
The Anti-Apoptotic Effects of Caspase Inhibitors in Propyl Gallate-Treated Lung Cancer Cells Are Related to Changes in Reactive Oxygen Species and Glutathione Levels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Cell Proliferation Assay
2.4. Cell Growth Inhibition Assay
2.5. Cell Cycle and Sub-G1 Cell Analysis
2.6. Detection of Apoptosis
2.7. Measurement of Mitochondrial Membrane Potential (MMP; ΔΨm)
2.8. Determination of Intracellular ROS and O2∙− Levels
2.9. Detection of Intracellular GSH Levels
2.10. Statistical Analysis
3. Results
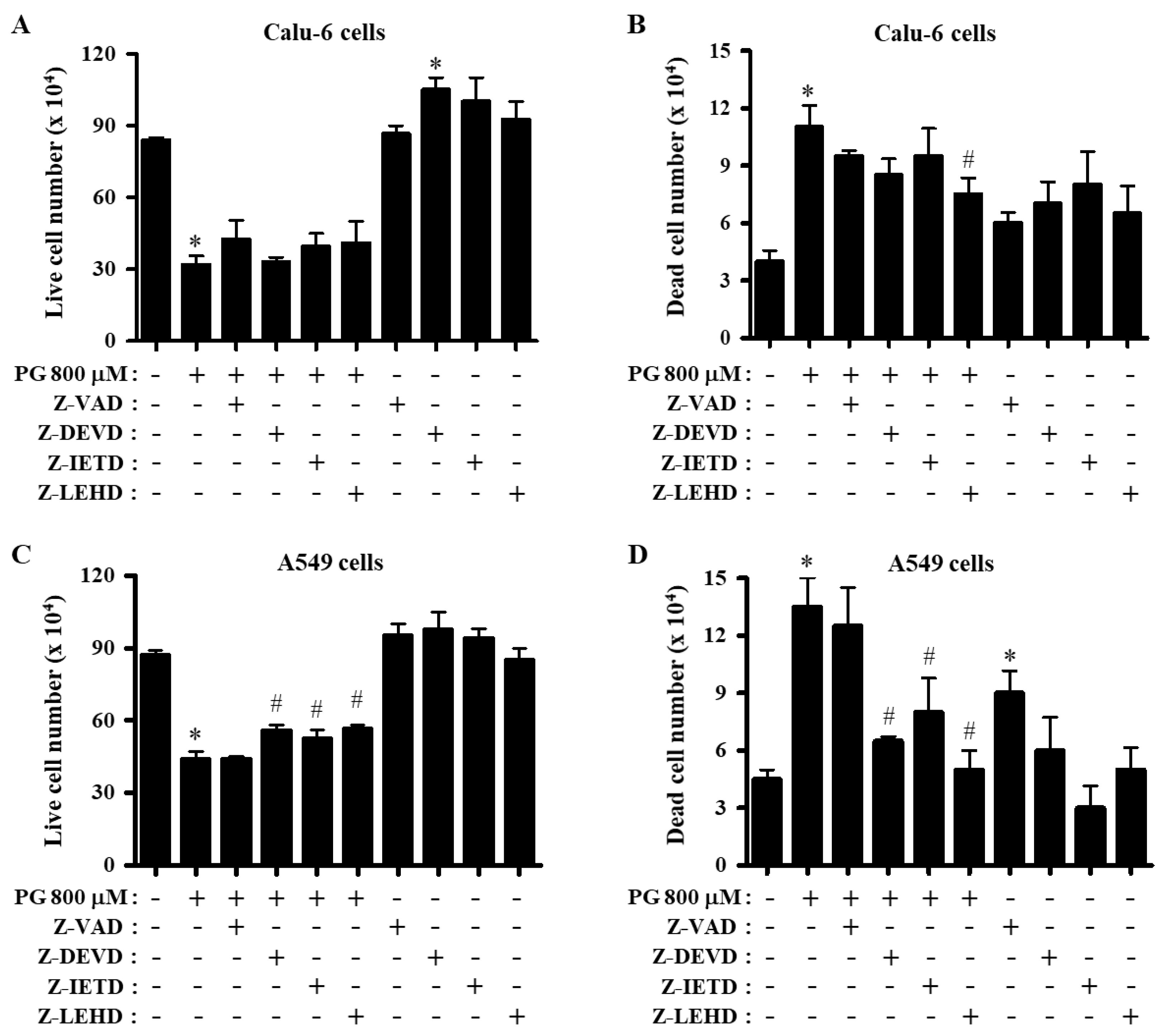
3.1. Effect of Caspase Inhibitors on Cell Proliferation in PG-Treated Lung Cancer Cells
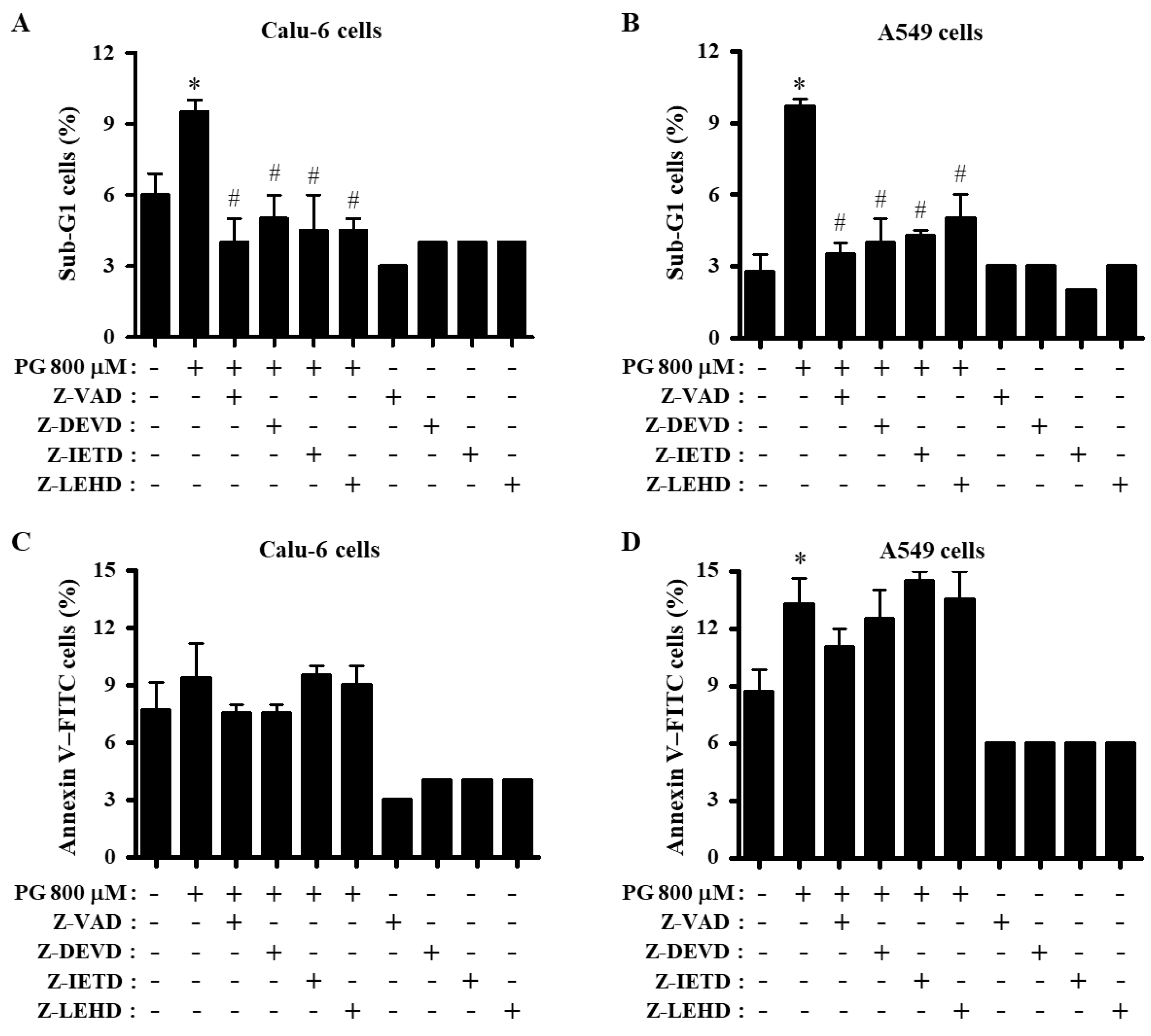
3.2. Effects of Caspase Inhibitors on Growth and Cell Cycle Distributions in PG-Treated Lung Cancer Cells
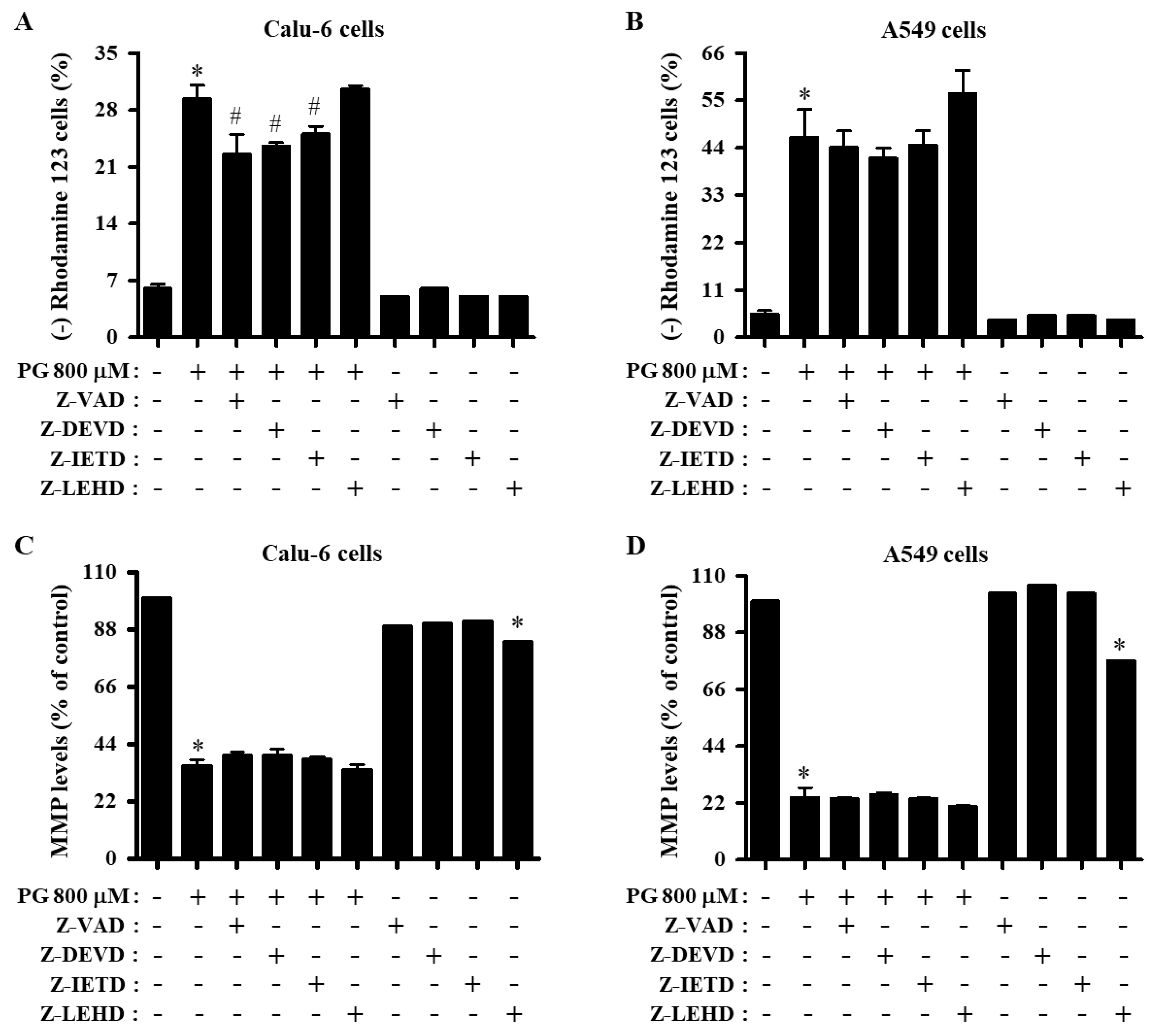
3.3. Effects of Caspase Inhibitors on Cell Death and MMP (∆Ψm) in PG-Treated Lung Cancer Cells
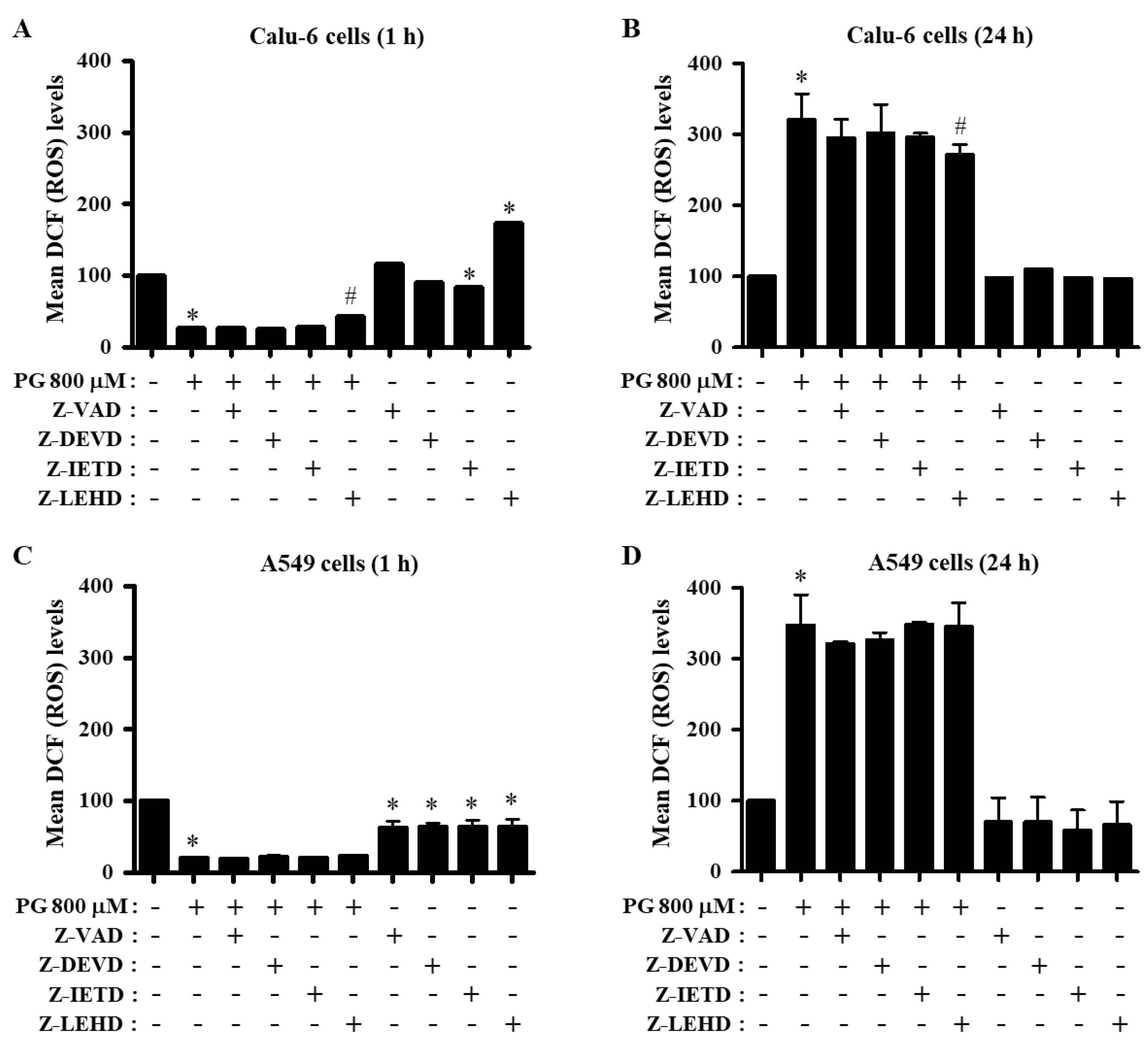
3.4. Effects of Caspase Inhibitors on ROS and O2∙− Levels in PG-Treated Lung Cancer Cells
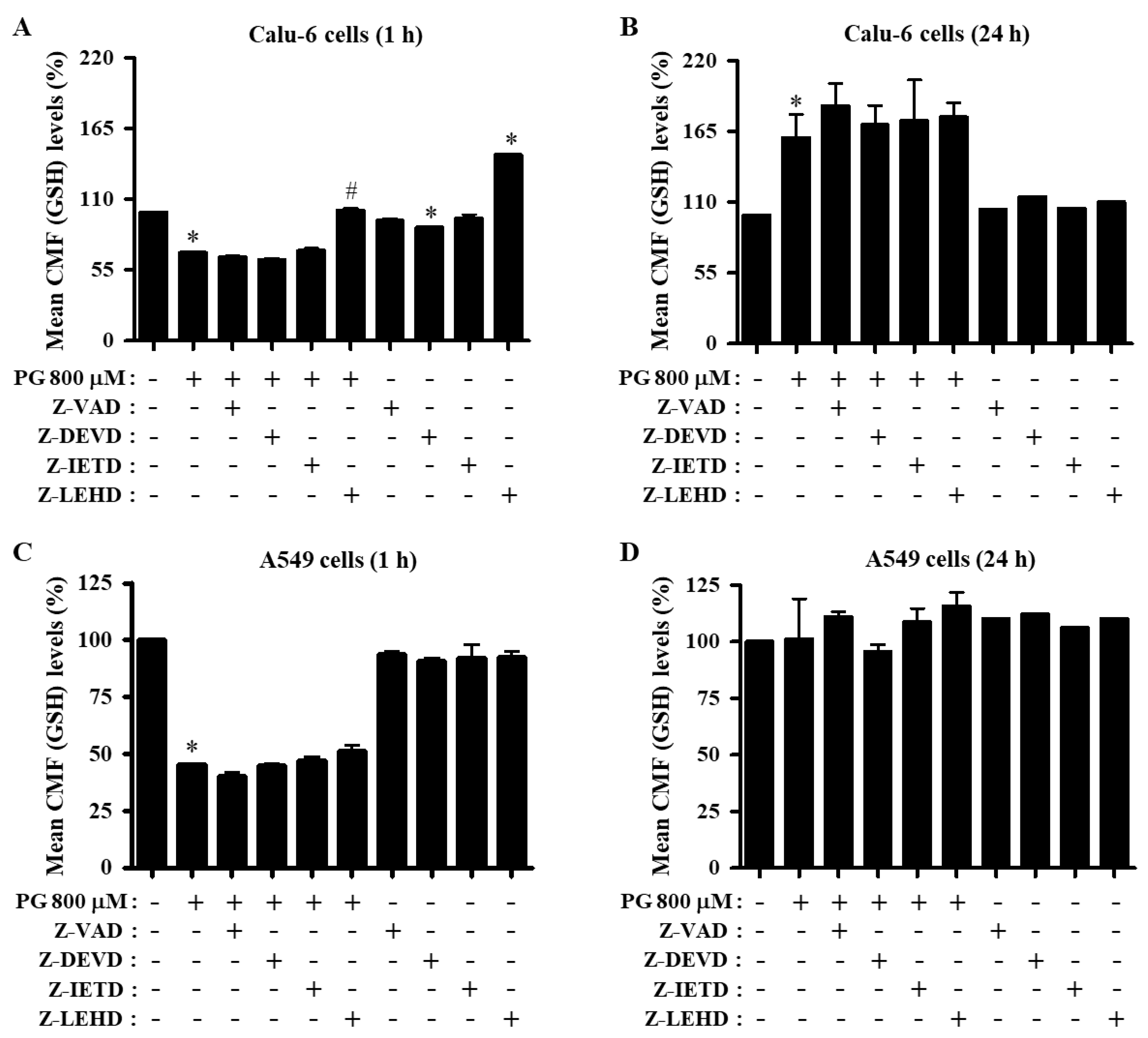
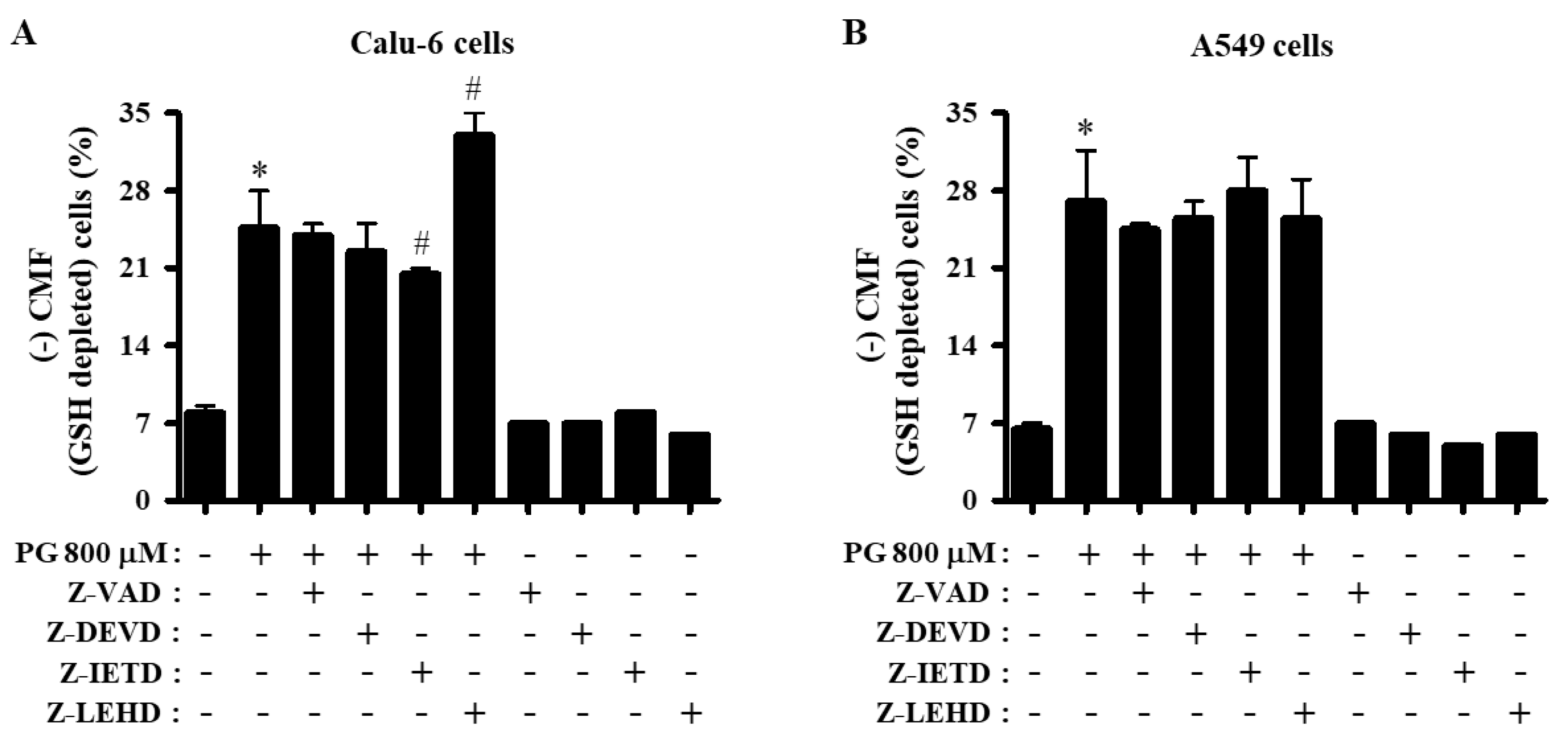
3.5. Effects of Caspase Inhibitors on GSH Levels in PG-Treated Lung Cancer Cells
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- Daniel, J.W. Metabolic aspects of antioxidants and preservatives. Xenobiotica Fate Foreign Compd. Biol. Syst. 1986, 16, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Becker, L. Final report on the amended safety assessment of Propyl Gallate. Int. J. Toxicol. 2007, 26, 89–118. Available online: https://pubmed.ncbi.nlm.nih.gov/18080874/ (accessed on 1 June 2022). [CrossRef] [PubMed]
- Wu, T.W.; Fung, K.P.; Zeng, L.H.; Wu, J.; Nakamura, H. Propyl gallate as a hepatoprotector in vitro and in vivo. Biochem. Pharmacol. 1994, 48, 419–422. [Google Scholar] [CrossRef]
- Reddan, J.R.; Giblin, F.J.; Sevilla, M.; Padgaonkar, V.; Dziedzic, D.C.; Leverenz, V.R.; Misra, I.C.; Chang, J.S.; Pena, J.T. Propyl gallate is a superoxide dismutase mimic and protects cultured lens epithelial cells from H2O2 insult. Exp. Eye Res. 2003, 76, 49–59. [Google Scholar] [CrossRef]
- Chen, C.H.; Liu, T.Z.; Chen, C.H.; Wong, C.H.; Chen, C.H.; Lu, F.J.; Chen, S.C. The efficacy of protective effects of tannic acid, gallic acid, ellagic acid, and propyl gallate against hydrogen peroxide-induced oxidative stress and DNA damages in IMR-90 cells. Mol. Nutr. Food Res. 2007, 51, 962–968. [Google Scholar] [CrossRef]
- Hirose, M.; Yada, H.; Hakoi, K.; Takahashi, S.; Ito, N. Modification of carcinogenesis by alpha-tocopherol, t-butylhydroquinone, propyl gallate and butylated hydroxytoluene in a rat multi-organ carcinogenesis model. Carcinogenesis 1993, 14, 2359–2364. [Google Scholar] [CrossRef]
- Kobayashi, H.; Oikawa, S.; Hirakawa, K.; Kawanishi, S. Metal-mediated oxidative damage to cellular and isolated DNA by gallic acid, a metabolite of antioxidant propyl gallate. Mutat. Res. 2004, 558, 111–120. [Google Scholar] [CrossRef]
- Kawanishi, S.; Oikawa, S.; Murata, M. Evaluation for safety of antioxidant chemopreventive agents. Antioxid. Redox Signal. 2005, 7, 1728–1739. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tayama, S. Cytotoxicity of propyl gallate and related compounds in rat hepatocytes. Arch. Toxicol. 1995, 69, 204–208. [Google Scholar] [CrossRef]
- Ham, J.; Lim, W.; Park, S.; Bae, H.; You, S.; Song, G. Synthetic phenolic antioxidant propyl gallate induces male infertility through disruption of calcium homeostasis and mitochondrial function. Environ. Pollut. 2019, 248, 845–856. [Google Scholar] [CrossRef]
- Han, Y.H.; Moon, H.J.; You, B.R.; Park, W.H. Propyl gallate inhibits the growth of calf pulmonary arterial endothelial cells via glutathione depletion. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2010, 24, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, H.; Eicke, B.; Witte, I. DNA strand break induction and enhanced cytotoxicity of propyl gallate in the presence of copper(II). Free Radic. Biol. Med. 1998, 24, 972–978. [Google Scholar] [CrossRef]
- Bettger, W.J.; Ham, R.G. Effects of non-steroidal anti-inflammatory agents and antioxidants on the clonal growth of human diploid fibroblasts. Prog. Lipid. Res. 1981, 20, 265–268. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, W.C.; Kuo, C.N.; Lu, F.J. Role of redox signaling regulation in propyl gallate-induced apoptosis of human leukemia cells. Food Chem. Toxicol. 2011, 49, 494–501. [Google Scholar] [CrossRef]
- Wei, P.L.; Huang, C.Y.; Chang, Y.J. Propyl gallate inhibits hepatocellular carcinoma cell growth through the induction of ROS and the activation of autophagy. PLoS ONE 2019, 14, e0210513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.H.; Park, W.H. Propyl gallate inhibits the growth of HeLa cells via regulating intracellular GSH level. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2009, 47, 2531–2538. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Moon, H.J.; You, B.R.; Park, W.H. The anti-apoptotic effects of caspase inhibitors on propyl gallate-treated HeLa cells in relation to reactive oxygen species and glutathione levels. Arch. Toxicol. 2009, 83, 825–833. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsuneoka, M. Gallic Acid Derivatives Propyl Gallate and Epigallocatechin Gallate Reduce rRNA Transcription via Induction of KDM2A Activation. Biomolecules 2021, 12, 30. [Google Scholar] [CrossRef]
- Han, Y.H.; Moon, H.J.; You, B.R.; Yang, Y.M.; Kim, S.Z.; Kim, S.H.; Park, W.H. Propyl gallate inhibits the growth of endothelial cells, especially calf pulmonary arterial endothelial cells via caspase-independent apoptosis. Int. J. Mol. Med. 2010, 25, 937–944. [Google Scholar]
- Chung, C. Restoring the switch for cancer cell death: Targeting the apoptosis signaling pathway. Am. J. Health Syst. Pharm. 2018, 75, 945–952. [Google Scholar] [CrossRef]
- Lopez, K.E.; Bouchier-Hayes, L. Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response. Cells 2022, 11, 1877. [Google Scholar] [CrossRef] [PubMed]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]
- Huska, J.D.; Lamb, H.M.; Hardwick, J.M. Overview of BCL-2 Family Proteins and Therapeutic Potentials. Methods Mol. Biol. 2019, 1877, 1–21. [Google Scholar] [PubMed]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 2012, 318, 1213–1220. [Google Scholar] [CrossRef]
- Liu, X.; Yue, P.; Zhou, Z.; Khuri, F.R.; Sun, S.Y. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J. Natl. Cancer Inst. 2004, 96, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Wilcox, C.S. Reactive oxygen species: Roles in blood pressure and kidney function. Curr. Hypertens. Rep. 2002, 4, 160–166. [Google Scholar] [CrossRef]
- Hu, Z.; Li, M.; Chen, Z.; Zhan, C.; Lin, Z.; Wang, Q. Advances in clinical trials of targeted therapy and immunotherapy of lung cancer in 2018. Transl. Lung Cancer Res. 2019, 8, 1091–1106. [Google Scholar] [CrossRef]
- Park, W.H. Propyl gallate reduces the growth of lung cancer cells through caspasedependent apoptosis and G1 phase arrest of the cell cycle. Oncol. Rep. 2020, 44, 2783–2791. [Google Scholar] [CrossRef]
- Park, W.H. Propyl gallate decreases the proliferation of Calu-6 and A549 lung cancer cells via affecting reactive oxygen species and glutathione levels. J. Appl. Toxicol. 2022, 42, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H. Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1phase arrest. Oncol. Rep. 2018, 40, 1787–1794. [Google Scholar] [PubMed]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Lett. 2008, 270, 40–55. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. The enhancement of propyl gallate-induced HeLa cell death by MAPK inhibitors is accompanied by increasing ROS levels. Mol. Biol. Rep. 2011, 38, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H.; Seol, J.G.; Kim, E.S.; Hyun, J.M.; Jung, C.W.; Lee, C.C.; Kim, B.K.; Lee, Y.Y. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21, and apoptosis. Cancer Res. 2000, 60, 3065–3071. [Google Scholar]
- Park, W.H. Upregulation of thioredoxin and its reductase attenuates arsenic trioxideinduced growth suppression in human pulmonary artery smooth muscle cells by reducing oxidative stress. Oncol. Rep. 2020, 43, 358–367. [Google Scholar]
- Han, Y.H.; Kim, S.H.; Kim, S.Z.; Park, W.H. Caspase inhibitor decreases apoptosis in pyrogallol-treated lung cancer Calu-6 cells via the prevention of GSH depletion. Int. J. Oncol. 2008, 33, 1099–1105. [Google Scholar]
- Han, Y.H.; Park, W.H. Proteasome inhibitor MG132 reduces growth of As4.1 juxtaglomerular cells via caspase-independent apoptosis. Arch. Toxicol. 2010, 84, 689–698. [Google Scholar] [CrossRef]
- Martinez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef]
- Dalton, S. Linking the Cell Cycle to Cell Fate Decisions. Trends Cell Biol. 2015, 25, 592–600. [Google Scholar] [CrossRef] [Green Version]
- Poot, M.; Teubert, H.; Rabinovitch, P.S.; Kavanagh, T.J. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J. Cell. Physiol. 1995, 163, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Induction of apoptosis in arsenic trioxide-treated lung cancer A549 cells by buthionine sulfoximine. Mol. Cells 2008, 26, 158–164. [Google Scholar] [PubMed]
- Park, W.H.; Han, B.R.; Park, H.K.; Kim, S.Z. Arsenic trioxide induces growth inhibition and death in human pulmonary artery smooth muscle cells accompanied by mitochondrial O2∙− increase and GSH depletion. Environ. Toxicol. 2018, 33, 833–840. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol. Rep. 2016, 35, 546–551. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, W.h. The Anti-Apoptotic Effects of Caspase Inhibitors in Propyl Gallate-Treated Lung Cancer Cells Are Related to Changes in Reactive Oxygen Species and Glutathione Levels. Molecules 2022, 27, 4587. https://doi.org/10.3390/molecules27144587
Park Wh. The Anti-Apoptotic Effects of Caspase Inhibitors in Propyl Gallate-Treated Lung Cancer Cells Are Related to Changes in Reactive Oxygen Species and Glutathione Levels. Molecules. 2022; 27(14):4587. https://doi.org/10.3390/molecules27144587
Chicago/Turabian StylePark, Woo hyun. 2022. "The Anti-Apoptotic Effects of Caspase Inhibitors in Propyl Gallate-Treated Lung Cancer Cells Are Related to Changes in Reactive Oxygen Species and Glutathione Levels" Molecules 27, no. 14: 4587. https://doi.org/10.3390/molecules27144587
APA StylePark, W. h. (2022). The Anti-Apoptotic Effects of Caspase Inhibitors in Propyl Gallate-Treated Lung Cancer Cells Are Related to Changes in Reactive Oxygen Species and Glutathione Levels. Molecules, 27(14), 4587. https://doi.org/10.3390/molecules27144587