capon, J.-F.; De Gioia, L.; Peétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Zampella, G. A diferrous dithiolate as a model of the elusive Hoxinact state of the [FeFe] hydrogenases: An electrochemical and theoretical dissection of its redox chemistry. Inorg. Chem. 2015, 54, 299–311. [Google Scholar] [CrossRef]
- Morvan, D.; Capon, J.-F.; Gloaguen, F.; Le Goff, A.; Marchivie, M.; Michaud, F.; Schollhammer, P.; Talarmin, J.; Yaouanc, J.-J.; Pichon, R.; et al. N-heterocyclic carbene ligands in nonsymmetric diiron models of hydrogenase active sites. Organometallics 2007, 26, 2042–2052. [Google Scholar] [CrossRef]
- Orain, P.-Y.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Zampella, G.; De Gioia, L.; Roisnel, T. Investigation on the protonation of a trisubstituted [Fe2(CO)3(PPh3)(κ2-phen)(μ-pdt)] complex: Rotated versus unrotated intermediate pathways. Inorg. Chem. 2010, 49, 5003–5008. [Google Scholar] [CrossRef]
- Orain, P.-Y.; Capon, J.-F.; Kervarec, N.; Gloaguen, F.; Pétillon, F.Y.; Pichon, R.; Schollhammer, P.; Talarmin, J. Use of 1,10-phenanthroline in diiron dithiolate derivatives related to the [Fe–Fe] hydrogenase active site. Dalton Trans. 2007, 3754–3756. [Google Scholar] [CrossRef]
- Arrigoni, F.; Elleouet, C.; Mele, A.; Pétillon, F.Y.; De Gioia, L.; Schollhammer, P.; Zampella, G. Insights into the two-electron reductive process of [FeFe]H2ase biomimetics: Cyclic voltammetry and DFT investigation on chelate control of redox properties of [Fe2(CO)4(κ2-chelate)-(µ-dithiolate)]. Chem. Eur. J. 2020, 26, 17536–17545. [Google Scholar] [CrossRef]
- Hu, S.-P.; Zhang, L.; Wu, Y.; Feng, J.-S.; Wu, S.; Xie, B.; Zou, L.-K. [FeFe]-hydrogenase active site mimics containing pyridyl-functionalized phosphine ligands: Synthesis, characterization and electrochemical investigation. Inorg. Chim. Acta 2020, 504, 119435–119441. [Google Scholar] [CrossRef]
- Schippers, E.C.F.; Nurttila, S.S.; Oudsen, J.-P.H.; Tromp, M.; Dzik, W.I.; van der Vlugt, J.I.; Reek, J.N.H. [FeFe]-hydrogenase mimic employing κ2-C, N-pyridine bridgehead catalyzes proton reduction at mild overpotential. Eur. J. Inorg. Chem. 2019, 2019, 2510–2517. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Groy, T.L.; Jones, A.K. Biomimetic model for [FeFe]-hydrogenase: Asymmetrically disubstituted diiron complex with a redox-active 2,2’-bipyridyl ligand. Dalton Trans. 2013, 42, 3843–3853. [Google Scholar] [CrossRef]
- Merinero, A.D.; Collado, A.; Casarrubios, L.; Gómez-Gallego, M.; Ramírez de Arellano, C.; Caballero, A.; Zapata, F.; Sierra, M.A. Triazole-containing [FeFe] hydrogenase mimics: Synthesis and electrocatalytic behaviour. Inorg. Chem. 2019, 58, 16267–16278. [Google Scholar] [CrossRef]
- Pandey, I.K.; Agarwal, T.; Mobin, S.M.; Stein, M.; Kaur-Ghumaan, S. Switching site reactivity in hydrogenase model systems by introducing a pendant amine ligand. ACS Omega 2021, 6, 4192–4203. [Google Scholar] [CrossRef]
- Becker, R.; Amirjalayer, S.; Li, P.; Woutersen, S.; Reek, J.N.H. An iron-iron hydrogenase mimic with appended electron reservoir for efficient proton reduction in aqueous media. Sci. Adv. 2016, 2, e1501014. [Google Scholar] [CrossRef] [Green Version]
- Camara, J.M.; Rauchfuss, T.B. Combining acid–base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat. Chem. 2012, 4, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-C.; Yen, T.-H.; Tseng, Y.-J.; Hu, C.-H.; Lee, G.H.; Chiang, M.-H. Electron delocalization from the fullerene attachment to the diiron core within the active-site mimics of [FeFe]hydrogenase. Inorg. Chem. 2012, 51, 5997–5999. [Google Scholar] [CrossRef]
- Tye, J.W.; Lee, J.; Wang, H.-W.; Mejia-Rodriguez, R.; Reibenspies, J.H.; Hall, M.B.; Darensbourg, M.Y. Dual electron uptake by simultaneous iron and ligand reduction in an N-Heterocyclic Carbene substituted [FeFe] hydrogenase model compound. Inorg. Chem. 2005, 44, 5550–5552. [Google Scholar] [CrossRef]
- Song, L.C.; Luo, X.; Wang, Y.-Z.; Gai, B.; Hu, Q.-M. Synthesis, characterization and electrochemical behavior of some N-heterocyclic carbene-containing active site models of [FeFe]-hydrogenases. J. Organomet. Chem. 2009, 694, 103–112. [Google Scholar] [CrossRef]
- Schwab, D.E.; Tard, C.; Brecht, E.; Peters, J.W.; Pickett, C.J.; Szilagyi, R.K. On the electronic structure of the hydrogenase H-cluster. Chem. Commun. 2006, 3696–3698. [Google Scholar] [CrossRef]
- Capon, J.-F.; El Hassnaoui, S.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. N-Heterocyclic Carbene Ligands as Cyanide Mimics in Diiron Models of the All-Iron Hydrogenase Active Site. Organometallics 2005, 24, 2020–2022. [Google Scholar] [CrossRef]
- Orton, G.R.F.; Ghosh, S.; Alker, L.; Sarker, J.C.; Pugh, D.; Richmond, M.G.; Hartl, F.; Hogarth, G. Biomimics of [FeFe]-hydrogenases incorporating redox-active ligands: Synthesis, redox properties and spectroelectrochemistry of diiron-dithiolate complexes with ferrocenyl-diphosphines as Fe4S4 surrogates. Dalton Trans. 2022, 51, 9748–9769. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Rana, S.; Hollingsworth, N.; Richmond, M.G.; Kabir, S.E.; Hogarth, G. Hydrogenase Biomimetics with Redox-Active Ligands: Synthesis, Structure, and Electrocatalytic Studies on [Fe2(CO)4(κ2-dppn)(µ-edt)] (edt = Ethanedithiolate; dppn = 1,8-bis(Diphenylphosphino)Naphthalene). Inorganics 2018, 6, 122. [Google Scholar] [CrossRef] [Green Version]
- Vivancos, A.; Segarra, C.; Albrecht, M. Mesoionic and related less heteroatom-stabilized N-Heterocyclic Carbene complexes: Synthesis, catalysis, and other applications. Chem. Rev. 2018, 118, 9493–9586. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. 1H-1,2,3-Triazol-5-ylidenes: Readily available mesoionic carbenes. Acc. Chem. Res. 2018, 51, 3236–3244. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Crystalline 1H-1,2,3-triazol-5-ylidenes: New stable mesoionic carbenes (MICs). Angew. Chem. Int. Ed. 2019, 49, 4759–4762. [Google Scholar] [CrossRef]
- Albrecht, M. C4-bound imidazolylidenes: From curiosities to high-impact carbene ligands. Chem. Commun. 2008, 3601–3610. [Google Scholar] [CrossRef] [Green Version]
- Mathew, P.; Neels, A.; Albrecht, M. 1,2,3-Triazolylidenes as versatile abnormal carbene ligands for late transition metals. J. Am. Chem. Soc. 2008, 130, 13534–13535. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M. Carbene in action. Science 2009, 326, 532–533. [Google Scholar] [CrossRef]
- Sanghi, S.; Willett, E.; Versek, C.; Tuominen, M.; Coughlin, E.B. Physicochemical properties of 1,2,3-triazolium ionic liquids. RSC Adv. 2012, 2, 848–853. [Google Scholar] [CrossRef]
- Mele, A.; Bertini, S.; Albrecht, M.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. A diiron hydrogenase mimic featuring a 1,2,3-triazolylidene. Chimia 2020, 74, 499–503. [Google Scholar] [CrossRef]
- Maity, R.; Sarkar, B. Chemistry of compounds based on 1,2,3-triazolylidene-type mesoionic carbenes. JACS Au 2022, 2, 22–57. [Google Scholar] [CrossRef]
- Huynh, H.V. Electronic properties of N-Heterocyclic Carbenes and their experimental determination. Chem. Rev. 2018, 118, 9457–9492. [Google Scholar] [CrossRef]
- Poulain, A.; Canseco-Gonzalez, D.; Hynes-Roche, R.; Muller-Bunz, H.; Schuster, O.; Stoeckli-Evans, H.; Neels, A.; Albrecht, M. Synthesis and tunability of abnormal 1,2,3-triazolylidene palladium and rhodium complexes. Organometallics 2011, 30, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Riener, K.; Haslinger, S.; Raba, A.; Högerl, M.P.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. Chemistry of iron N-Heterocyclic Carbene complexes: Syntheses, structures, reactivities, and catalytic applications. Chem. Rev. 2014, 114, 5215–5272. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, J.; Shi, Y.; Wang, Z.; Åkermark, B.; Sun, L. Preparation, characteristics and crystal structures of novel N-heterocyclic carbene substituted furan- and pyridine-containing azadithiolate Fe–S complexes. Polyhedron 2007, 26, 1499–1504. [Google Scholar] [CrossRef]
- Lyon, E.J.; Georgakaki, I.P.; Reibenspies, J.H.; Darensbourg, M.Y. Carbon monoxide and cyanide Ligands in a classical organometallic complex model for Fe-only hydrogenase. Angew. Chem. Int. Ed. 1999, 38, 3178–3180. [Google Scholar] [CrossRef]
- Arrigoni, F.; Mohamed Bouh, S.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; De Gioia, L.; Zampella, G. Electrochemical and theoretical investigations of the oxidatively induced reactivity of the complex [Fe2(CO)4(κ2-dmpe)(µ-adtBn)] related to the active site of [FeFe] hydrogenases. Chem. Eur. J. 2018, 24, 15036–15051. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A At. Mol. Opt. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Arrigoni, F.; Bertini, L.; Bruschi, M.; Greco, C.; De Gioia, L.; Zampella, G. H2 Activation in [FeFe]-Hydrogenase cofactor versus diiron dithiolate models: Factors underlying the catalytic success of Nature and implications for an improved bio-mimicry. Chem. Eur. J. 2019, 25, 1227–1241. [Google Scholar] [CrossRef]
- Basu, D.; Bailey, T.S.; Lalaoui, N.; Richers, C.P.; Woods, T.J.; Rauchfuss, T.B.; Arrigoni, F.; Zampella, G. Synthetic designs and structural investigations of biomimetic Ni–Fe thiolates. Inorg. Chem. 2019, 58, 2430–2443. [Google Scholar] [CrossRef]
- Breglia, R.; Arrigoni, F.; Sensi, M.; Greco, C.; Fantucci, P.; De Gioia, L.; Bruschi, M. First-principles calculations on Ni,Fe-containing carbon monoxide dehydrogenases reveal key stereoelectronic features for binding and release of CO2 to/ from the C-cluster. Inorg. Chem. 2021, 60, 387–402. [Google Scholar] [CrossRef]
- Almazahreh, L.R.; Arrigoni, F.; Abul-Futouh, H.; El-Khateeb, M.; Görls, H.; Elleouet, C.; Schollhammer, P.; Bertini, L.; De Gioia, L.; Rudolph, M.; et al. Proton shuttle mediated by (SCH2)2P═O moiety in [FeFe]-hydrogenase mimics: Electrochemical and DFT studies. ACS Catalysis 2021, 11, 7080–7098. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab ini-tio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A. Calculation of UV/Vis spectra in solution. J. Phys. Chem. 1996, 100, 3349–3353. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mele, A.; Arrigoni, F.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; Zampella, G. Insights into Triazolylidene Ligands Behaviour at a Di-Iron Site Related to [FeFe]-Hydrogenases. Molecules 2022, 27, 4700. https://doi.org/10.3390/molecules27154700
Mele A, Arrigoni F, Elleouet C, Pétillon FY, Schollhammer P, Zampella G. Insights into Triazolylidene Ligands Behaviour at a Di-Iron Site Related to [FeFe]-Hydrogenases. Molecules. 2022; 27(15):4700. https://doi.org/10.3390/molecules27154700
Chicago/Turabian StyleMele, Andrea, Federica Arrigoni, Catherine Elleouet, François Y. Pétillon, Philippe Schollhammer, and Giuseppe Zampella. 2022. "Insights into Triazolylidene Ligands Behaviour at a Di-Iron Site Related to [FeFe]-Hydrogenases" Molecules 27, no. 15: 4700. https://doi.org/10.3390/molecules27154700
APA StyleMele, A., Arrigoni, F., Elleouet, C., Pétillon, F. Y., Schollhammer, P., & Zampella, G. (2022). Insights into Triazolylidene Ligands Behaviour at a Di-Iron Site Related to [FeFe]-Hydrogenases. Molecules, 27(15), 4700. https://doi.org/10.3390/molecules27154700