
Noble Gas—Silicon Cations: Theoretical Insights into the Nature of the Bond


Abstract
:1. Introduction
2. Method of Bonding Analysis
3. Computational Details
4. Results and Discussion
4.1. The MP2/aVTZ Predicted Data
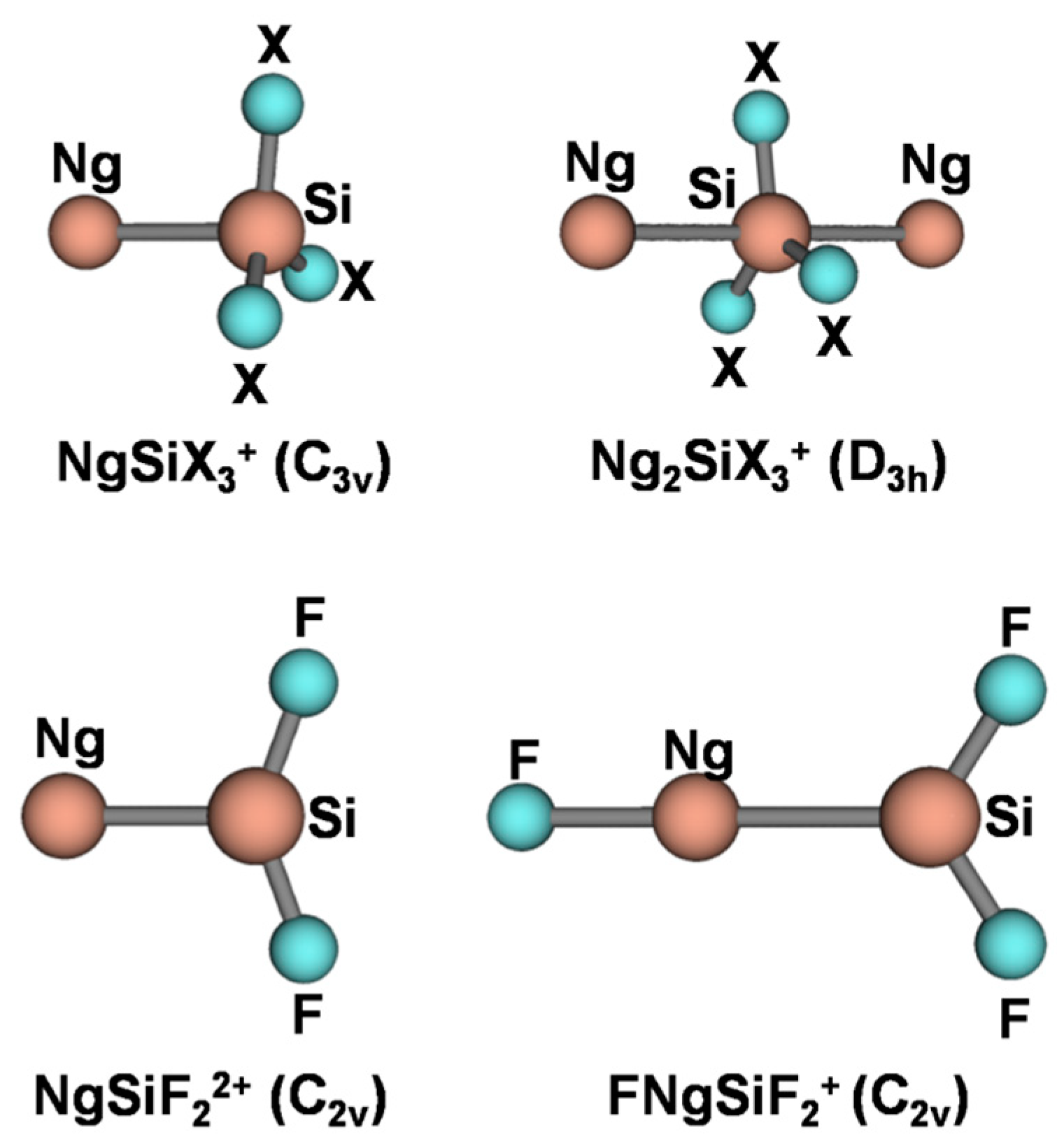
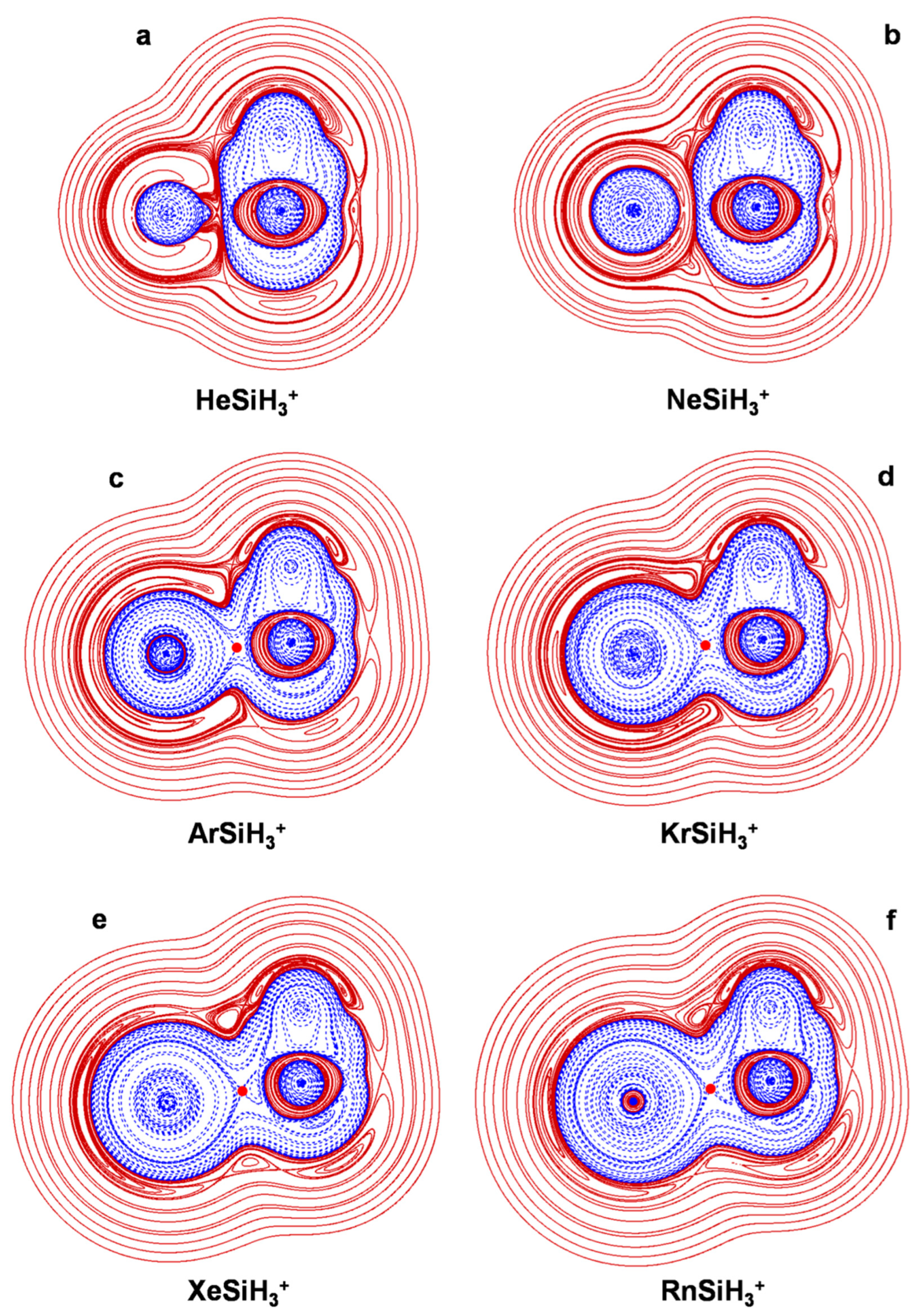
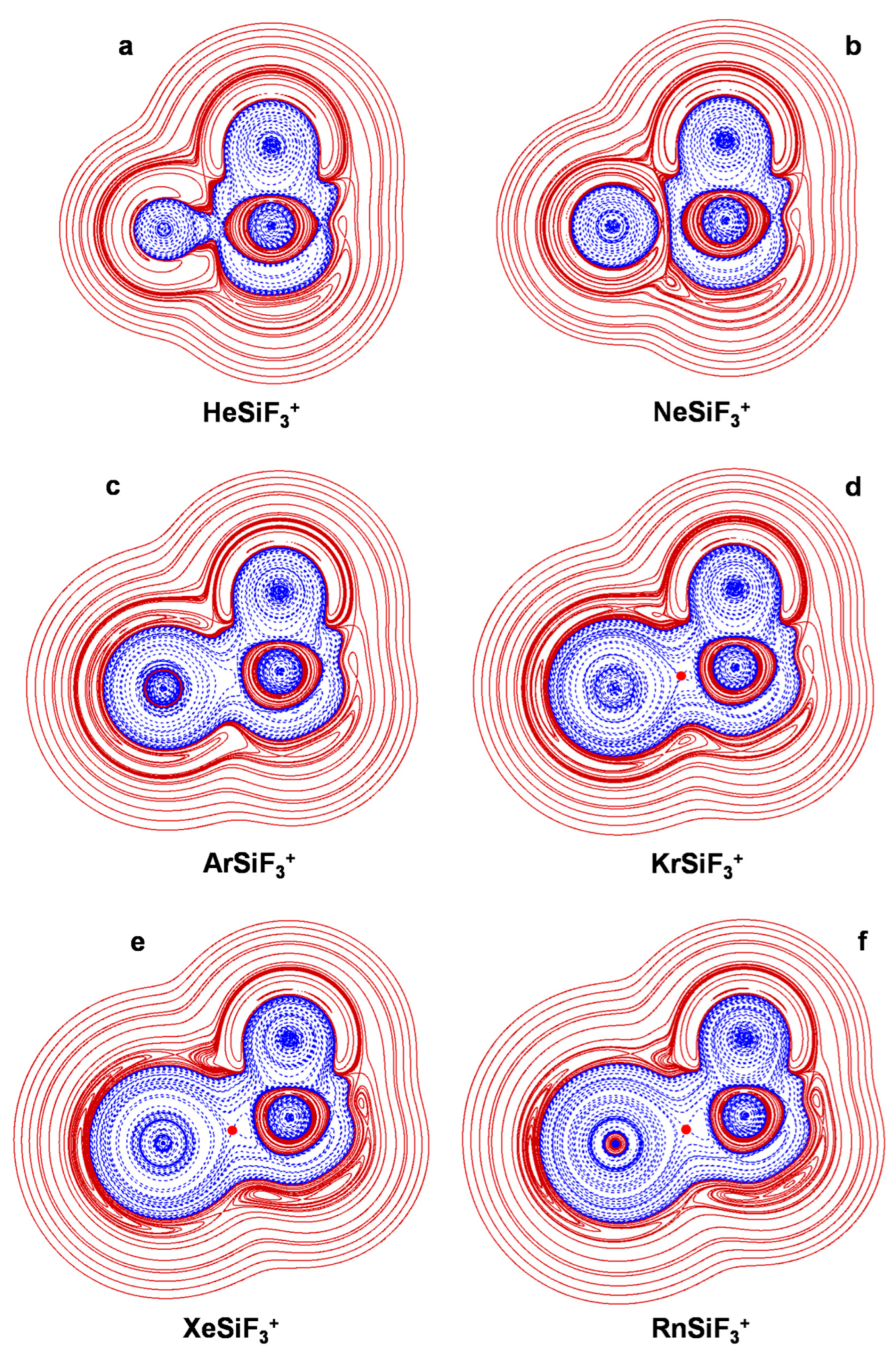
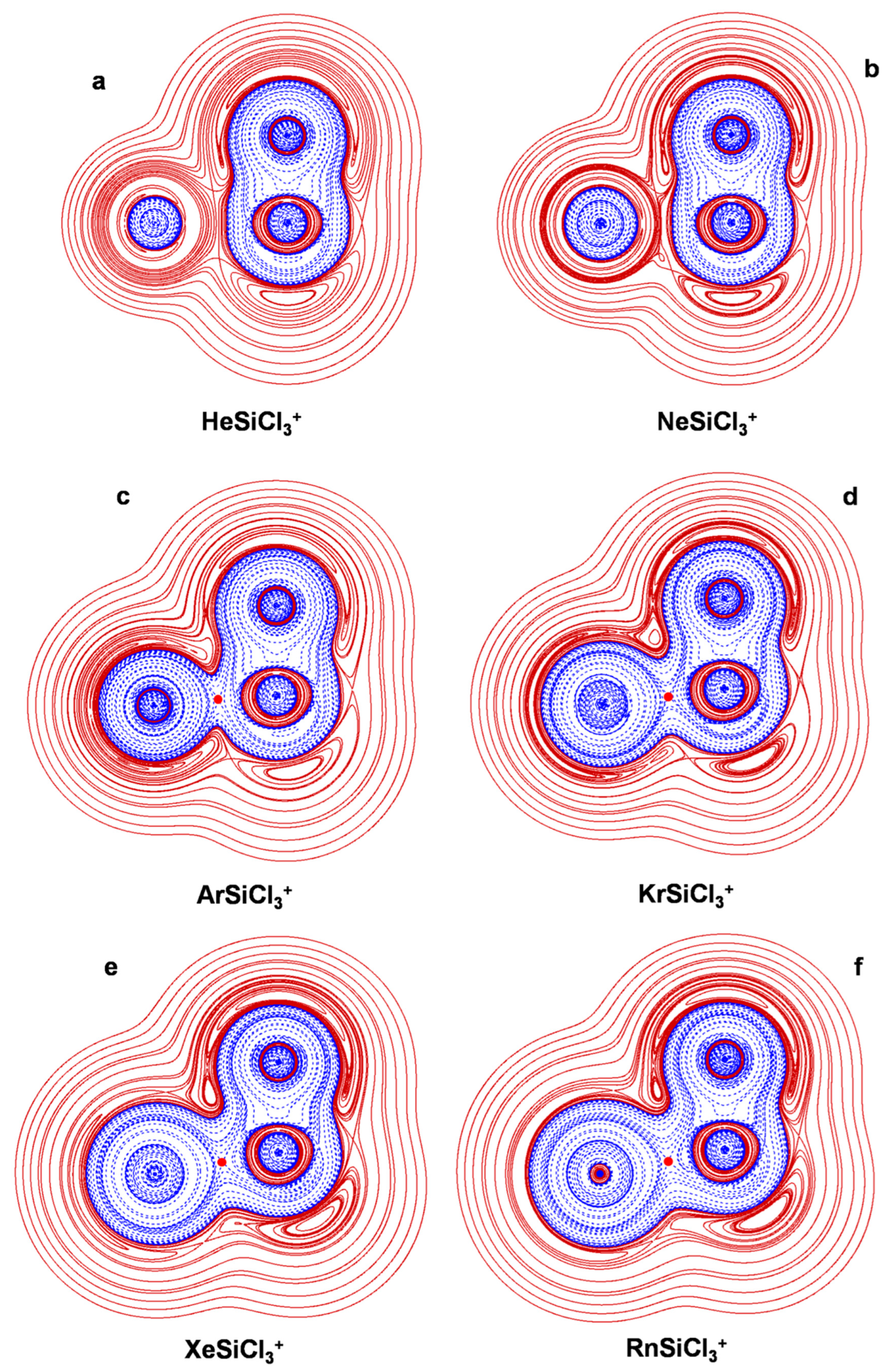
4.2. The NgSiX3+ (Ng = He-Rn; X = H, F, Cl): The Ng-Si Bond in Mono-Coordinated SinglyCharged Complexes
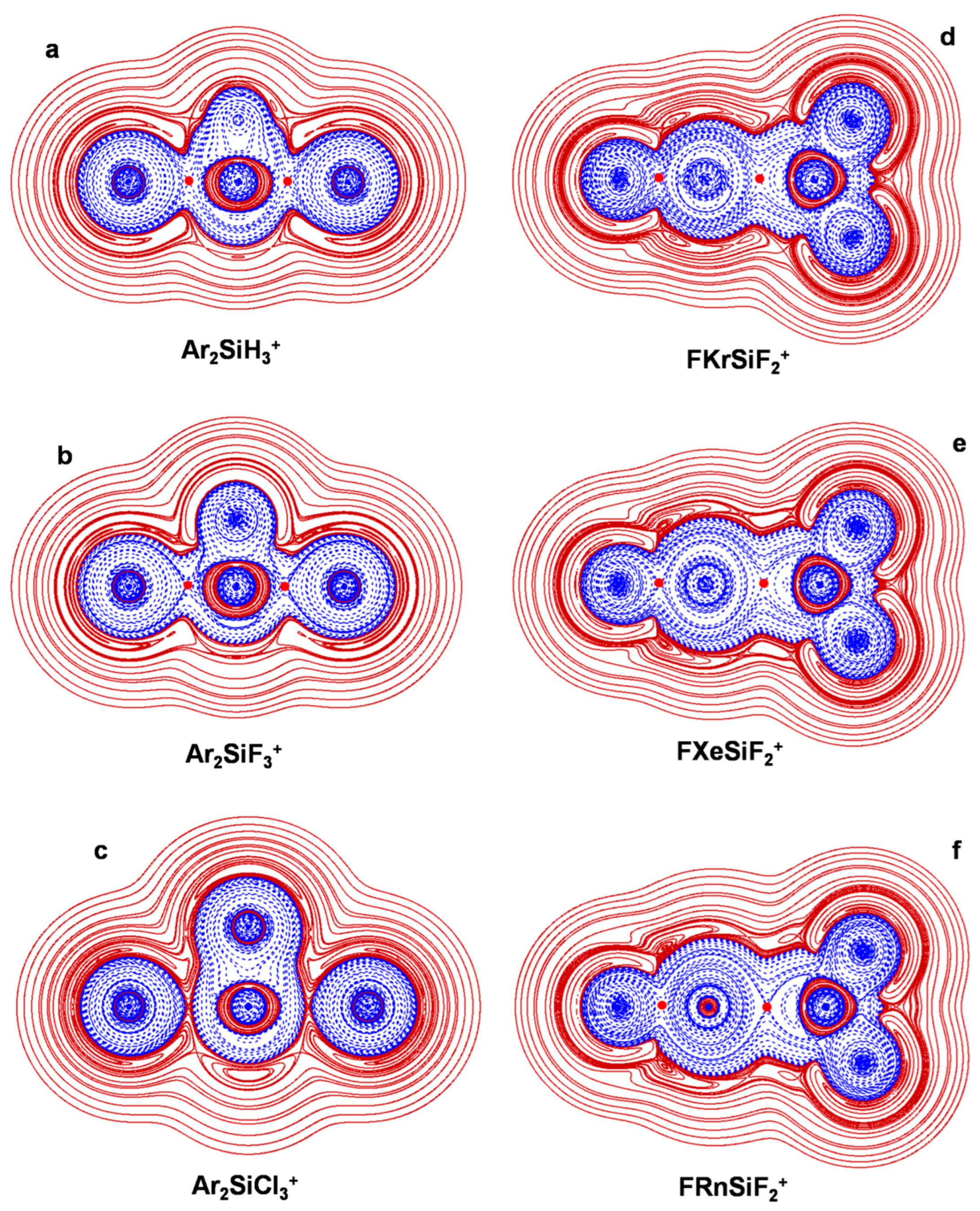
4.3. The Ar2(SiX3+) (X = H, F, Cl): The Ng-Si Bond in Di-Coordinated SinglyCharged Complexes
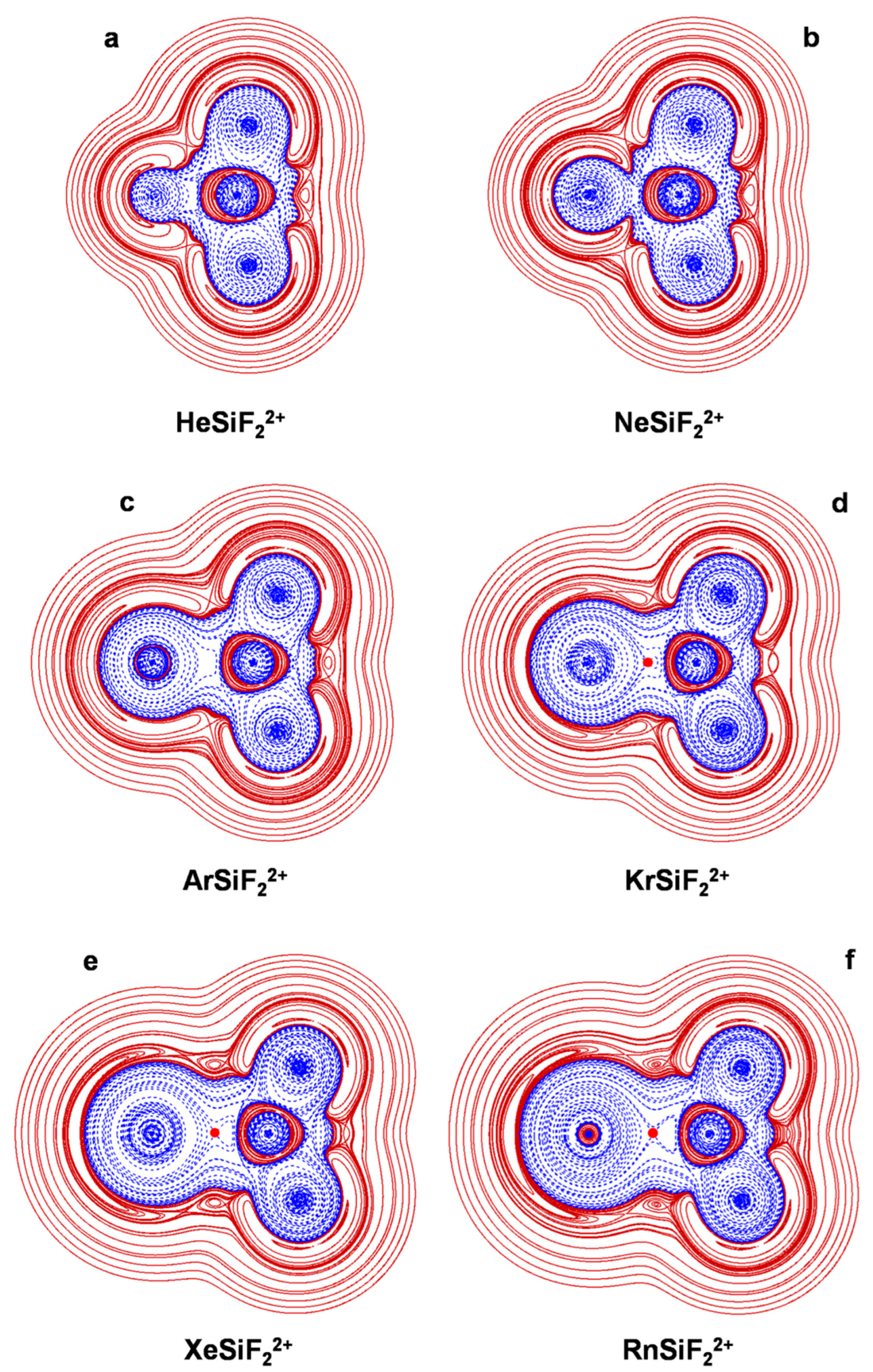
4.4. The NgSiF22+ (Ng = He-Rn): The Ng-Si Bond in Mono-Coordinated DoublyCharged Complexes
4.5. The FNgSiF2+ (Ng = Kr, Xe, Rn): The Ng-Si Bond in Inserted Cations
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rayleigh, L.; Ramsay, W. Argon, a new constituent of the atmosphere. Philos. Trans. R. Soc. Lond. A 1985, 186, 187–241. [Google Scholar] [CrossRef]
- Grandinetti, F. Noble Gas Chemistry: Structure, Bonding, and Gas-Phase Chemistry; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Brock, D.S.; Schrobilgen, G.J.; Žemva, B. Comprehensive Inorganic Chemistry II; Reedijk, J., Poepplemeier, K., Eds.; Elsevier: Oxford, UK, 2013; Volume. 1, pp. 755–822. [Google Scholar]
- Haner, J.; Schrobilgen, G.J. The chemistry of xenon (IV). Chem. Rev. 2015, 115, 1255–1295. [Google Scholar] [CrossRef] [PubMed]
- Grochala, W. Atypical compounds of gases, which have been called ‘noble’. Chem. Soc. Rev. 2007, 36, 1632–1655. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.F.; Mercier, H.P.A.; Schrobilgen, G.J. The chemistry of krypton. Coord. Chem. Rev. 2002, 233–234, 1–39. [Google Scholar] [CrossRef]
- Mazej, Z. Noble-gas chemistry more than half a century after the first report of the noble-gas compound. Molecules 2020, 25, 3014. [Google Scholar] [CrossRef]
- Grandinetti, F. Cationic noble-gas hydrides: From ion sources to outer space. Front. Chem. 2020, 8, 462. [Google Scholar] [CrossRef]
- Nunzi, F.; Pannacci, G.; Tarantelli, F.; Belpassi, L.; Cappelletti, D.; Falcinelli, S.; Pirani, F. Leading interaction components in the structure and reactivity of noble gases compounds. Molecules 2020, 25, 2367. [Google Scholar] [CrossRef]
- Wu, L.; Sun, X.Z.; Wu, X.; George, M.W. Formation of organometallic xenon complexes in conventional fluids: A time-resolved infrared (TRIR) study of the photochemistry of W(CO)5(4AcPyr) (4-AcPyr = 4-Acetylpyridine) in perfluoromethylcyclohexane (PFMCH). Vibr. Spectrosc. 2020, 108, 103053. [Google Scholar] [CrossRef]
- Grochala, W.; Khriachtchev, L.; Räsänen, M. Noble-Gas Chemistry, in Physics and Chemistry at Low Temperatures; Khriachtchev, L., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 419–446. [Google Scholar]
- Miao, M. Noble gases in solid compounds show a rich display of chemistry with enough pressure. Front. Chem. 2020, 8, 570492. [Google Scholar] [CrossRef]
- Frohn, H.J.; Bardin, V.V. Preparation and reactivity of compounds containing a carbon− xenon bond. Organometallics 2001, 20, 4750–4762. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Organic chemistry of noble gases. Russ. Chem. Bull. 1993, 42, 1763–1771. [Google Scholar] [CrossRef]
- Lundell, J.; Panek, J.; Latajka, Z. Quantum chemical calculations on FXeSiF. Chem. Phys. Lett. 2001, 348, 147–154. [Google Scholar] [CrossRef]
- Cohen, A.; Lundell, J.; Gerber, R.B. First compounds with argon-carbon and argon-silicon chemical bonds. J. Chem. Phys. 2003, 119, 6415–6417. [Google Scholar] [CrossRef] [Green Version]
- Yockel, S.; Garg, A.; Wilson, A.K. The existence of FKrCF3, FKrSiF3, and FKrGeF3: A theoretical study. Chem. Phys. Lett. 2005, 411, 91–97. [Google Scholar] [CrossRef]
- Pan, S.; Saha, R.; Chattaraj, P.K. Exploring the nature of silicon-noble gas bonds in H3SiNgNSi and HSiNgNSi compounds (Ng = Xe, Rn). Int. J. Mol. Sci. 2015, 16, 6402–6418. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Maitra, A.; Kuntar, S.P.; Ghanty, T.K. Stability-order reversal in FSiY and FYSi (Y = N and P) molecules after the Insertion of a Noble Gas Atom. J. Phys. Chem. A 2022, 126, 1132–1143. [Google Scholar] [CrossRef]
- Walker, J.C.; Klare, H.F.; Oestreich, M. Cationic silicon Lewis acids in catalysis. Nat. Rev. Chem. 2020, 4, 54–62. [Google Scholar] [CrossRef]
- Lyon, J.T.; Gruene, P.; Fielicke, A.; Meijer, G.; Janssens, E.; Claes, P.; Lievens, P. Structures of silicon cluster cations in the gas phase. J. Am. Chem. Soc. 2009, 131, 1115–1121. [Google Scholar] [CrossRef]
- Savoca, M.; Langer, J.; Harding, D.J.; Dopfer, O.; Fielicke, A. Incipient chemical bond formation of Xe to a cationic silicon cluster: Vibrational spectroscopy and structure of the Si4Xe+ complex. Chem. Phys. Lett. 2013, 557, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Jansen, H.; Gardeniers, H.; de Boer, M.; Elwenspoek, M.; Fluitman, J. A survey on the reactive ion etching of silicon in microtechnology. J. Micromech. Microeng. 1996, 6, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Cipollini, R.; Grandinetti, F. The gaseous trifluorosilylxenon cation, F3SiXe+: A stable species with a silicon-xenon bond. J. Chem. Soc. Chem. Commun. 1995, 7, 773–774. [Google Scholar] [CrossRef]
- Cunje, A.; Baranov, V.I.; Ling, Y.; Hopkinson, A.C.; Bohme, D.K. Bonding of rare-gas atoms to Si in reactions of rare gases with SiF3+. J. Phys. Chem. A 2001, 105, 11073–11079. [Google Scholar] [CrossRef]
- Roithová, J.; Schröder, D. Silicon compounds of neon and argon. Angew. Chem. Int. Ed. 2009, 48, 8788–8790. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Moreno, D.; Merino, G.; Chattaraj, P.K. Stability of noble-gas-bound SiH3+ clusters. ChemPhysChem 2014, 15, 3554–3564. [Google Scholar] [CrossRef] [PubMed]
- Borocci, S.; Giordani, M.; Grandinetti, F. Bonding motifs of noble-gas compounds as described by the local electron energy density. J. Phys. Chem. A 2015, 119, 6528–6541. [Google Scholar] [CrossRef] [PubMed]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Antoniotti, P.; Nunzi, F. Non-covalent complexes of the noble-gas atoms: Analyzing the transition from physical to chemical interactions. J. Comput. Chem. 2019, 40, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Nunzi, F. Classifying the chemical bonds involving the noble-gas atoms. N. J. Chem. 2020, 44, 14536–14550. [Google Scholar] [CrossRef]
- Borocci, S.; Grandinetti, F.; Sanna, N. Noble gas compounds: A general procedure of bonding analysis. J. Chem. Phys. 2022, 156, 014104. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density-does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narth, C.; Maroun, Z.; Boto, R.A.; Chaudret, R.; Bonnet, M.L.; Piquemal, J.-P.; Contreras-García, J. A complete NCI perspective: From new bonds to reactivity. In Applications of Topological Methods in Molecular Chemistry; Springer: Berlin/Heidelberg, Germany, 2016; pp. 491–527. [Google Scholar]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Antoniotti, P.; Nunzi, F. Complexes of helium with neutral molecules: Progress toward a quantitative scale of bonding character. J. Comput. Chem. 2020, 41, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian, version09, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Lo Presti, L. Energetics of non-covalent interactions from electron and energy density distributions. Comput. Theor. Chem. 2015, 1053, 53–59. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Nori-Shargh, D.; Boggs, J. On the covalent character of rare gas bonding interactions: A new kind of weak interaction. J. Phys. Chem. A 2013, 117, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H. Prediction of a metastable helium compound: HHeF. J. Am. Chem. Soc. 2000, 122, 6289–6290. [Google Scholar] [CrossRef]
- Li, T.-H.; Mou, C.-H.; Chen, H.-R.; Hu, W.-P. Theoretical prediction of noble gas containing anions FNgO− (Ng = He, Ar, and Kr). J. Am. Chem. Soc. 2005, 127, 9241–9245. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H(r) at Around the BCP | ||
|---|---|---|
| Bond type | Ng side | X side |
| A | negative | negative |
| Bl or Cl | positive | positive |
| Bt or Ct | positive | negative |
| negative | positive | |
| negative | negative | |
| ρs(ave) a | H(Ωs) | Notation | |
|---|---|---|---|
| Cov | ≥0.08 | invariably negative | H− |
| pCov | <0.08 | invariably negative | H− |
| any value | from negative to positive | H+/− (positive on the average) | |
| H−/+ (negative on the average) | |||
| nCov | any value | invariably positive | H+ |
| Bond | R | De | Type | Ωs | N(Ωs) | ρs(ave) | Hs(ave/max/min) | H(Ωs) a | BDs(ave) | Assignment | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HeSiH3+ | He-Si | 2.122 | 2.0 | Ct | 0.3120 | 4.86 | 0.0156 | −0.000075/0.00067/−0.00097 | H−/+ | 0.0042 | pCov(Ct/H−/+) |
| NeSiH3+ | Ne-Si | 2.330 | 3.1 | Ct | 0.4510 | 7.57 | 0.0168 | −0.0013/−0.00008/−0.0025 | H− | 0.0747 | pCov(Ct/H−) |
| ArSiH3+ | Ar-Si | 2.406 | 14.1 | A | 0.9603 | 31.2 | 0.0325 | −0.0092/−0.0043/−0.0123 | H− | 0.283 | pCov(A/H−) |
| KrSiH3+ | Kr-Si | 2.514 | 19.3 | A | 1.2968 | 47.0 | 0.0363 | −0.0124/−0.0063/−0.0167 | H− | 0.341 | pCov(A/H−) |
| XeSiH3+ | Xe-Si | 2.664 | 26.5 | A | 1.9199 | 78.1 | 0.0407 | −0.0156/−0.0082/−0.0215 | H− | 0.382 | pCov(A/H−) |
| RnSiH3+ | Rn-Si | 2.736 | 30.1 | A | 2.2688 | 95.6 | 0.0421 | −0.0164/−0.0089/−0.0233 | H− | 0.389 | pCov(A/H−) |
| HeSiF3+ | He-Si | 2.060 | 2.5 | A | 0.3645 | 6.35 | 0.0174 | −0.00075/0.00028/−0.0019 | H−/+ | 0.043 | pCov(A/H−/+) |
| NeSiF3+ | Ne-Si | 2.188 | 4.7 | Ct | 0.5476 | 11.7 | 0.0215 | −0.0029/−0.00082/−0.0049 | H− | 0.136 | pCov(Ct/H−) |
| ArSiF3+ | Ar-Si | 2.297 | 21.7 | Bt | 0.7289 | 29.0 | 0.0398 | −0.0142/−0.0064/−0.0220 | H− | 0.354 | pCov(Bt/H−) |
| KrSiF3+ | Kr-Si | 2.415 | 29.3 | A | 0.9947 | 45.0 | 0.0452 | −0.0196/−0.0101/−0.0275 | H− | 0.431 | pCov(A/H−) |
| XeSiF3+ | Xe-Si | 2.576 | 39.6 | A | 1.6579 | 88.6 | 0.0534 | −0.0249/−0.0145/−0.0393 | H− | 0.470 | pCov(A/H−) |
| RnSiF3+ | Rn-Si | 2.652 | 44.9 | A | 2.0403 | 113.0 | 0.0556 | −0.0259/−0.0170/−0.0392 | H− | 0.471 | pCov(A/H−) |
| HeSiCl3+ | He-Si | 2.986 | 0.4 | Cl | 0.0880 | 0.30 | 0.0035 | 0.00095/0.0011/0.00087 | H− | −0.272 | nCov(Cl) |
| NeSiCl3+ | Ne-Si | 2.916 | 1.0 | Cl | 0.2278 | 1.53 | 0.0067 | 0.00057/0.00075/0.00042 | H− | −0.086 | nCov(Cl) |
| ArSiCl3+ | Ar-Si | 2.690 | 6.1 | A | 0.9792 | 21.6 | 0.0220 | −0.0035/−0.00030/−0.0074 | H− | 0.157 | pCov(A/H−) |
| KrSiCl3+ | Kr-Si | 2.670 | 11.2 | A | 1.4951 | 46.0 | 0.0308 | −0.0092/−0.0033/−0.0133 | H− | 0.295 | pCov(A/H−) |
| XeSiCl3+ | Xe-Si | 2.746 | 19.3 | A | 2.1271 | 83.0 | 0.0390 | −0.0146/−0.0072/−0.0215 | H− | 0.373 | pCov(A/H−) |
| RnSiCl3+ | Rn-Si | 2.793 | 23.9 | A | 2.4402 | 102.9 | 0.0422 | −0.0165/−0.0090/−0.0252 | H− | 0.391 | pCov(A/H−) |
| Ar2SiH3+ | Ar-Si | 2.570 | 8.1 | A | 0.9572 | 23.8 | 0.0248 | −0.0048/−0.0014/−0.0075 | H− | 0.190 | pCov(A/H−) |
| Ar2SiF3+ | Ar-Si | 2.491 | 10.6 | A | 1.0771 | 30.9 | 0.0287 | −0.0077/−0.0013/−0.0108 | H− | 0.265 | pCov(A/H−) |
| Ar2SiCl3+ | Ar-Si | 2.961 | 3.9 | Ct | 0.7456 | 9.96 | 0.0134 | −0.000078/0.0012/−0.0014 | H−/+ | 0.0036 | pCov(Ct/H−/+) |
| HeSiF22+ | He-Si | 1.721 | 14.0 | Bt | 0.1253 | 4.8 | 0.0383 | −0.0057/0.0037/−0.0094 | H−/+ | 0.155 | pCov(Bt/H−/+) |
| NeSiF22+ | Ne-Si | 1.888 | 23.2 | Bt | 0.1466 | 6.36 | 0.0434 | −0.0065/0.0034/−0.0116 | H−/+ | 0.159 | pCov(Bt/H−/+) |
| ArSiF22+ | Ar-Si | 2.112 | 73.0 | Bt | 0.3182 | 23.6 | 0.0743 | −0.0374/−0.0165/−0.0541 | H− | 0.500 | pCov(Bt/H−) |
| KrSiF22+ | Kr-Si | 2.247 | 94.3 | A | 0.9095 | 75.5 | 0.0830 | −0.0455/−0.0268/−0.0791 | H− | 0.552 | Cov(A) |
| XeSiF22+ | Xe-Si | 2.428 | 123.2 | A | 2.4498 | 213.3 | 0.0871 | −0.0477/−0.0287/−0.1095 | H− | 0.545 | Cov(A) |
| RnSiF22+ | Rn-Si | 2.518 | 137.4 | A | 2.7699 | 217.7 | 0.0786 | −0.0403/−0.0239/−0.0660 | H− | 0.515 | pCov(A/H−) |
| FKrSiF2+ | Kr-F | 1.927 | A | 0.3675 | 44.0 | 0.1197 | −0.0476/−0.0264/−0.0973 | H− | 0.392 | Cov(A) | |
| Kr-Si | 2.477 | A | 2.1834 | 156.5 | 0.0717 | −0.0356/−0.0158/−0.0656 | H− | 0.487 | pCov(A/H−) | ||
| FXeSiF2+ | Xe-F | 1.983 | A | 0.5258 | 61.2 | 0.1164 | −0.0553/−0.0337/−0.0825 | H− | 0.471 | Cov(A) | |
| Xe-Si | 2.631 | A | 3.1721 | 223.3 | 0.0704 | −0.0339/−0.0132/−0.0707 | H− | 0.464 | pCov(A/H−) | ||
| FRnSiF2+ | Rn-F | 2.054 | A | 0.4141 | 44.3 | 0.1071 | −0.0403/−0.0254/−0.0591 | H− | 0.373 | Cov(A) | |
| Rn-Si | 2.735 | A | 3.2828 | 215.2 | 0.0656 | −0.0307/−0.0104/−0.0701 | H− | 0.441 | pCov(A/H−) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borocci, S.; Grandinetti, F.; Sanna, N. Noble Gas—Silicon Cations: Theoretical Insights into the Nature of the Bond. Molecules 2022, 27, 4592. https://doi.org/10.3390/molecules27144592
Borocci S, Grandinetti F, Sanna N. Noble Gas—Silicon Cations: Theoretical Insights into the Nature of the Bond. Molecules. 2022; 27(14):4592. https://doi.org/10.3390/molecules27144592
Chicago/Turabian StyleBorocci, Stefano, Felice Grandinetti, and Nico Sanna. 2022. "Noble Gas—Silicon Cations: Theoretical Insights into the Nature of the Bond" Molecules 27, no. 14: 4592. https://doi.org/10.3390/molecules27144592
APA StyleBorocci, S., Grandinetti, F., & Sanna, N. (2022). Noble Gas—Silicon Cations: Theoretical Insights into the Nature of the Bond. Molecules, 27(14), 4592. https://doi.org/10.3390/molecules27144592