
Identifying Terpenoid Biosynthesis Genes in Euphorbia maculata via Full-Length cDNA Sequencing

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Illumina RNA-Seq Library Construction and Sequencing
2.3. Full-Length cDNA Sequencing
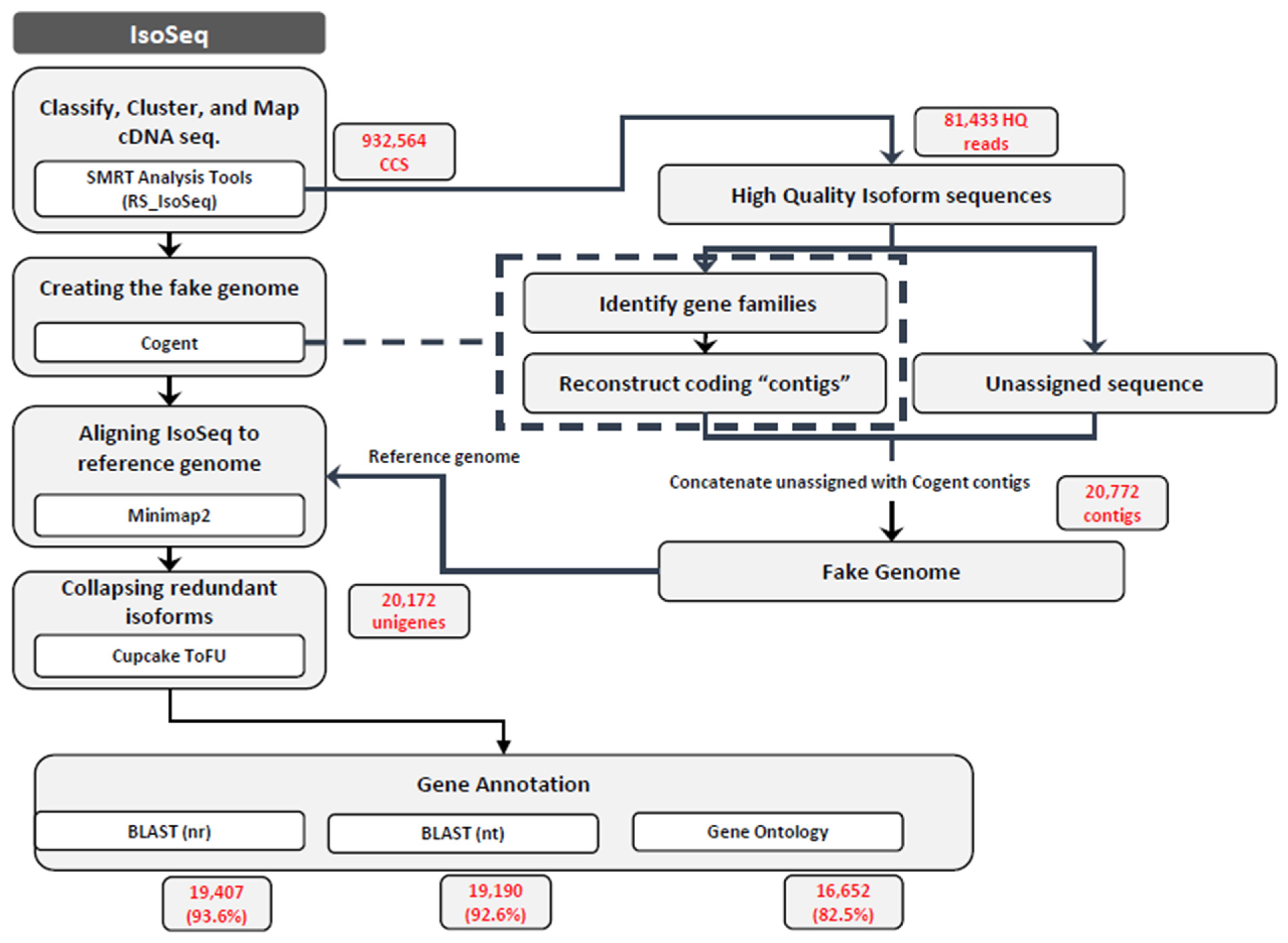
2.4. De Novo Assembly and Iso-Seq Data Analysis Using a Bioinformatics Pipeline
2.5. Full-Length Unique Transcript Model Reconstruction
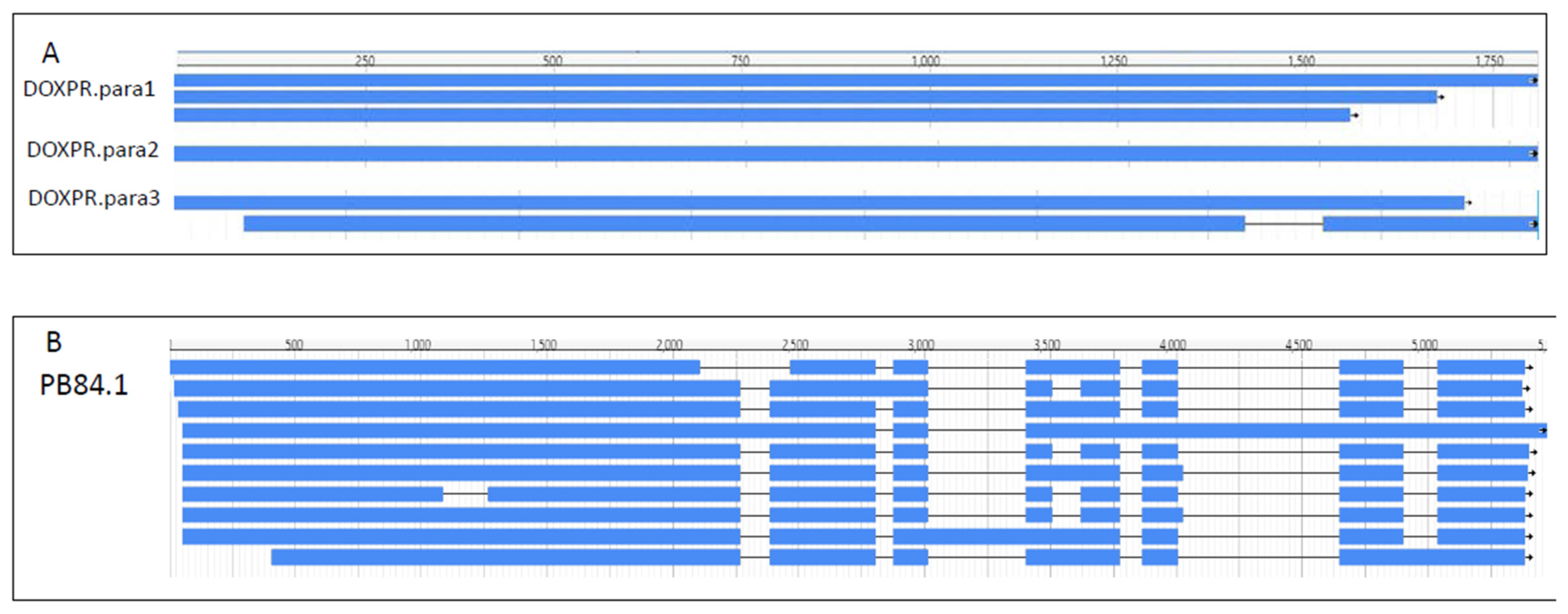
2.6. Isoform and Paralog Identification
2.7. Functional Annotation
2.8. Differential Gene Expression Analysis
3. Results
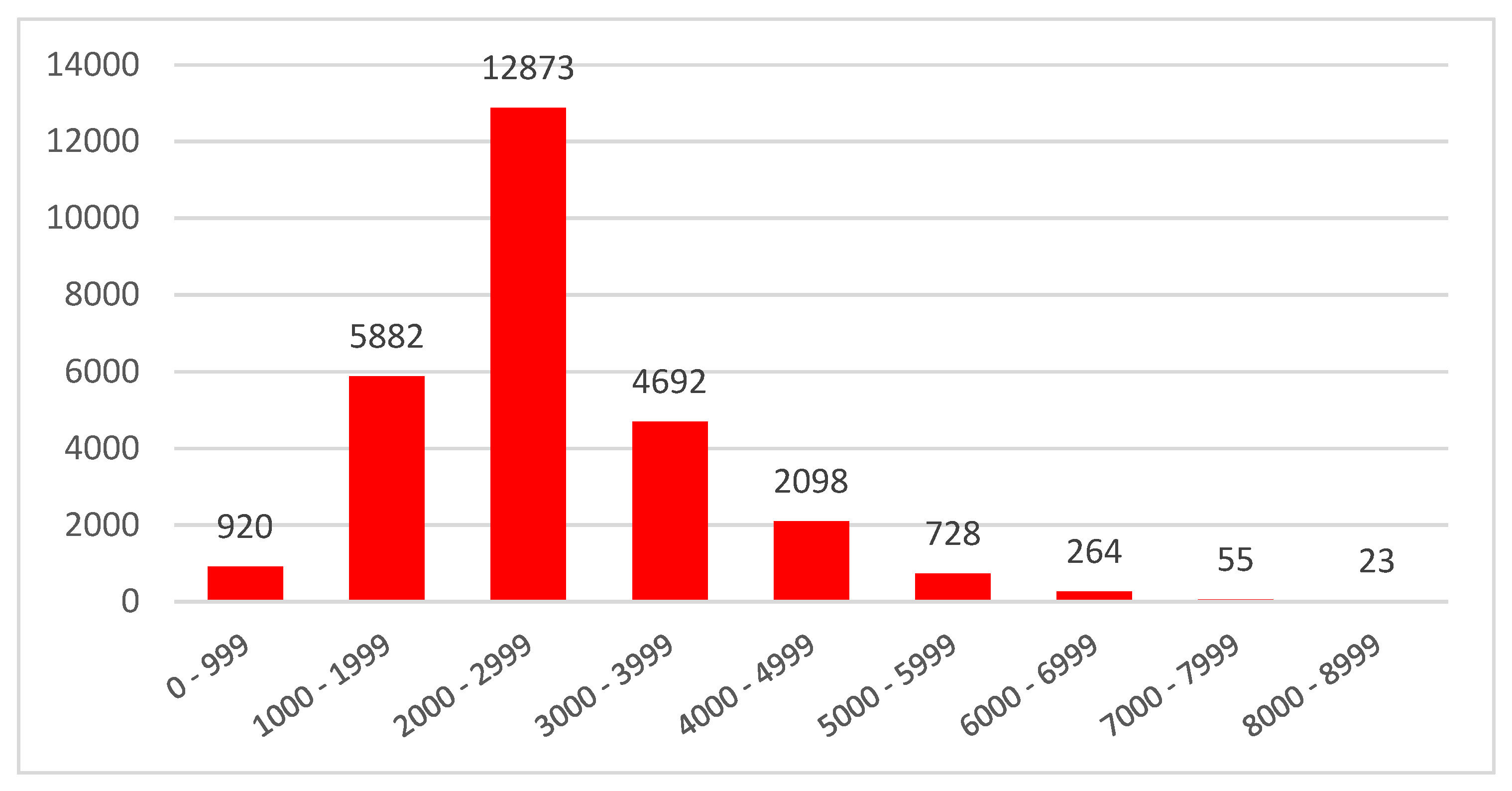
3.1. E. maculata Transcriptome Analysis Using PacBio Iso-Seq
3.2. Isoforms and Paralogs
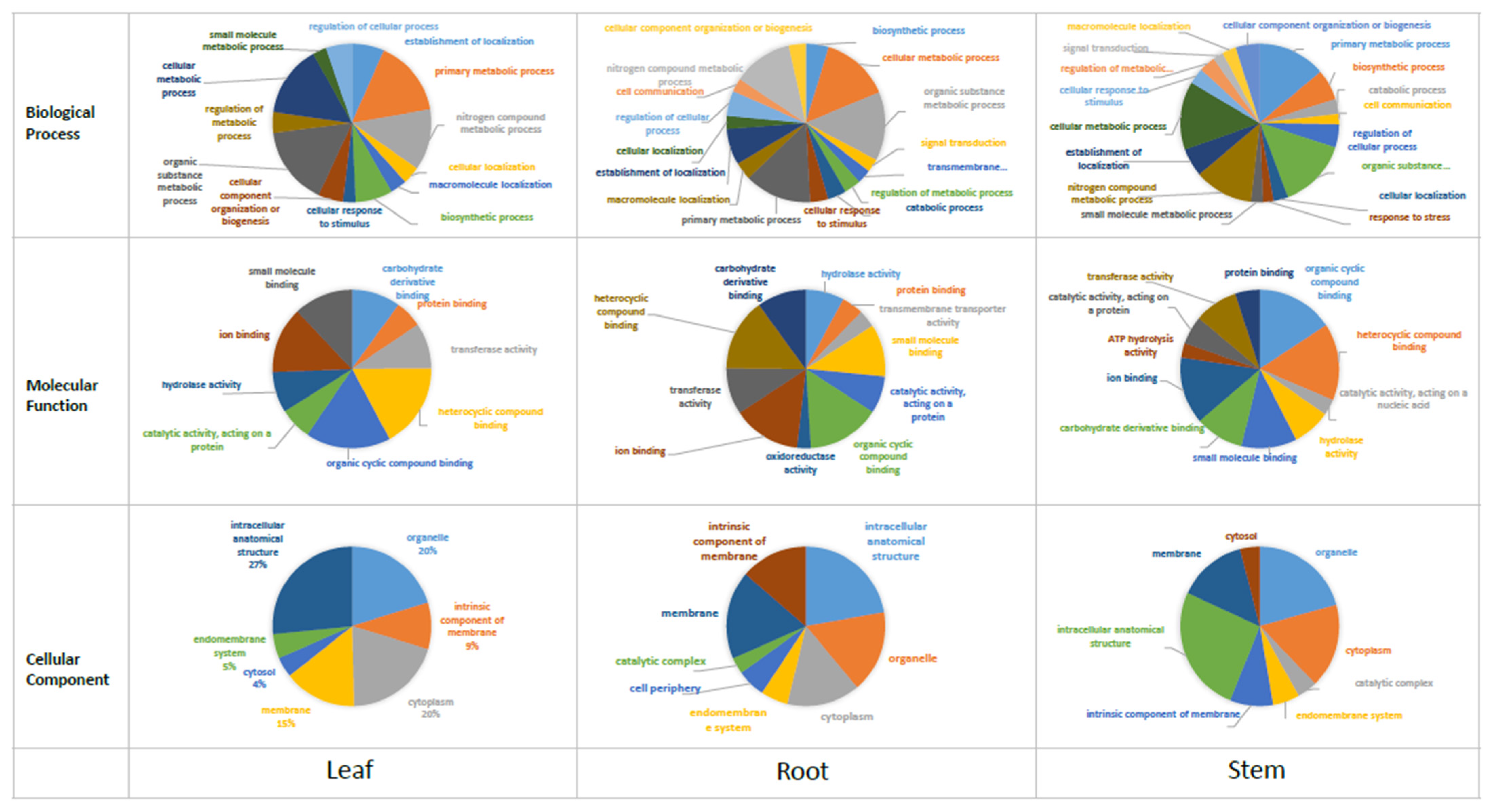
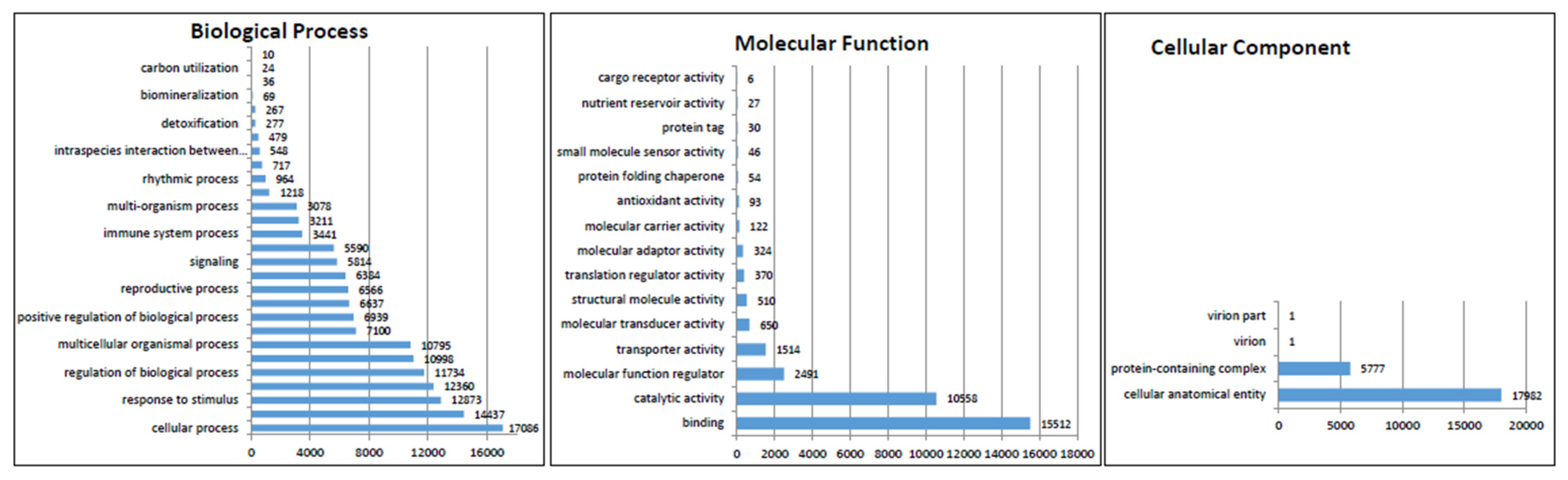
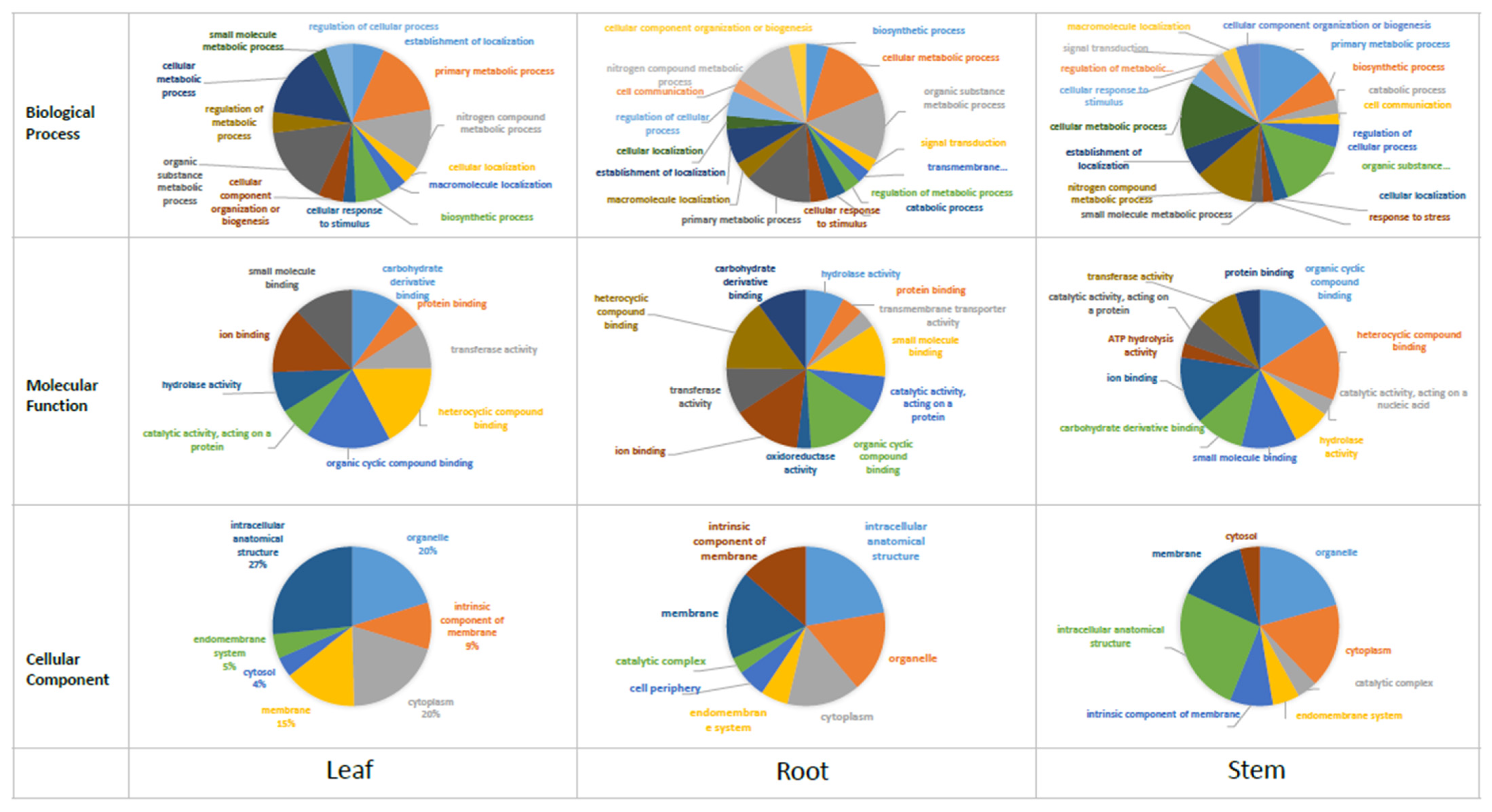
3.3. Functional Annotation
3.4. Gene Expression Analysis across Different Tissues
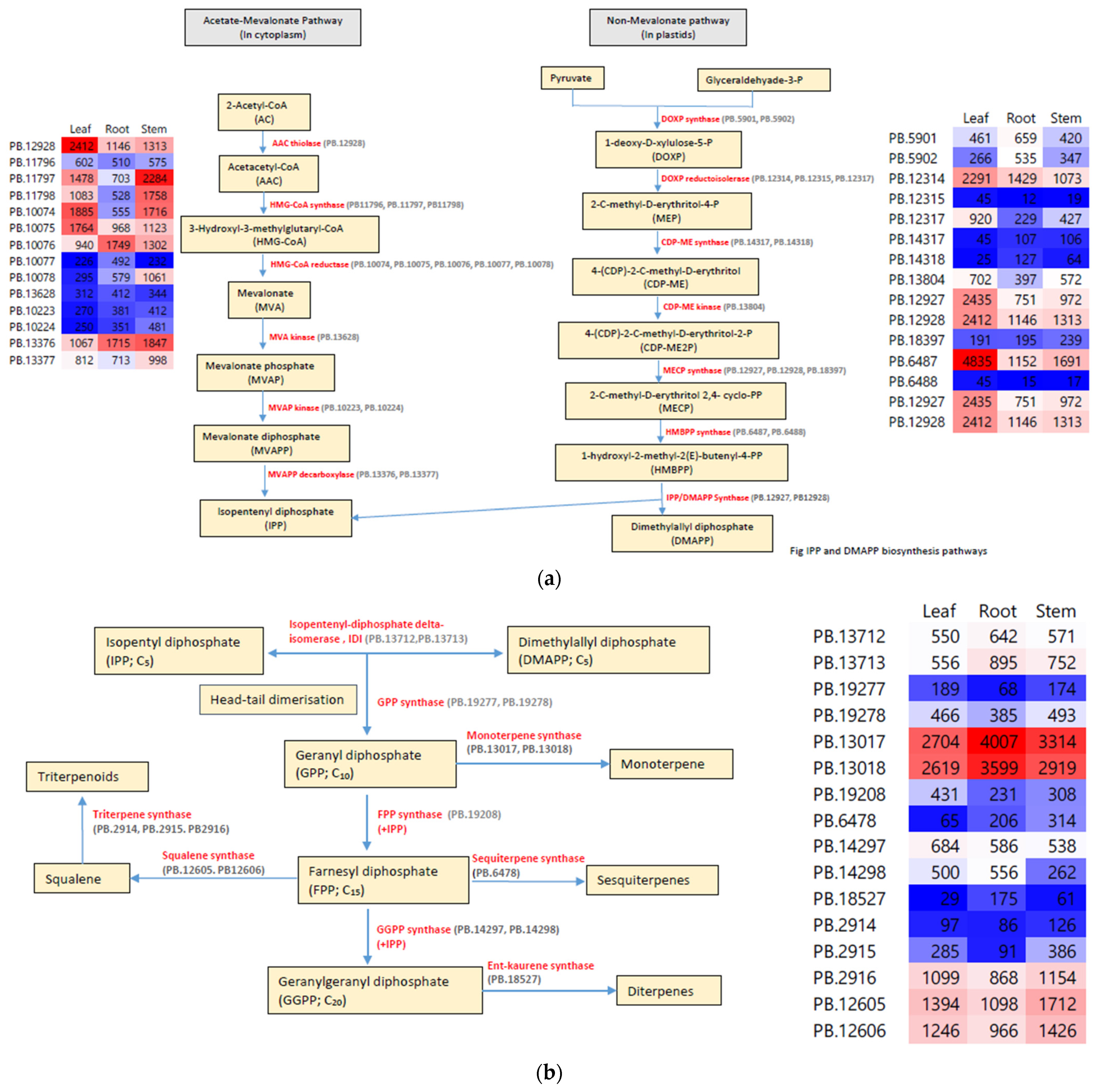
3.5. Terpenoid Biosynthesis Pathway Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- The Plant List. Available online: http://www.theplantlist.org/1.1/browse/A/Compositae/Inula/ (accessed on 12 April 2022).
- Shi, Q.; Su, X.; Kiyota, H. Chemical and pharmacological research of the plants in genus Euphorbia. Chem. Rev. 2008, 108, 4295–4327. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Iriti, M.; Vitalini, S.; Antolak, H.; Pawlikowska, E.; Kregel, D.; Sharifi-Rad, J.; Pyeleye, S.; Ademiluyi, A.; Czpek, K. Euphorbia-derived natural products with potential for use in health maintenance. BioMol. 2019, 9, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Gao, L.; Tang, M.; Feng, B.; Pei, Y.; Yasukawa, K. Triterpenoids from Euphorbia maculata and their anti-inflammatory effects. Molecules 2018, 23, 2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agata, I.; Hatano, T.; Nakaya, Y.; Sugaya, T.; Nishibe, S.; Yoshida, T. Tannins and related polyphenols of Euphorbiaceous plants. 8. Emaculin A and Eusupinin A, and accompanying polyphenols from Euphorbia maculata L. and E. supine Rafin. Chem. Pharm. Bull. 1991, 39, 881–883. [Google Scholar] [CrossRef] [Green Version]
- Akamura, Y.; Kawada, K.; Hatano, T.; Agata, I.; Sugaya, T.; Nishibe, S.; Okuda, T.; Yoshida, T. Four new hydrolysable tannins and an acylated flavonol glycoside from Euphorbia maculata. Can. J. Chem. 1997, 75, 727–733. [Google Scholar]
- Matsunaga, S.; Tanaka, R.; Akagi, M. Triterpinoids from Euphorbia maculata. Phytochemistry 1988, 27, 535–537. [Google Scholar] [CrossRef]
- Sun, L.; Li, S.; Wang, F.; Xin, F. Research progresses in the synthetic biology of terpenoids. Biotechnol. Bull. 2017, 33, 64–75. [Google Scholar] [CrossRef]
- Ludwiczuk, A.; Skalika-Woziak, K.; Georgiev, M. Terpenoids Pharmacogsy; Badal, S., Delgoda, R., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 233–266. [Google Scholar] [CrossRef]
- Dubey, V.; Bhalla, R.; Luthra, R. An overview of the non-mevalonate pathway for terpenoid biosyntheis in plants. J. Biosci. 2003, 28, 637–646. [Google Scholar] [CrossRef]
- Bochar, D.; Friesen, J.; Stauffacher, C.; Rodwell, V. Biosynthesis of mevalonic acid from acetyl—CoA. In Comprehensive Natural Product Chemistry; Cane, D.E., Ed.; Pergamon: Oxford, UK, 1999; pp. 15–44. [Google Scholar]
- Eisenreich, W.; Bacher, A.; Arigoni, D.; Rodhdich, F. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell. Mol. Life Sci. 2004, 61, 1401–1426. [Google Scholar] [CrossRef]
- Chang, W.; Song, H.; Liu, H.; Liu, P. Current development in isoprenoid precursor biosynthesis and regulation. Curr. Opin. Chem. Biol. 2013, 17, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Sawai, S.; Saito, K. Triterpenoid biosynthesis and engineering in plants. Front. Plant. Sci. 2011, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Tsopmo, A.; Kamnaing, P. Terpenoids constituents of Euphobia sapinii. Phytochem. Letters 2011, 4, 218–221. [Google Scholar] [CrossRef]
- Ferreira, R.; Kincses, A.; Gajdacs, M.; Spengler, G.; Dos Santos, D.; Molnar, J.; Ferreira, M. Terpenoids from Euphorbia pedroi as multidrug-resistance reversers. J. Nat. Prod. 2018, 81, 2032–2040. [Google Scholar] [CrossRef]
- Lima, E.; Medeiros, J. Terpenoid compounds in the latex of Euphorbia azorica from Azores. BioMed. J. Sci. Tech. Res. 2020, 26, 19680–19682. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, X.; Wei, W.; Fan, Z.; Huang, T.; Pan, X. Analaysis of leaf morphology, secondary metabolites nd proteins related to the resisytance to Tetranychus cinnabarinus in Cassaba (Manihot esculenta Crantz). Sci. Rep. 2020, 10, 14197. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Wang, B.; Kumar, V.; Olson, A.; Ware, D. Reviving the transcriptome studies: An insght of single-molecule transcriptome sequencing. Front. Genet. 2019, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Ban, Y.; Roy, N.; Yang, H.; Choi, H.; Kim, J.; Babu, P.; Ha, K.; Ham, J.; Park, K.; Choi, I. Comparative transcriptome analysis reveals higher expression of stress and defense responsive genes in dwarf soybeans obtained from the crossing of G. max and G. soja. Genes Genom. 2019, 41, 1315–1327. [Google Scholar] [CrossRef]
- Mitu, S.; Cummins, S.; Reddell, P.; Ogbourne, S. Transcriptome analysis of the medicinally significant plant Fontainea picrosperma (Euphorbiaceae) reveals conserved biosynthetic pathways. Fitoterapia 2020, 146, 104680. [Google Scholar] [CrossRef]
- Tilgner, H.; Jahanbani, F.; Blauwkamp, T.; Moshrefi, A.; Jaeger, E.; Chen, F.; Harel, I.; Bustamante, C.; Rasmussen, M.; Snyder, M. Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat. Biotechnol. 2015, 33, 736–742. [Google Scholar] [CrossRef]
- Zimin, A.; Puiu, D.; Luo, M.C.; Zhu, T.; Koren, S.; Marcais, G.; Yorke, J.; Dvorak, J.; Salzberg, S. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Roy, N.; Lee, I.; Choi, A.; Choi, B.; Yu, Y.; Park, N.; Park, K.; Kim, S.; Yang, H.; et al. Genome-wide transcriptome profiling of the medicinal plant Zanthoxylum planispinum using a single-molecule direct RNA sequencing approach. Genomics 2019, 111, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Lee, I.; Kim, J.; Ramekar, R.; Park, K.; Park, N.; Yeo, J.; Choi, I.; Kim, S. De novo assembly and characterization of transcriptome in the medicinal plant Euphorbia jolkini. Genes Genom. 2020, 42, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Choi, I.; Um, T.; Jeon, M.; Kim, B.; Kim, Y.; Yu, J.; Kim, S.; Kim, N. Gene Expression and Isoform Identification of PacBio Full-Length cDNA Sequences for Berberine Biosynthesis in Berberis koreana. Plants 2021, 10, 1314. [Google Scholar] [CrossRef]
- Qiao, W.; Li, C.; Mosongo, I.; Liang, Q.; Liu, M.; Wang, X. Comparative Transcriptome Analysis Identifies Putative Genes Involved in Steroid Biosynthesis in Euphorbia tirucalli. Genes 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wang, M.; Chai, J.; Li, Q.; Zhou, Y.; Li, Y.; Cai, X. De novo assembly and characterization of the transcriptome and development of microsatellite markers in a Chinese endemic Euphorbia kansui. Biotechnol. Biotechnol. Equipm. 2020, 34, 562–574. [Google Scholar] [CrossRef]
- Chin, C.; Alexander, H.; Marks, P.; Klammer, A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; McCarthy, D.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McGettigan, P. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Jo, I.; Lee, J.; Hong, C.; Lee, D.; Bae, W.; Park, S.; Ahn, Y.; Kim, Y.; Kim, J.; Lee, J.; et al. Isoform sequencing provides a more comprehensive view of the Panax ginng transcriptome. Gene 2017, 8, 228. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Zhang, H.; Sun, X.; Zhao, H.; Wu, L.; Zhu, D.; Yang, G.; Shao, Y.; Zhang, X.; Mao, X.; et al. A comprehensive review of the structure elucidation and biological activity of triterpenoids from Ganoderma spp. Molecules 2014, 19, 17478–17535. [Google Scholar] [CrossRef]
- AGI 2020. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2020, 408, 796–815. [Google Scholar] [CrossRef] [Green Version]
- Abdelgardir, H.; Van Staden, J. Ethnobotany, ethnopharmacology and toxicity of Jatropha curcas L. (Euphorbiaceae): A review. South African J. Bot. 2013, 88, 204–218. [Google Scholar] [CrossRef] [Green Version]
- Gracz-Bernaciak, J.; Mazur, O.; Nawrot, R. Functional studies of plant latex as a rich source of bioactive compounds: Focus on proeins and alkaloids. Int. J. Mol. Sci. 2021, 22, 12427. [Google Scholar] [CrossRef]
- Montoro, P.; Wu, S.; Favreau, B.; Herlinawati, E.; Labrune, C.; Martin-Magniette, M.-L.; Pointet, S.; Rio, M.; Leclercq, J.; Ismawanto, S.; et al. Transcriptome analysis in Hevea brasiliensis latex revealed changes in hormone signalling pathways during ethephon stimulation and consequent Tapping Panel Dryness. Sci. Rep. 2018, 8, 8483. [Google Scholar] [CrossRef]
- Bakar, M.; Kamerker, U.; Rahman, S.; Sakaff, M.; Othgman, A. Transcriptome dataset from bark and latex tissues of three Havea brasilensis clones. Data Brief 2020, 32, 106188. [Google Scholar] [CrossRef]
- Liu, X.; Li, R.; Lu, W.; Zhou, Z.; Jiang, X.; Zhao, H.; Yang, B.; Lu, S. Transcriptome analysis identifies key genes involed in the regulation of epidermal lupeol biosynthesis in Ricinus communis. Indus. Crops Product. 2021, 160, 113100. [Google Scholar] [CrossRef]
- Li, S.; Yu, X.; Cheng, Z.; Zeng, C.; Li, W.; Zhang, L.; Peng, M. Large-scale analysis of the cassava transcriptome freveals the impact of cold stress on alternative splicing. J. Exp. Bot. 2020, 71, 422–434. [Google Scholar] [CrossRef]
- Kamsen, R.; Kalapanulak, S.; Chiewchanaset, P.; Saithong, T. Transcriptome integrated metabolic modeling of carbon assimilation underlying storage root development in cassava. Sci. Rep. 2021, 11, 8758. [Google Scholar] [CrossRef]
- Kwon, E.; Basnet, P.; Roy, N.; Kim, J.; Heo, K.; Park, K.; Um, T.; Kim, N.; Choi, I. Identification of resurrection genes from the transcription of dehydrated and rehydrated Selaginella tamaricina. Plant Signal. Behav. 2021, 16, 1973703. [Google Scholar] [CrossRef]
- Sahlin, K.; Tomaszkiewicz, M.; Makova, K.; Meddev, P. Desiphering highly similar multigene family transcripts from Iso-Seq data with IsoCon. Nat. Commun. 2018, 9, 4601. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E. Orthologs, paralogs, and evolutionary genomics. Ann. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef] [Green Version]
- Ibn-Salem, J.; Muro, E.; Andrade-Navarro, M. Co-regultion of paralog genes in the three-dimensional chromatin architecture. Nucleic Acids Res. 2017, 45, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Lambrosino, L.; Bostan, H.; di Salle, P.; Sangiovanni, M.; Vigilante, A.; Chiusano, M. pATsi:paralogs and singlton genes from Arabidopsis thaliana. Evol. Bioinform. 2016, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.; Frey, B.; Blencowe, B. Deep surveying of alternative splicing compexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Filchkin, S.; Priest, H.; Megraw, M.; Mockler, T. Alternative splcing in plants: Direct traffic at the crossroad of adatation and environmental stresses. Genome Res. 2015, 20, 45–58. [Google Scholar] [CrossRef]
- Rodwell, V.; Beach, M.; Bischoff, K.; Bochar, D.; Darnay, B.; Friesen, J.; Gill, J.; Hedl, M.; Jordan-Starck, T.; Kennely, P.; et al. 3-Hydroxy-3-methylglutaryl-CoA reductase. Methods Enzymol. 2000, 324, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Yeo, H.; Park, Y.; Kim, Y.; Park, C.; Kim, J.; Park, S. Integrated analysis of transcriptome and metabolome and evaluation of antioxidant activities in Lavendula pubescence. Antioxidants 2021, 10, 1027. [Google Scholar] [CrossRef]
- Hale, I.; O’Neill, P.; Berry, N.; Odom, A.; Sharma, R. The MEP pathway and development of inhibotors as potential antiinfective agents. Med. Chem. Comm. 2012, 3, 418–433. [Google Scholar] [CrossRef]
- Obiol-Pardo, C.; Rubio-Martinez, J.; Impeial, S. The methy;erythritol phosphate (MEP) pathway for isopreniod biosynthesis as a target for the deelopemnt of new drugs against tuberculosis. Curr. Med. Chem. 2011, 18, 1325–1338. [Google Scholar] [CrossRef]
- Demirkiran, O.; Topcu, G.; Hussain, J.; Ahamd, V.; Choudhary, M. Structure elucidation of two new unusal monoterpene glycoside from Euphorbia decipiens, by 1D and 2D NMR experiments. Mag. Reson. Chem. 2011, 49, 673–677. [Google Scholar] [CrossRef]
- Wang, A.; Huo, X.; Feng, L.; Sun, C.; Deng, S.; Zhang, H.; Zhang, B.; Ma, X.; Jia, J.; Wang, C. Phenolic glycosides and monterpenoids from roots of Euphobia ebracteolata and their bioectivities. Fitoterapia 2017, 121, 175–182. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, L.; Wu, M.; Xia, G.; Lin, P.; Zi, J. Chracyerization of a sequiterpene synthase catalyzing formation of Cedrol and two diasteroisomers of Trichoacorenol from Euphobia fischeriana. J. Nat. Prod. 2021, 84, 1780–1786. [Google Scholar] [CrossRef]
- Fais, A.; Delogi, G.; Floris, S.; Era, B.; Medda, R.; Pintus, F. Euphorbia characias: Phytochemistry and biological activities. Plants 2021, 10, 1468. [Google Scholar] [CrossRef]
- Bloch, K. Sterol, structure and membrane function. Critical. Rev. Biochem. 2008, 14, 47–92. [Google Scholar] [CrossRef]
- Zheng, Z.; Cao, X.; Li, C.; Yuan, B.; Jiang, J. Molecular cloning and expression of a squalene synthase gene from a medicinal plant, Euphorbia pekinensis Rupr. Acta Physiol. Plant. 2013, 35, 3007–3014. [Google Scholar] [CrossRef]
- Uchida, H.; Yamashita, H.; Kajikawa, M.; Ohyama, K.; Nakayachi, O.; Sugiyama, R.; Yamato, K.; Muranaka, T.; Fukazawa, H.; Takemura, M.; et al. Cloning and characterization of a squalene synthase gene from a petroleum plant, Euphorbia tirucalli L. Planta 2009, 229, 1243–1252. [Google Scholar] [CrossRef]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene biosynthesis in plants. Ann. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef]
- Forestier, E.; Romero-Segura, C.; Pateraki, I.; Centeno, E.; Compagnon, V.; Preiss, M.; Berna, A.; Boronat, A.; Bach, T.; Darnet, S.; et al. Distinct triterpene synthases in laticifer of Euphorbia lathyris. Sci. Rep. 2019, 9, 4840. [Google Scholar] [CrossRef] [Green Version]
- Zerbe, P.; Bohlmann, J. Plant diterpene synthases: Exploring modularity and metabolic diversity for bioengineering. Trends Biotechnol. 2015, 33, 419–428. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis Metric | Under 4 kb | Over 4 kb |
|---|---|---|
| Polymerase reads | ||
| Total Polymerase Read length (bp) | 31,143,923,142 | 31,036,246,900 |
| Total Polymerase Reads | 548,527 | 601,659 |
| Average Polymerase Read Length (bp) | 56,777 | 51,584 |
| Subreads | ||
| Total Subreads | 18,525,814 | 8,597,836 |
| N50 | 2504 | 3893 |
| Average Subread Length (bp) | 1630 | 3739 |
| Circular consensus sequence (CCS) reads | ||
| Total CCS reads | 467,479 | 465,085 |
| Total CCS read length (bp) | 1,155,280,061 | 1,879,756,017 |
| Average CCS read length (bp) | 2471 | 4040 |
| Transcript clustering | ||
| Number of polished high-quality isoforms | 47,860 | 33,573 |
| Number of polished low-quality isoforms | 405 | 993 |
| Iso Seq Result | Number of Reads | Length (bp) |
|---|---|---|
| High-quality consensus Seq. | 76,631 | 216,086,311 |
| Reconstructed Coding Contig | 19,902 | 60,494,776 |
| Unassigned Seq | 3344 | 10,608,597 |
| Fake Genome | 20,722 | 71,103,373 |
| Minimum read length | 100 | |
| Maximum read length | 13,544 | |
| Average read length | 3059 | |
| Number of Isoforms | Number of Transcripts | Percentage (%) |
| 1 | 13,492 | 66.9 |
| 2 | 3946 | 19.6 |
| 3 | 1269 | 6.3 |
| 4 | 630 | 3.1 |
| 5 | 381 | 1.9 |
| 6 | 185 | 0.9 |
| 7 | 116 | 0.6 |
| 8–25 | 153 | 0.8 |
| Total | 20,172 | 100 |
| Number of Paralogs | Number of Transcripts |
|---|---|
| 1 | 20,246 |
| 2 | 84 |
| 3 | 14 |
| 4 | 18 |
| 5–20 | 27 |
| Mapping Information | Leaf | Root | Stem |
|---|---|---|---|
| No. of total reads | 25,971,888 | 29,095,594 | 26,009,774 |
| No. of mapped Paired-end reads | 18,411,506 | 17,458,816 | 16,843,542 |
| % Mapped Paired-end reads | 70.9 | 60 | 64.8 |
| No. of expressed genes | |||
| 0 | 2987 | 3642 | 2714 |
| >0 | 17,735 | 17,260 | 18,008 |
| Differential Expression | Leaf vs. Root | Root vs. Stem | Leaf vs. Stem |
| Up | 447 | 1049 | 87 |
| Down | 1660 | 177 | 266 |
| Enzymes | Abbreviation | Pathway | No of Paralogs | Range of Isoform |
|---|---|---|---|---|
| Acetate-Mevalonate | ||||
| Acetoacetyl CoA thiolase | AAC thiolase | 1 | 1 | |
| 3-Hydroxy-3-methylglutaryl synthase | HMG-CoA Synthase | 3 | 1 | |
| 3-Hydroxy-3-methylglutaryl reductase | HMG-CoA Reductase | 5 | 1–3 | |
| Mevalonate kinase | MVA kinase | 1 | 1 | |
| Mevalonate phosphate kinase | MVAP kinase | 2 | 1–2 | |
| Mevalonate diphosphate decarboxylase | MVAPP carboxylase | 2 | 1–2 | |
| Non-Mevalonate | ||||
| 1-deoxy-D-xylulose-5-phophate synthase | DOXP synthase | 2 | 1–3 | |
| 1-deoxy-D-xylulose-5-phophate reductoisomerase | DOXP reductoisomerase | 3 | 1–3 | |
| Cytidine diphosphate 2-C-methyl-D-erythritol synthase | CDP-ME synthase | 2 | 1 | |
| Cytidine diphosphate 2-C-methyl-D-erythritol kinase | CDP-ME kinase | 1 | 1 | |
| 2C-methyl-D-erythritol synthase | MECP synthase | 4 | 1 | |
| 1-hydroxy-2-methyl-2-D-butenyl-4-diphosphate synthase | HMBPP synthase | 2 | 2 | |
| IPP/MDAPP synthase | IspH | 2 | 1 | |
| Terpenoid synthesis | ||||
| Isopentenyl-diphosphate delta-isomerase | IDI | 2 | 1–2 | |
| Geranyl diphosphate synthase | GPP synthase | 2 | 1 | |
| Farnesyl diphosphate synthase | FPP synthase | 1 | 2 | |
| Geranyl geranyl diphosphate synthase | GGPP synthase | 2 | 1 | |
| Monoterpene synthase | Monoterpene synthase | 2 | 1 | |
| Sesquiterpene synthase | Sesquiterpene synthase | 1 | 1 | |
| Diterpene synthase | Ent-Kaurene synthase | 1 | 1 | |
| Squalene synthase | Squalene synthase | 2 | 1 | |
| Triterpene synthase | Triterpene synthase | 3 | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, M.J.; Roy, N.S.; Choi, B.-S.; Oh, J.Y.; Kim, Y.-I.; Park, H.Y.; Um, T.; Kim, N.-S.; Kim, S.; Choi, I.-Y. Identifying Terpenoid Biosynthesis Genes in Euphorbia maculata via Full-Length cDNA Sequencing. Molecules 2022, 27, 4591. https://doi.org/10.3390/molecules27144591
Jeon MJ, Roy NS, Choi B-S, Oh JY, Kim Y-I, Park HY, Um T, Kim N-S, Kim S, Choi I-Y. Identifying Terpenoid Biosynthesis Genes in Euphorbia maculata via Full-Length cDNA Sequencing. Molecules. 2022; 27(14):4591. https://doi.org/10.3390/molecules27144591
Chicago/Turabian StyleJeon, Mi Jin, Neha Samir Roy, Beom-Soon Choi, Ji Yeon Oh, Yong-In Kim, Hye Yoon Park, Taeyoung Um, Nam-Soo Kim, Soonok Kim, and Ik-Young Choi. 2022. "Identifying Terpenoid Biosynthesis Genes in Euphorbia maculata via Full-Length cDNA Sequencing" Molecules 27, no. 14: 4591. https://doi.org/10.3390/molecules27144591
APA StyleJeon, M. J., Roy, N. S., Choi, B.-S., Oh, J. Y., Kim, Y.-I., Park, H. Y., Um, T., Kim, N.-S., Kim, S., & Choi, I.-Y. (2022). Identifying Terpenoid Biosynthesis Genes in Euphorbia maculata via Full-Length cDNA Sequencing. Molecules, 27(14), 4591. https://doi.org/10.3390/molecules27144591