
Cancer-Associated Mutations of the Adenosine A2A Receptor Have Diverse Influences on Ligand Binding and Receptor Functions

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Selection of Cancer-Associated A2AAR Mutations
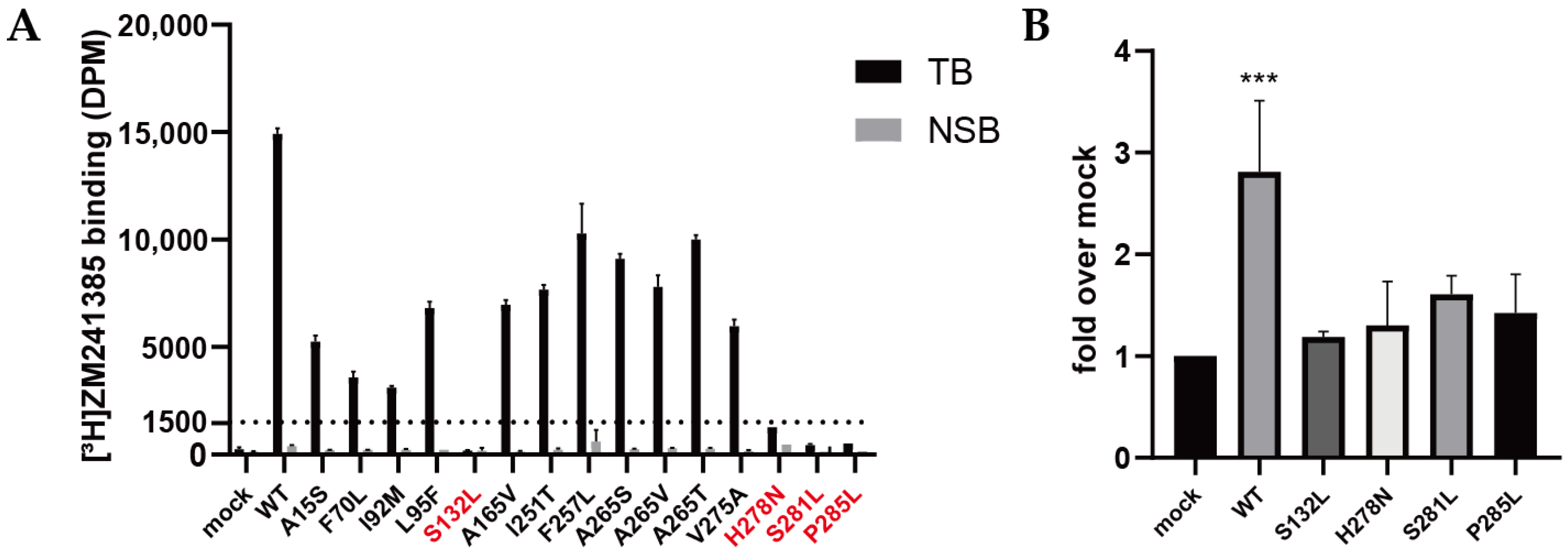
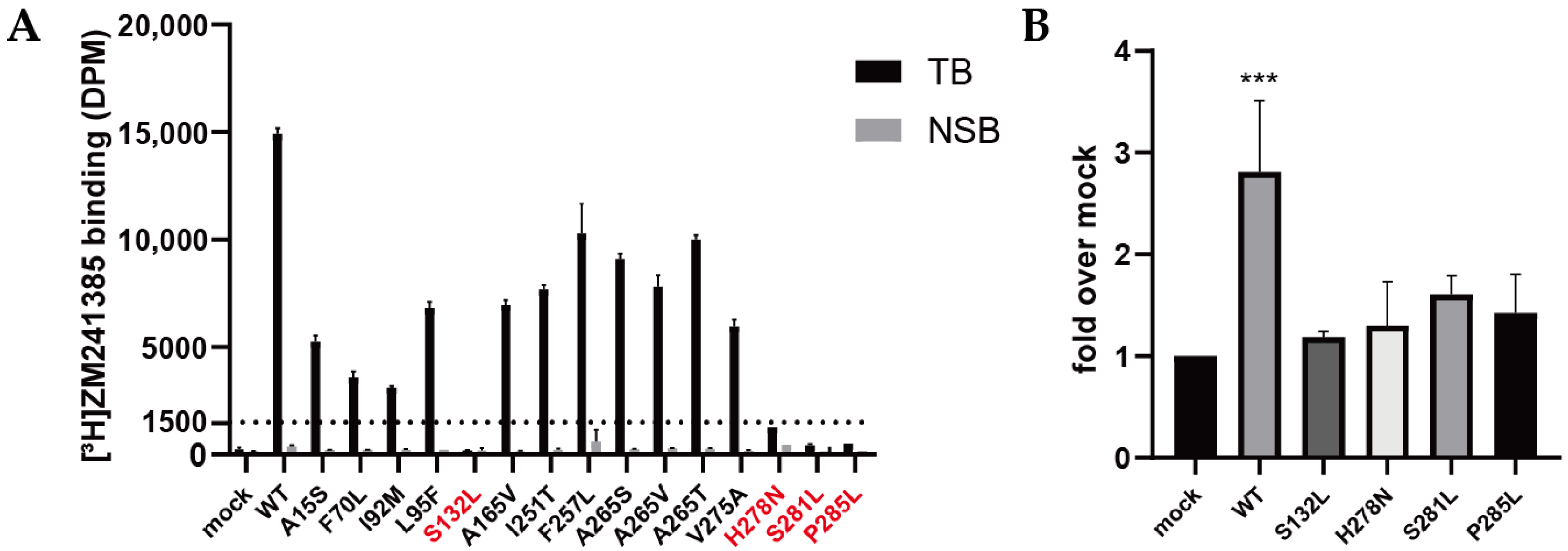
2.2. Validation of Expression and Radioligand Binding of Wild-Type and Mutant A2AARs
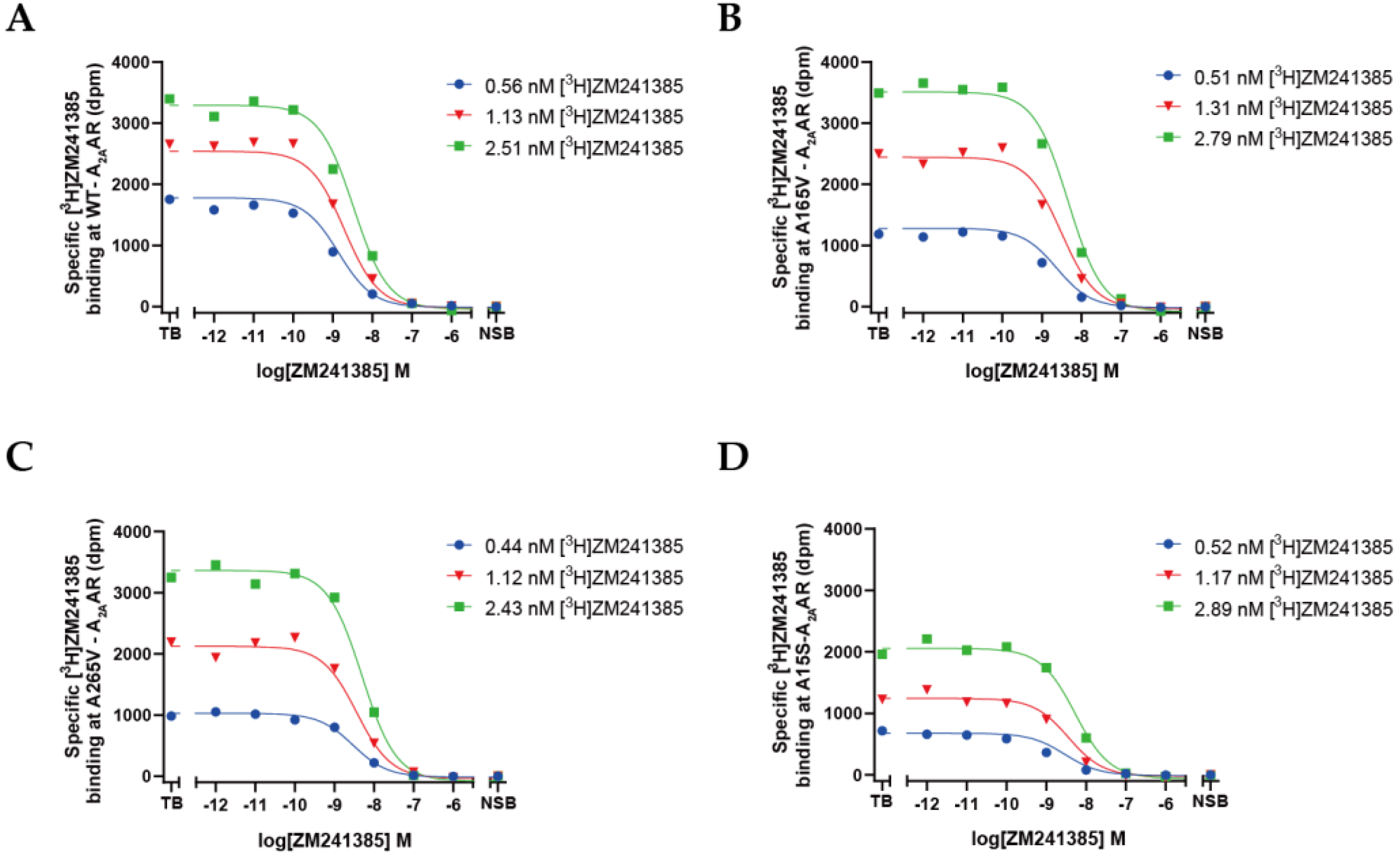
2.3. Quantification of Expression Levels (Bmax) and ZM241385 Binding Affinity (KD) for A2AARs
2.4. Quantification of NECA Binding Affinity (Ki) for A2AARs
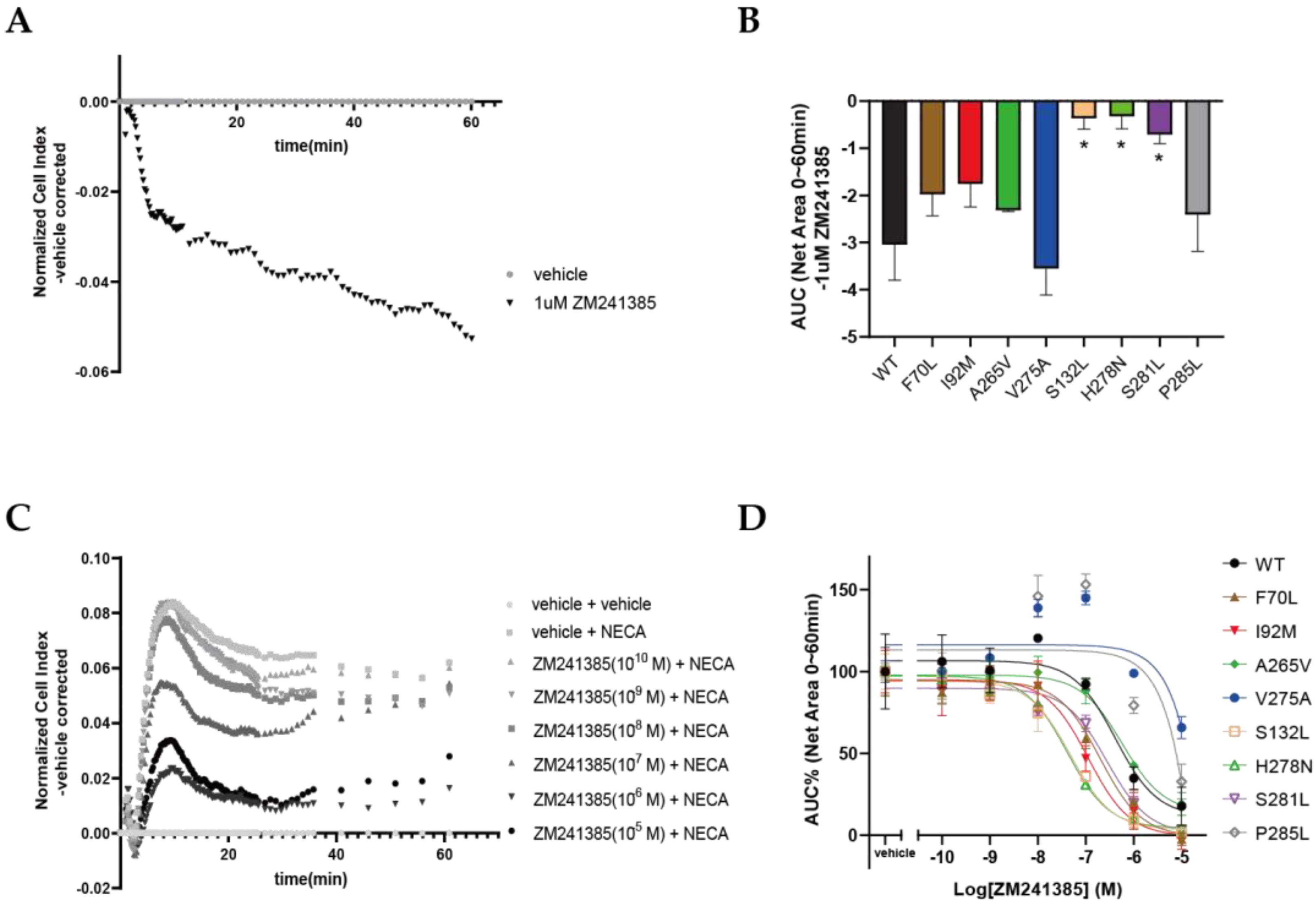
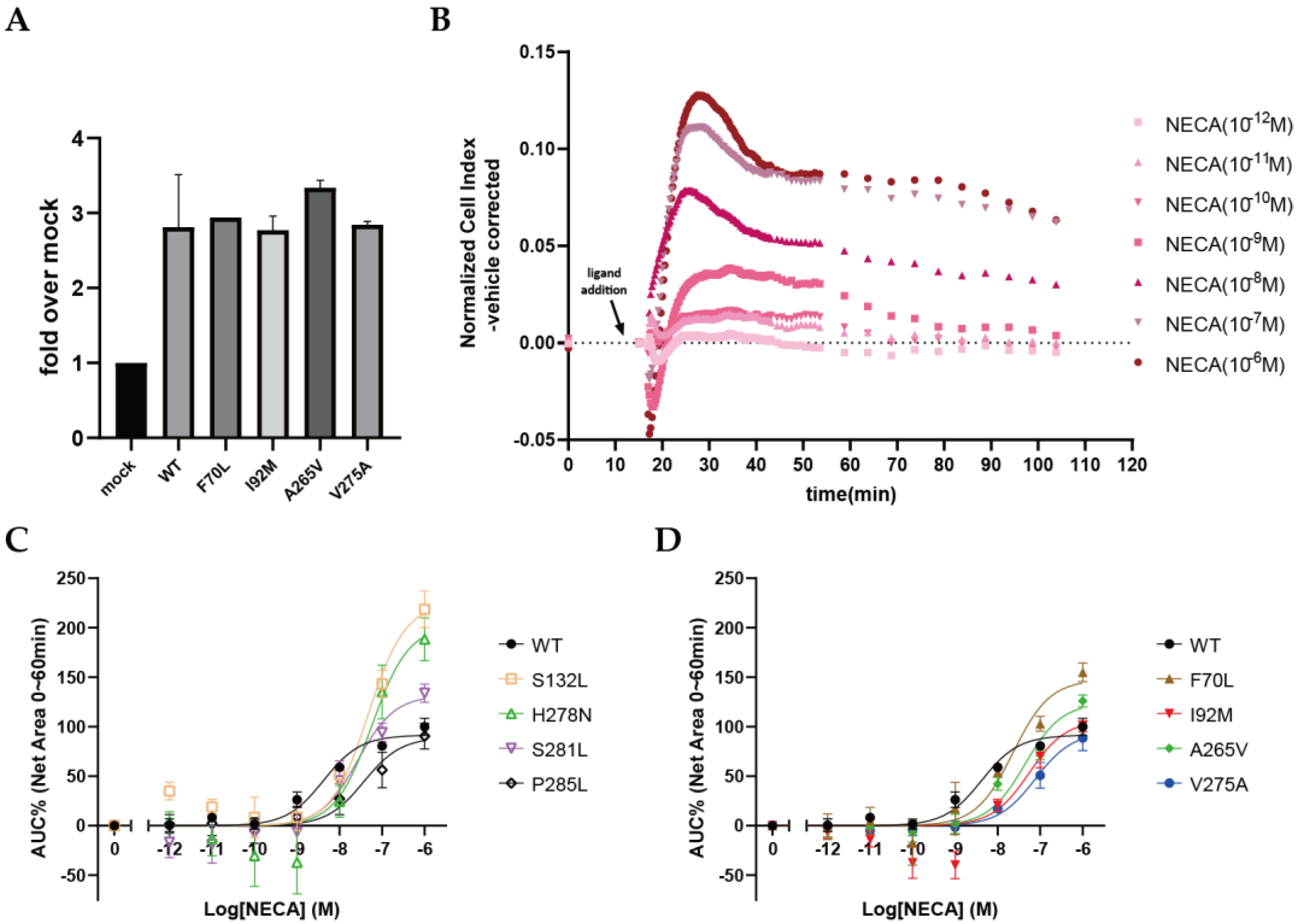
2.5. Functional Effects of Cancer-Associated Mutations on A2AAR in a Label-Free Whole-Cell Assay
2.6. Mapping of Cancer-Associated Mutations in the Crystal Structures of A2AAR
3. Discussion
3.1. Some Cancer-Associated Mutations Cause a Conformational Change in A2AAR
3.2. Some Cancer-Associated Mutations Affect Ligand Binding and Functioning of A2AAR
3.3. Potential Effects of A2AAR Mutations on Cancer Development and Treatment
3.4. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Site-Directed Mutagenesis
4.3. Cell Culture and Transfection
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Membrane Preparation and Determination of Specific [3H]ZM241385 Binding
4.6. Radioligand-Homologous- and -Heterologous-Displacement Assays
4.7. Label-Free Whole-Cell Assay (xCELLigence RTCA System)
4.8. Data Analysis
4.9. Structural Mapping
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- de Lera Ruiz, M.; Lim, Y.H.; Zheng, J. Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Ann. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Linden, J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. 2014, 74, 7239–7249. [Google Scholar] [CrossRef] [Green Version]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood J. Am. Soc. Hematol. 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.W. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congreve, M.; Brown, G.A.; Borodovsky, A.; Lamb, M.L. Targeting adenosine A2A receptor antagonism for treatment of cancer. Expert Opin. Drug Discov. 2018, 13, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A 2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Zhukov, A.; Andrews, S.P.; Errey, J.C.; Robertson, N.; Tehan, B.; Mason, J.S.; Marshall, F.H.; Weir, M.; Congreve, M. Biophysical mapping of the adenosine A2A receptor. J. Med. Chem. 2011, 54, 4312–4323. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef]
- Guo, D.; Pan, A.C.; Dror, R.O.; Mocking, T.; Liu, R.; Heitman, L.H.; Shaw, D.E.; IJzerman, A.P. Molecular basis of ligand dissociation from the adenosine A2A receptor. Mol. Pharmacol. 2016, 89, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Segala, E.; Guo, D.; Cheng, R.K.; Bortolato, A.; Deflorian, F.; Dore, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef]
- O’Hayre, M.; Vazquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.A.; Ferretti, V.; Grossman, R.L.; Staudt, L.M. The NCI genomic data commons as an engine for precision medicine. Blood 2017, 130, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Bongers, B.J.; González, M.G.; Wang, X.; van Vlijmen, H.W.T.; Jespers, W.; Gutiérrez-de-Terán, H.; Ye, K.; IJzerman, A.P.; Heitman, L.H.; van Westen, G.J.P. Pan-cancer in silico analysis of somatic mutations in G-protein coupled receptors: The effect of evolutionary conservation and natural variance. bioRxiv 2010. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Receptor Molecular Biology; Sealfon, S.C., Ed.; Methods in Neurosciences; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Consortium, G.P. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Mulder-Krieger, T.; IJzerman, A.P.; Heitman, L.H. Functional efficacy of adenosine A2A receptor agonists is positively correlated to their receptor residence time. Br. J. Pharmacol. 2012, 166, 1846–1859. [Google Scholar] [CrossRef] [Green Version]
- Jespers, W.; Schiedel, A.C.; Heitman, L.H.; Cooke, R.M.; Kleene, L.; van Westen, G.J.; Gloriam, D.E.; Müller, C.E.; Sotelo, E.; Gutiérrez-de-Terán, H. Structural mapping of adenosine receptor mutations: Ligand binding and signaling mechanisms. Trends Pharmacol. Sci. 2018, 39, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Wess, J.; van Rhee, A.M.; Schöneberg, T.; Jacobson, K.A. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem. 1995, 270, 13987–13997. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Lee, B.X.; Glashofer, M.; van Rhee, A.M.; Jacobson, K.A. Mutagenesis reveals structure—Activity parallels between human A2A adenosine receptors and biogenic amine G protein-coupled receptors. J. Med. Chem. 1997, 40, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Stoy, H.; Gurevich, V.V. How genetic errors in GPCRs affect their function: Possible therapeutic strategies. Genes Dis. 2015, 2, 108–132. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Jakubík, J.; Randáková, A.; Rudajev, V.; Zimčík, P.; El-Fakahany, E.E.; Doležal, V. Applications and limitations of fitting of the operational model to determine relative efficacies of agonists. Sci. Rep. 2019, 9, 4637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, K.A.; Tehan, B.; Lebon, G.; Tate, C.G.; Weir, M.; Marshall, F.H.; Langmead, C.J. Pharmacology and structure of isolated conformations of the adenosine A2A receptor define ligand efficacy. Mol. Pharmacol. 2013, 83, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrisimovic, E.; Drobny, H.; Yang, Q.; Höfer, T.; Boehm, S.; Nanoff, C.; Schicker, K. Constitutive activity of the A2A adenosine receptor and compartmentalised cyclic AMP signalling fine-tune noradrenaline release. Purinergic Signal. 2012, 8, 677–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespers, W.; Heitman, L.H.; IJzerman, A.P.; Sotelo, E.; van Westen, G.J.; Åqvist, J.; Gutiérrez-de-Terán, H. Deciphering conformational selectivity in the A2A adenosine G protein-coupled receptor by free energy simulations. PLoS Comput. Biol. 2021, 17, e1009152. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Jiang, Q.; Jacobson, K.A.; IJzerman, A.P. Site-directed mutagenesis studies of human A2A adenosine receptors: Involvement of glu13 and his278 in ligand binding and sodium modulation. Biochem. Pharmacol. 2000, 60, 661–668. [Google Scholar] [CrossRef]
- Gutiérrez-de-Terán, H.; Massink, A.; Rodríguez, D.; Liu, W.; Han, G.W.; Joseph, J.S.; Katritch, I.; Heitman, L.H.; Xia, L.; Ijzerman, A.P.; et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure 2013, 21, 2175–2185. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Van Rhee, A.M.; Kim, J.; Yehle, S.; Wess, J.; Jacobson, K.A. Hydrophilic side chains in the third and seventh transmembrane helical domains of human A2A adenosine receptors are required for ligand recognition. Mol. Pharmacol. 1996, 50, 512–521. [Google Scholar]
- Nygaard, R.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Schwartz, T.W. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009, 30, 249–259. [Google Scholar] [CrossRef]
- Massink, A.; Gutiérrez-de-Terán, H.; Lenselink, E.B.; Zacarías, N.V.O.; Xia, L.; Heitman, L.H.; Katritch, V.; Stevens, R.C.; IJzerman, A.P. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol. Pharmacol. 2015, 87, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Dawson, E.S.; Wells, J.N. Determination of amino acid residues that are accessible from the ligand binding crevice in the seventh transmembrane-spanning region of the human A1 adenosine receptor. Mol. Pharmacol. 2001, 59, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, V.P.; Lane, J.R.; Lin, J.Y.; Katritch, V.; IJzerman, A.P.; Stevens, R.C. Ligand binding and subtype selectivity of the human A(2A) adenosine receptor: Identification and characterization of essential amino acid residues. J. Biol. Chem. 2010, 285, 13032–13044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hua, T.; Liu, Z.-J. Structural features of activated GPCR signaling complexes. Curr. Opin. Struct. Biol. 2020, 63, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Black, J.W.; Leff, P.; Shankley, N.P.; Wood, J. An operational model of pharmacological agonism: The effect of E/[A] curve shape on agonist dissociation constant estimation. Br. J. Pharmacol. 1985, 84, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular pharmacology of VEGF-A isoforms: Binding and signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [Green Version]
- Galandrin, S.; Bouvier, M. Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol. Pharmacol. 2006, 70, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Mediavilla-Varela, M.; Luddy, K.; Noyes, D.; Khalil, F.K.; Neuger, A.M.; Soliman, H.; Antonia, S.J. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol. Ther. 2013, 14, 860–868. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Bencivenni, S.; Battistello, E.; Vincenzi, F.; Colotta, V.; Catarzi, D.; Varano, F.; Merighi, S.; Borea, P.A.; Varani, K. Inhibition of A2A adenosine receptor signaling in cancer cells proliferation by the novel antagonist TP455. Front. Pharmacol. 2017, 8, 888. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Yu, N.; Atienza, J.M.; Bernard, J.; Blanc, S.; Zhu, J.; Wang, X.; Xu, X.; Abassi, Y.A. Real-time monitoring of morphological changes in living cells by electronic cell sensor arrays: An approach to study G protein-coupled receptors. Anal. Chem. 2006, 78, 35–43. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Black, J.W.; Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1983, 220, 141–162. [Google Scholar] [CrossRef]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.W.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | ZM241385 pKD a (KD (nM)) | NECA pKi b (Ki (nM)) | Bmax a (pmol/mg) |
|---|---|---|---|
| WT | 9.0 ± 0.1 (0.98) | 6.9 ± 0.0 (134) | 37 ± 5 |
| A15S1.41 | 8.9 ± 0.1 (1.5) | 6.6 ± 0.1 ** (277) | 12 ± 2 **** |
| F70LECL1 | 8.8 ± 0.1 (1.7) | 6.2 ± 0.1 **** (595) | 8 ± 1 **** |
| I92M3.40 | 8.9 ± 0.1 (1.3) | 6.2 ± 0.1 **** (606) | 9 ± 1 **** |
| L95F3.43 | 9.1 ± 0.0 (0.83) | 6.9 ± 0.1 (144) | 10 ± 1 **** |
| A165VECL2 | 8.7 ± 0.1 ** (2.2) | 6.7 ± 0.0 (210) | 17 ± 1 *** |
| I251T6.53 | 8.8 ± 0.1 (1.9) | 6.9 ± 0.1 (134) | 14 ± 1 *** |
| F257L6.59 | 8.8 ± 0.0 (1.8) | 6.8 ± 0.0 (151) | 27 ± 1 |
| A265SECL3 | 8.9 ± 0.0 (1.3) | 6.6 ± 0.1 * (246) | 27 ± 6 |
| A265VECL3 | 8.5 ± 0.1 *** (3.1) | 6.2 ± 0.0 **** (632) | 39 ± 1 |
| A265TECL3 | 8.9 ± 0.0 (1.2) | 6.6 ± 0.0 ** (266) | 37 ± 6 |
| V275A7.40 | 8.9 ± 0.1 (1.4) | 7.4 ± 0.0 **** (42) | 20 ± 2 ** |
| Mutant | Potency pEC50 a | Efficacy Emax% (AUC-1 μM) a | Relative Efficacy τ b |
|---|---|---|---|
| WT | 8.4 ± 0.2 | 100 ± 5 (5.7) | 37 ± 13 |
| F70LECL1 | 7.6 ± 0.2 | 164 ± 19 (8.9) | 28 ± 11 |
| I92M3.40 | 7.2 ± 0.2 ** | 119 ± 4 (5.8) | 10 ± 3 |
| A265VECL3 | 7.4 ± 0.2 * | 135 ± 2 (7.2) | 21 ± 8 |
| V275A7.40 | 7.1 ± 0.1 ** | 106 ± 19 (5.1) | 1 ± 0 * |
| S132L4.53 | 7.3 ± 0.0 * | 248 ± 30 *** (12.6) | n.a. |
| H278N7.43 | 7.3 ± 0.2 ** | 223 ± 33 ** (10.8) | n.a. |
| S281L7.46 | 7.7 ± 0.3 | 146 ± 25 (7.7) | n.a. |
| P285L7.50 | 7.4 ± 0.5 * | 101 ± 8 (5.2) | n.a. |
| Mutant | Inverse Agonism of | Inhibition |
|---|---|---|
| ZM241385-1 μM a | pIC50 b | |
| WT | −3.0 ± 0.8 | 6.4 ± 0.3 |
| F70LECL1 | −2.0 ± 0.5 | 6.7 ± 0.2 |
| I92M3.40 | −1.8 ± 0.5 | 6.9 ± 0.1 |
| A265VECL3 | −2.3 ± 0.0 | 6.3 ± 0.2 |
| V275A7.40 | −3.6 ± 0.6 | n.d. |
| S132L4.53 | −0.4 ± 0.2 ** | 7.3 ± 0.1 ** |
| H278N7.43 | −0.3 ± 0.3 ** | 7.4 ± 0.1 ** |
| S281L7.46 | −0.7 ± 0.2 * | 6.6 ± 0.1 |
| P285L7.50 | −2.4 ± 0.8 | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, C.; Wang, X.; Jespers, W.; Liu, R.; Zamarbide Losada, S.D.; Gorostiola González, M.; van Westen, G.J.P.; Danen, E.H.J.; Heitman, L.H. Cancer-Associated Mutations of the Adenosine A2A Receptor Have Diverse Influences on Ligand Binding and Receptor Functions. Molecules 2022, 27, 4676. https://doi.org/10.3390/molecules27154676
Feng C, Wang X, Jespers W, Liu R, Zamarbide Losada SD, Gorostiola González M, van Westen GJP, Danen EHJ, Heitman LH. Cancer-Associated Mutations of the Adenosine A2A Receptor Have Diverse Influences on Ligand Binding and Receptor Functions. Molecules. 2022; 27(15):4676. https://doi.org/10.3390/molecules27154676
Chicago/Turabian StyleFeng, Chenlin, Xuesong Wang, Willem Jespers, Rongfang Liu, Sofía Denise Zamarbide Losada, Marina Gorostiola González, Gerard J. P. van Westen, Erik H. J. Danen, and Laura H. Heitman. 2022. "Cancer-Associated Mutations of the Adenosine A2A Receptor Have Diverse Influences on Ligand Binding and Receptor Functions" Molecules 27, no. 15: 4676. https://doi.org/10.3390/molecules27154676
APA StyleFeng, C., Wang, X., Jespers, W., Liu, R., Zamarbide Losada, S. D., Gorostiola González, M., van Westen, G. J. P., Danen, E. H. J., & Heitman, L. H. (2022). Cancer-Associated Mutations of the Adenosine A2A Receptor Have Diverse Influences on Ligand Binding and Receptor Functions. Molecules, 27(15), 4676. https://doi.org/10.3390/molecules27154676