
Conformational Properties and Putative Bioactive Targets for Novel Thiosemicarbazone Derivatives


 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results
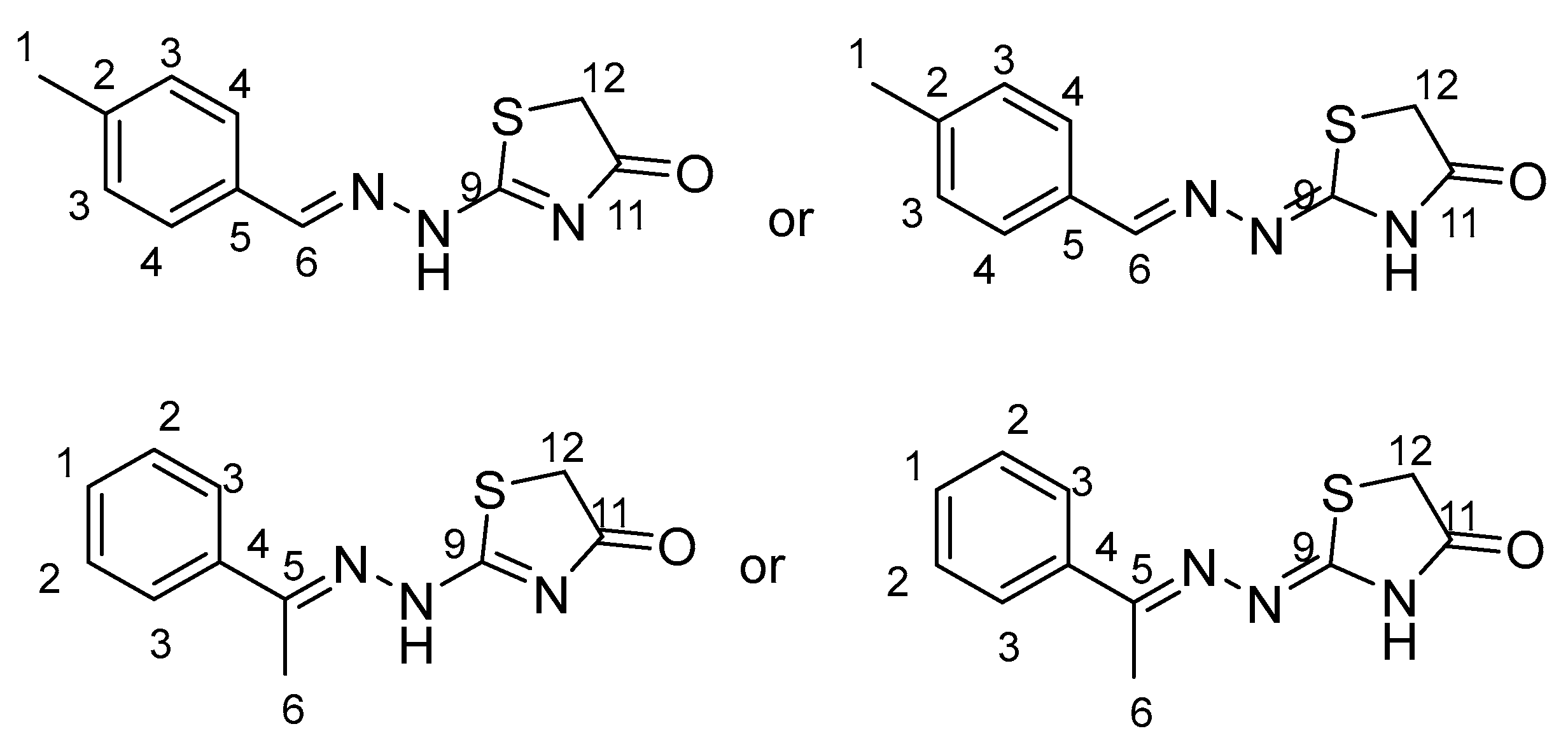
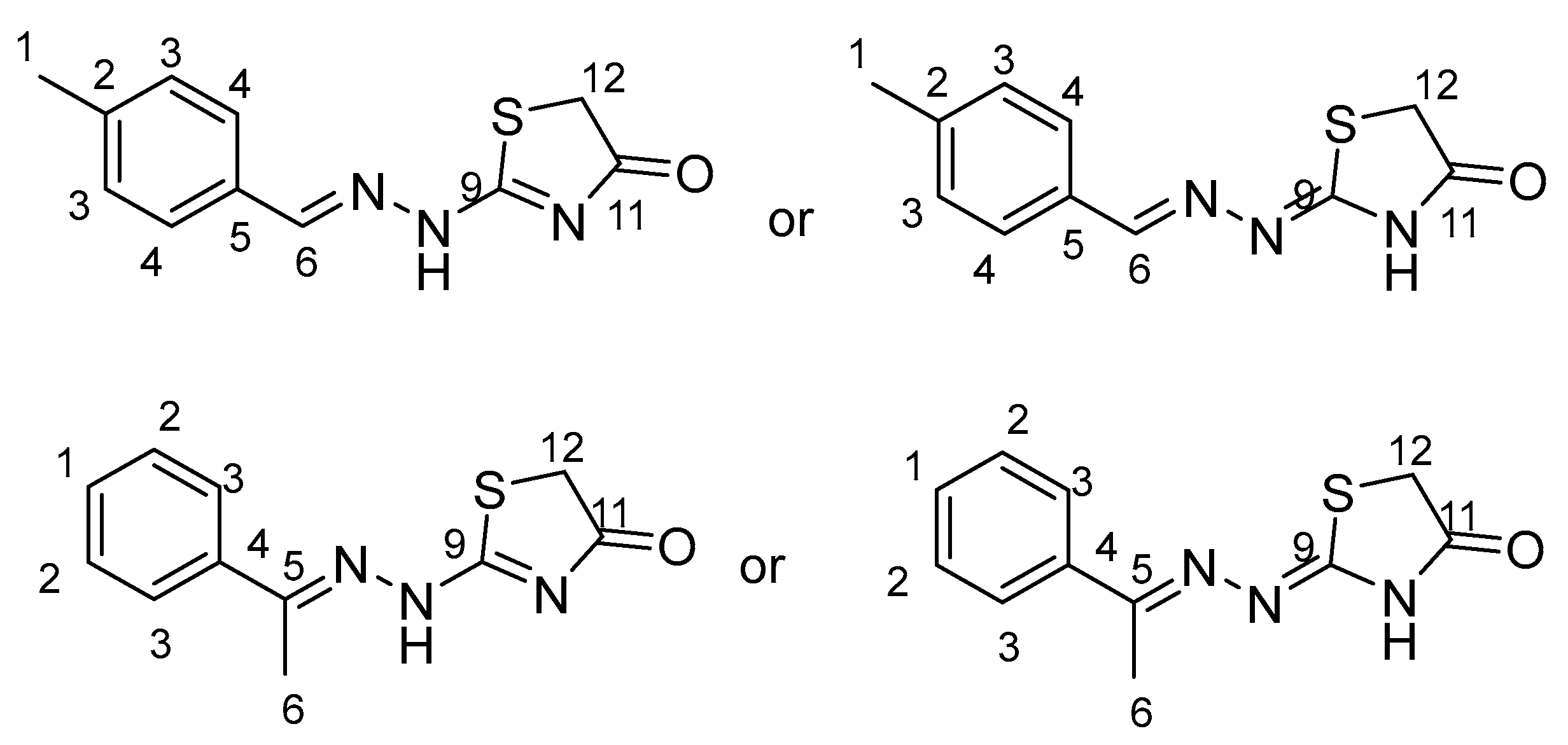
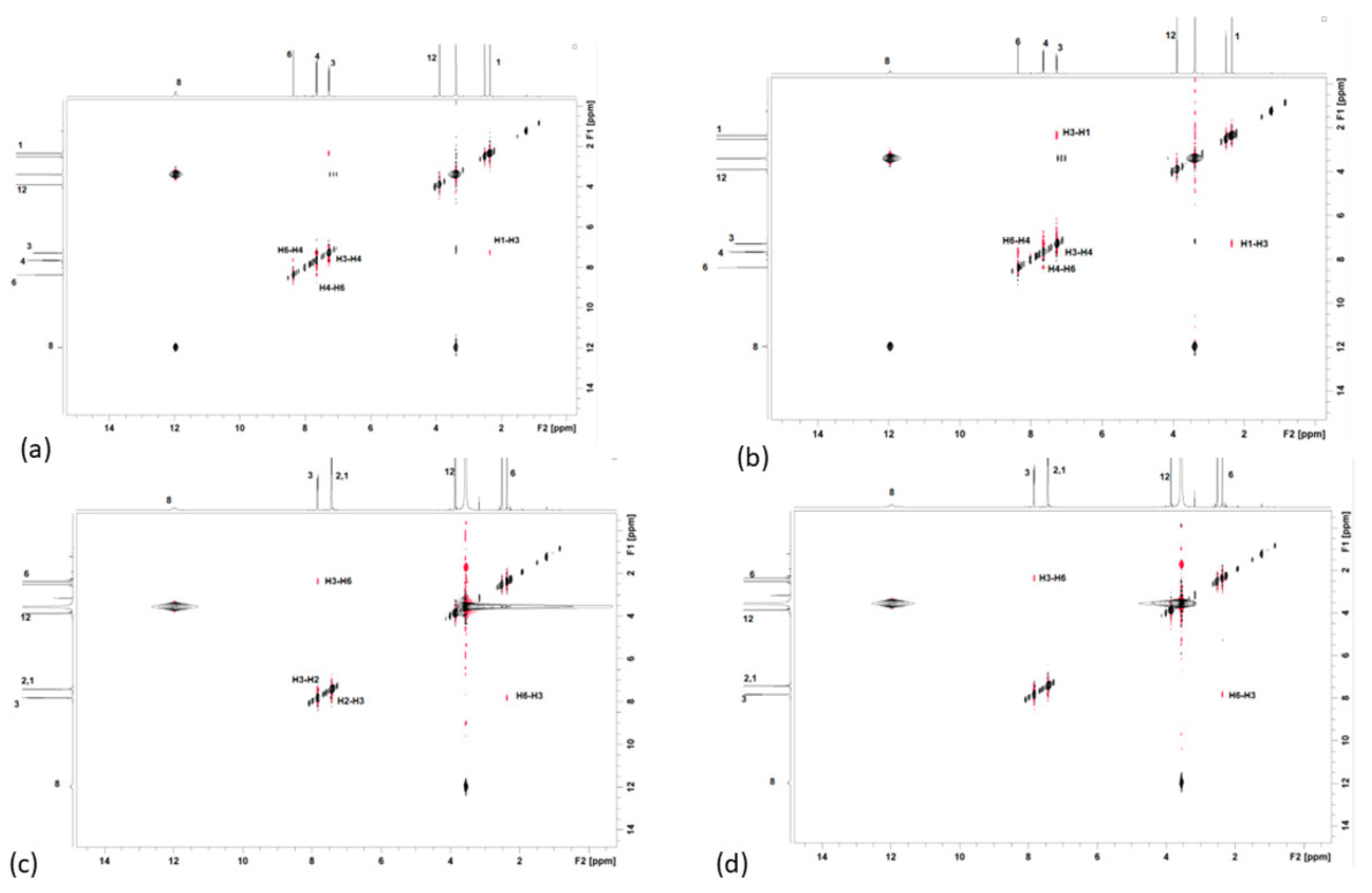
2.1. Structure Assignment
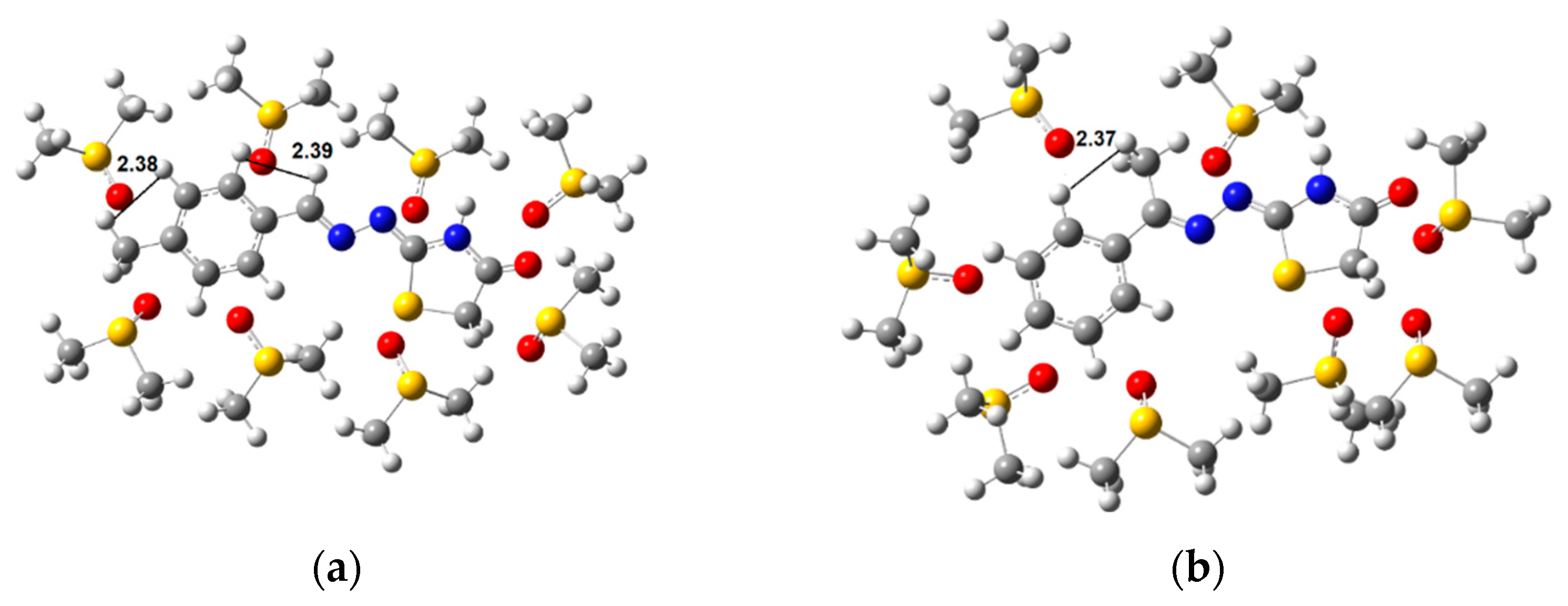
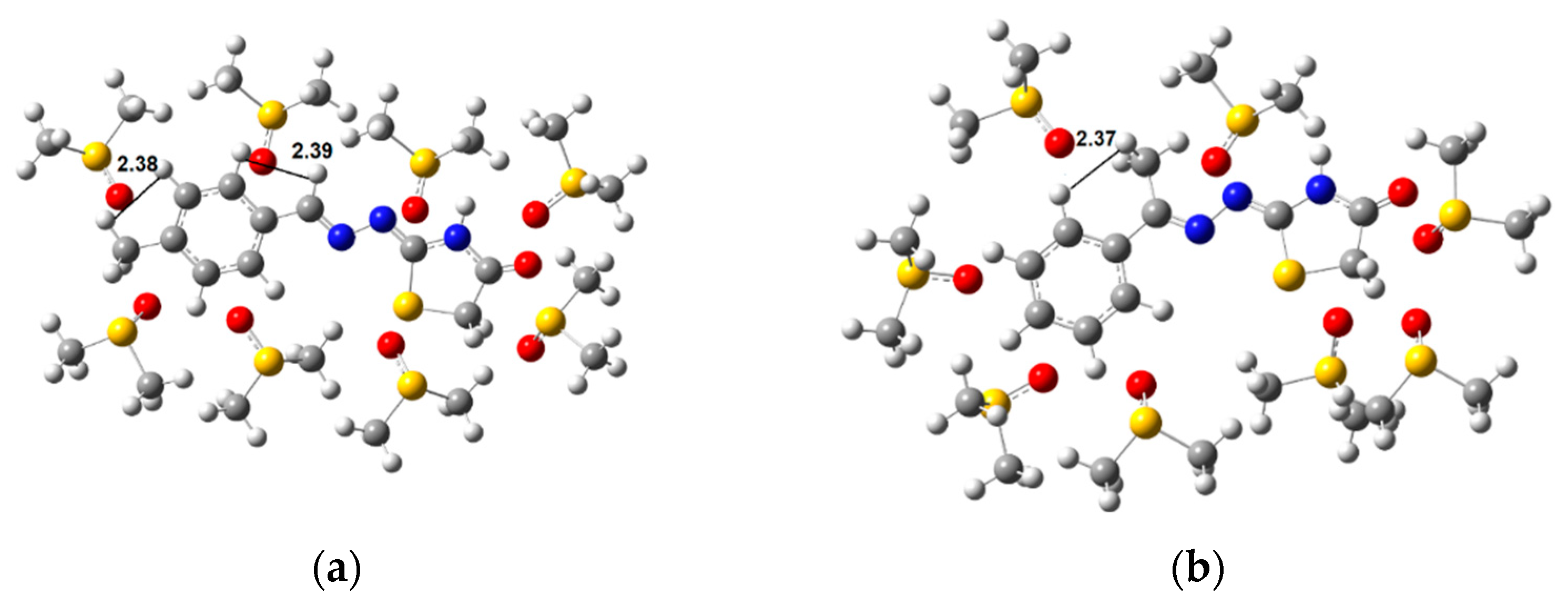
2.2. Conformational Analysis
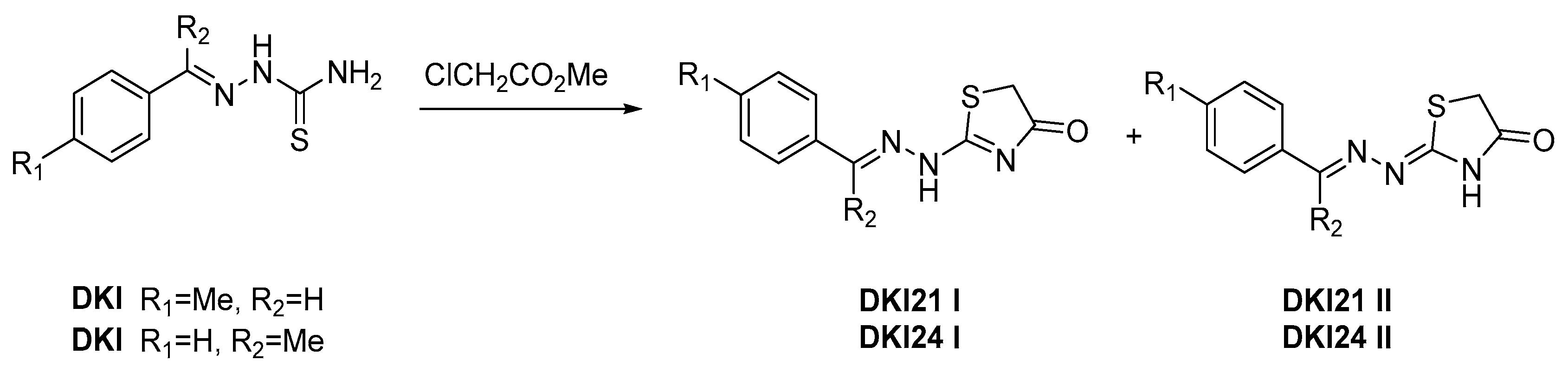
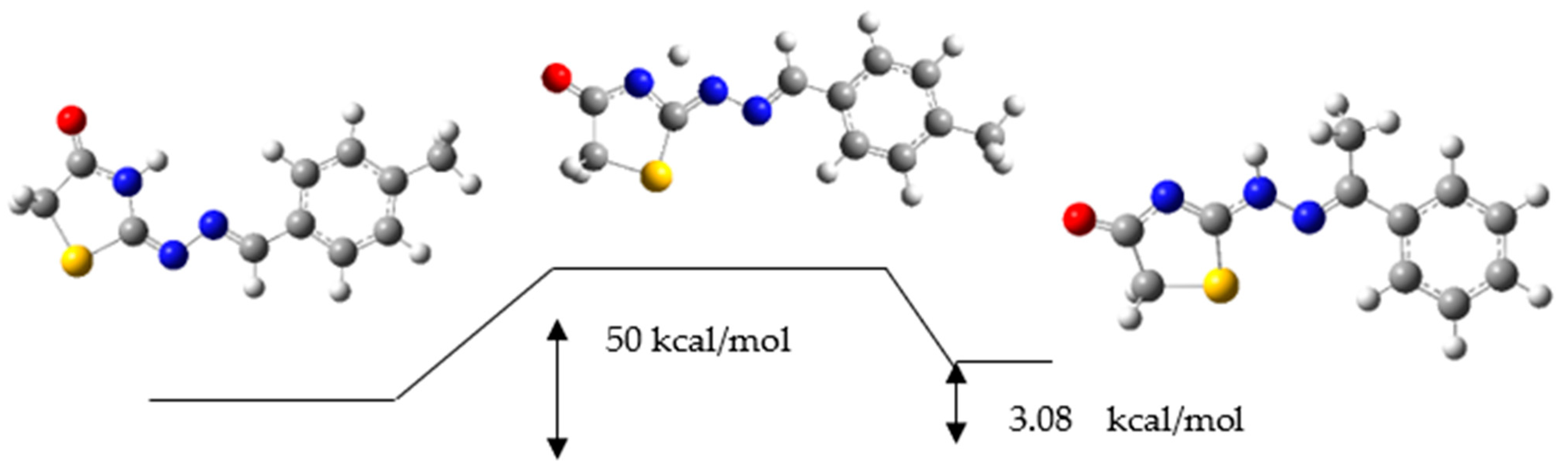
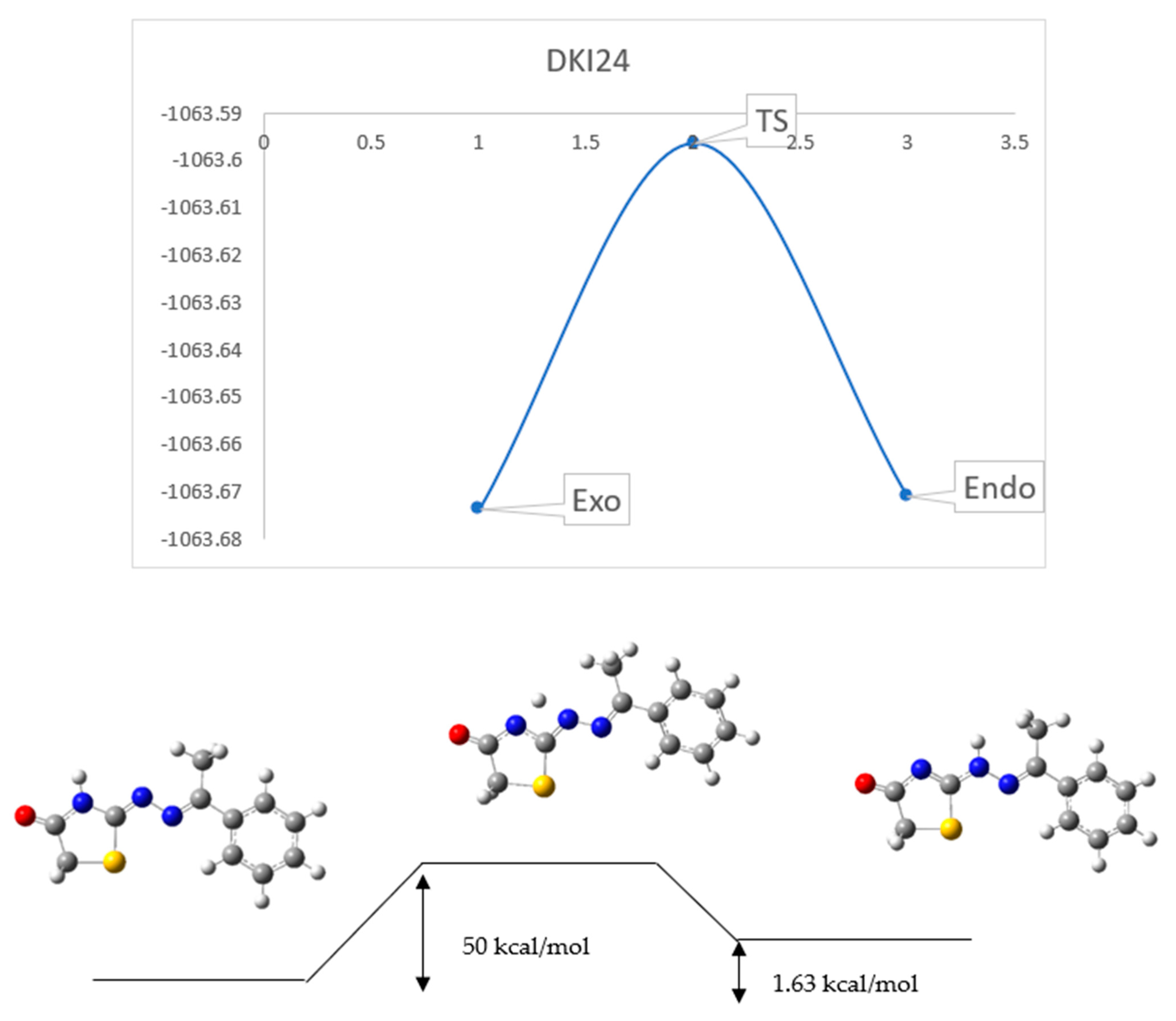
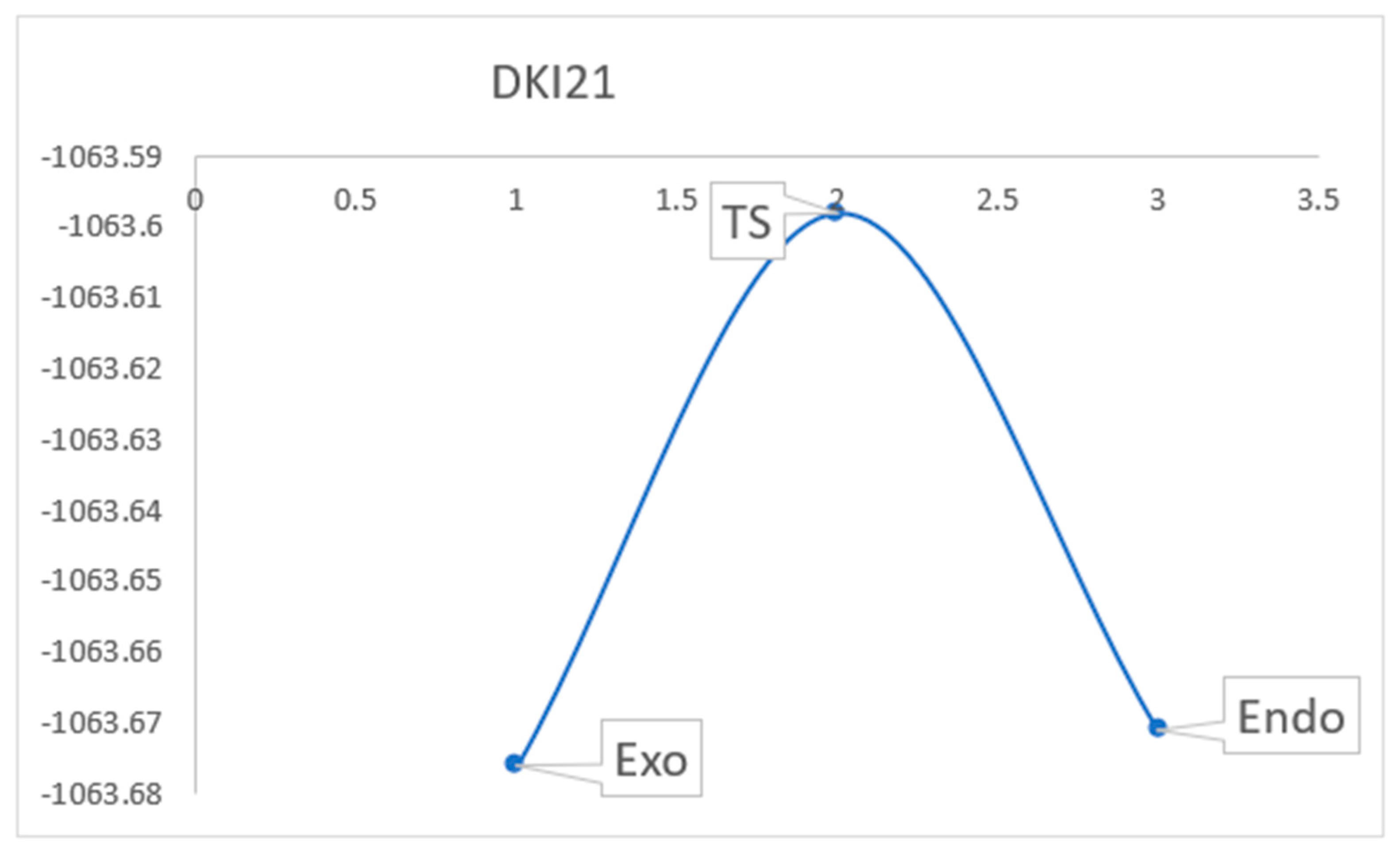
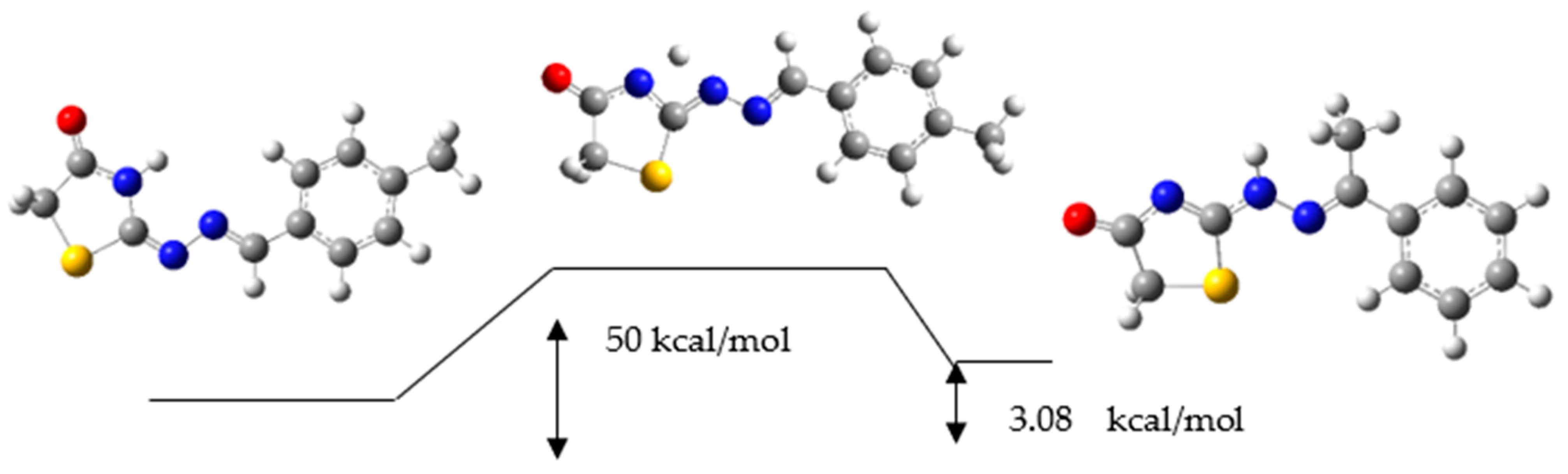
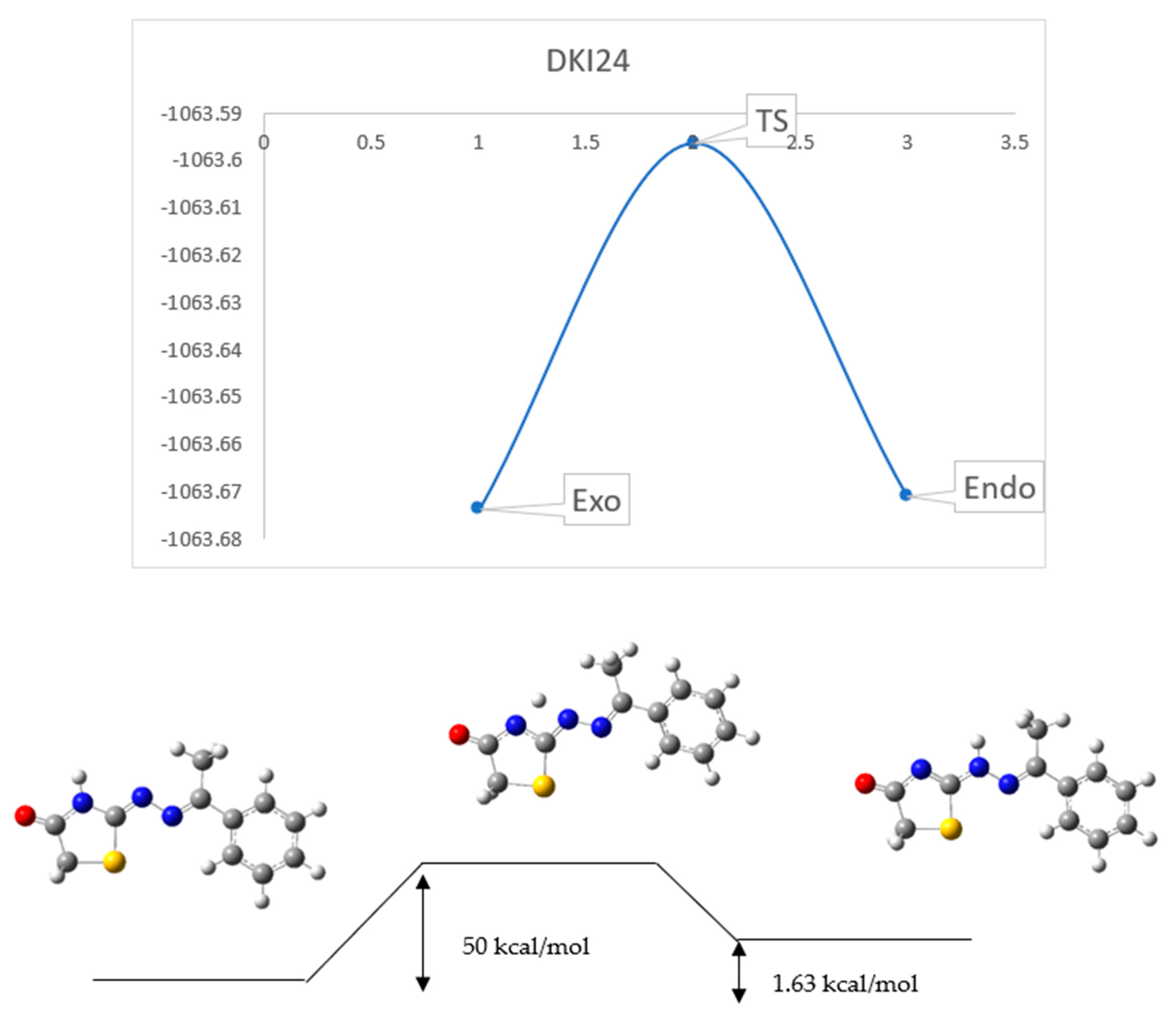
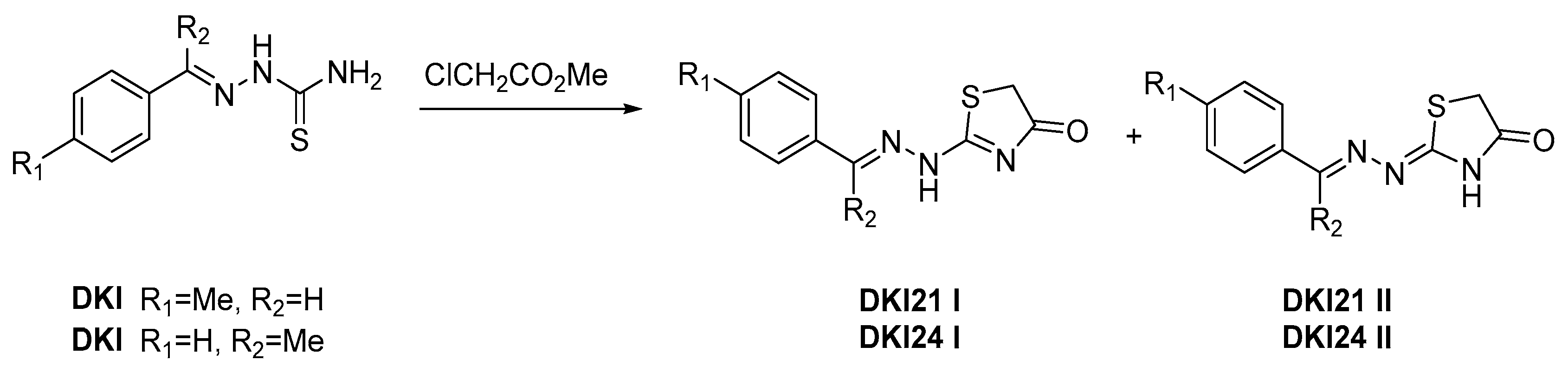
2.3. Reaction Mechanism
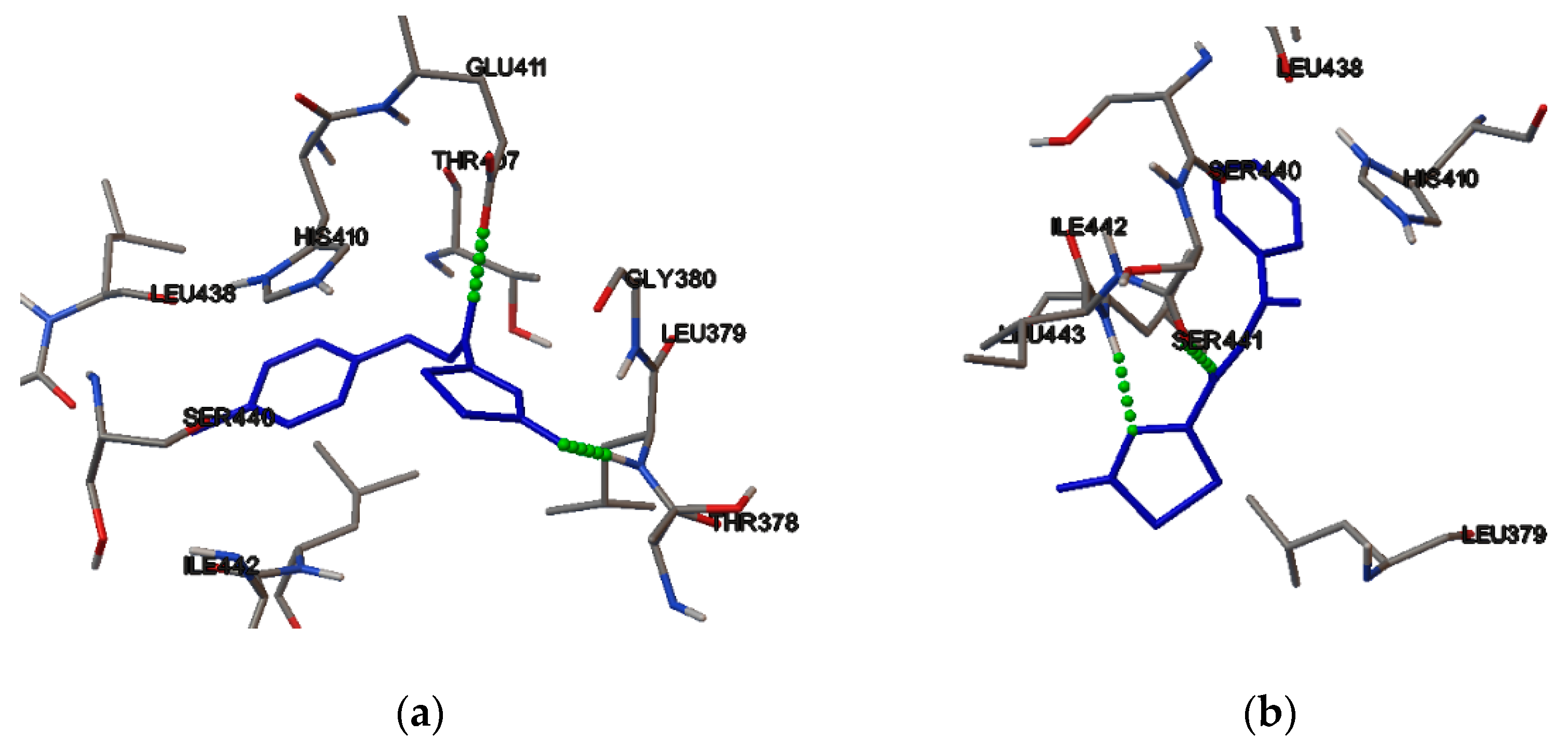
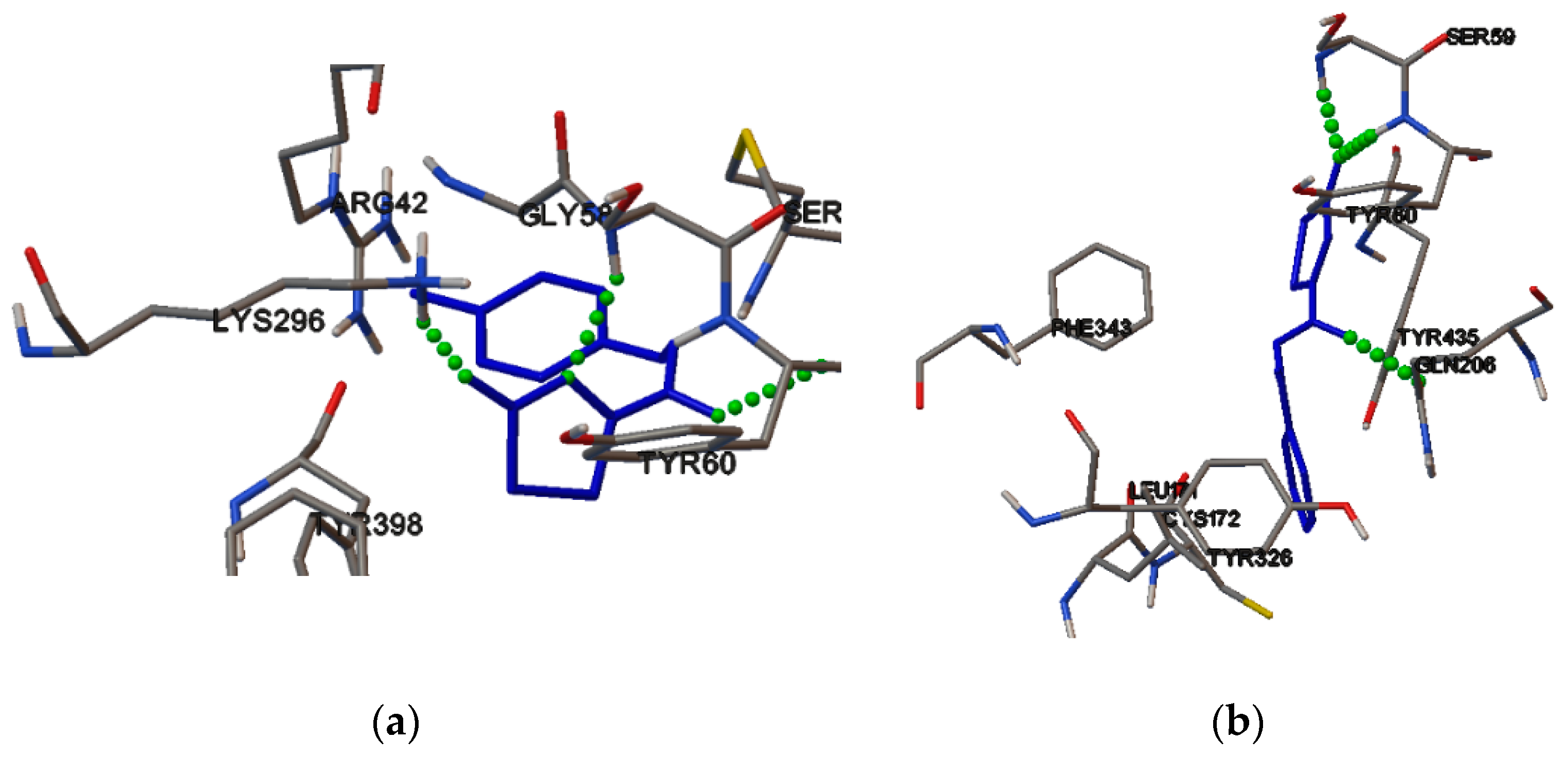
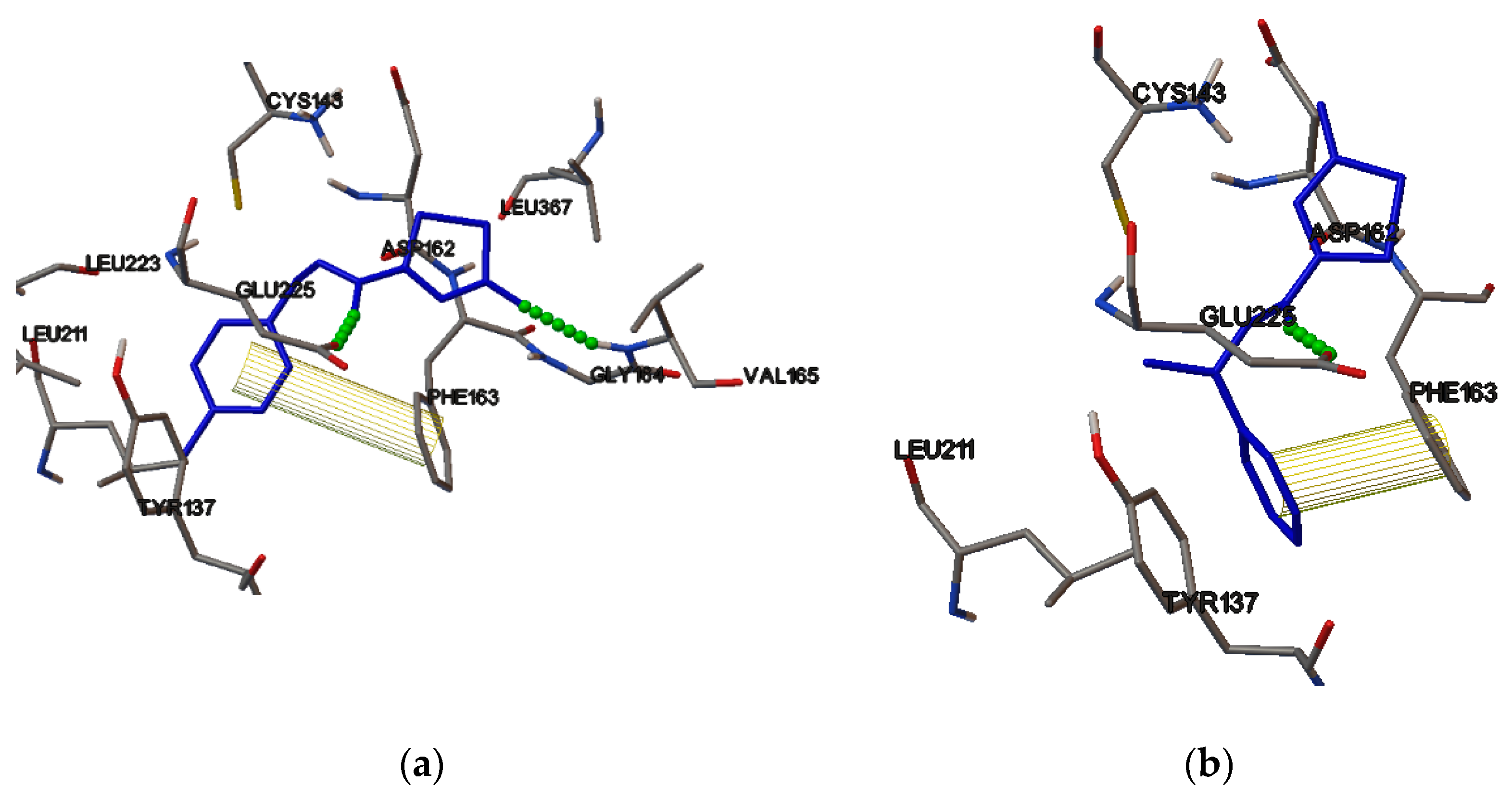
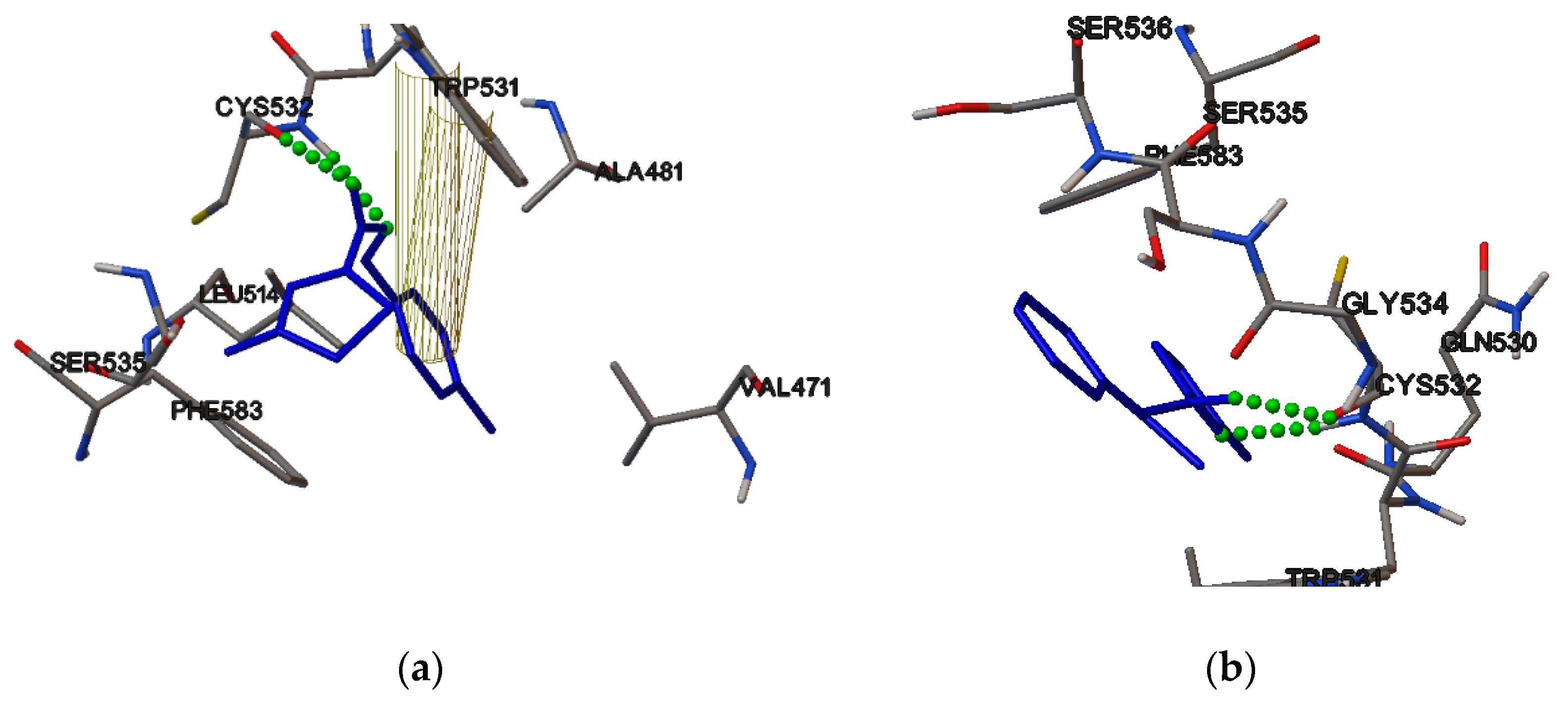
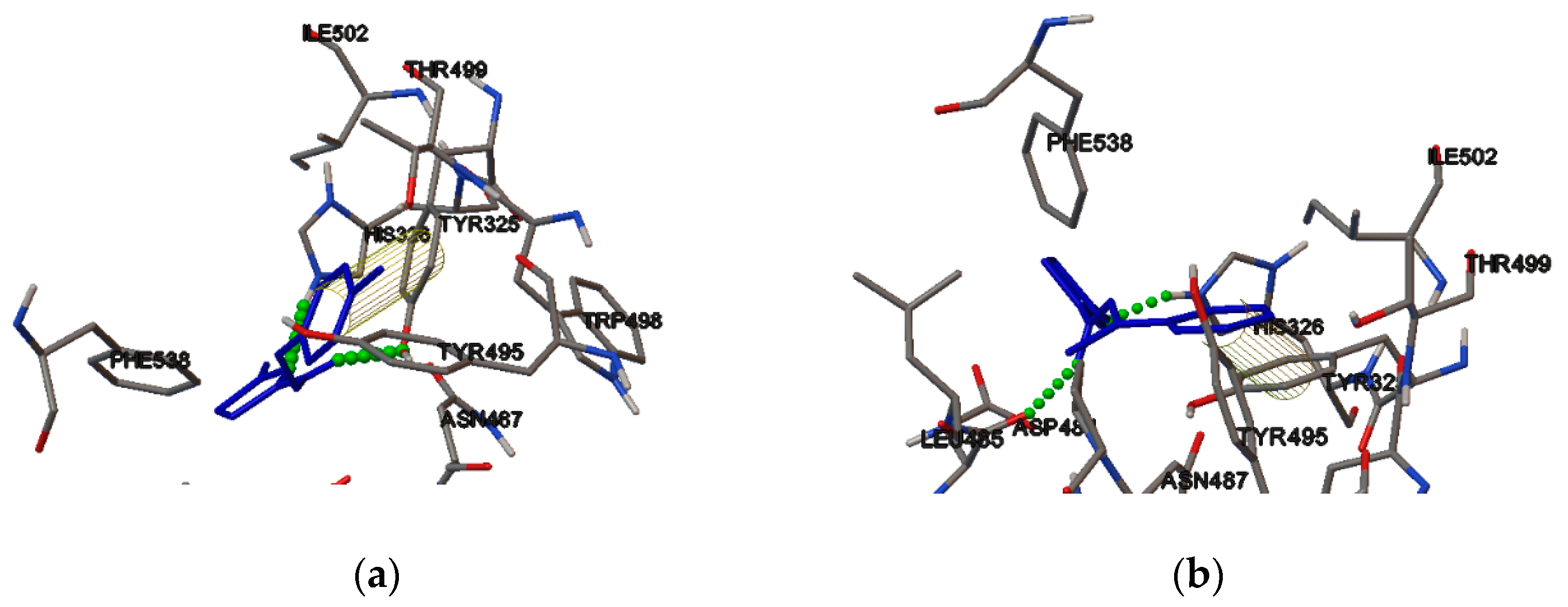
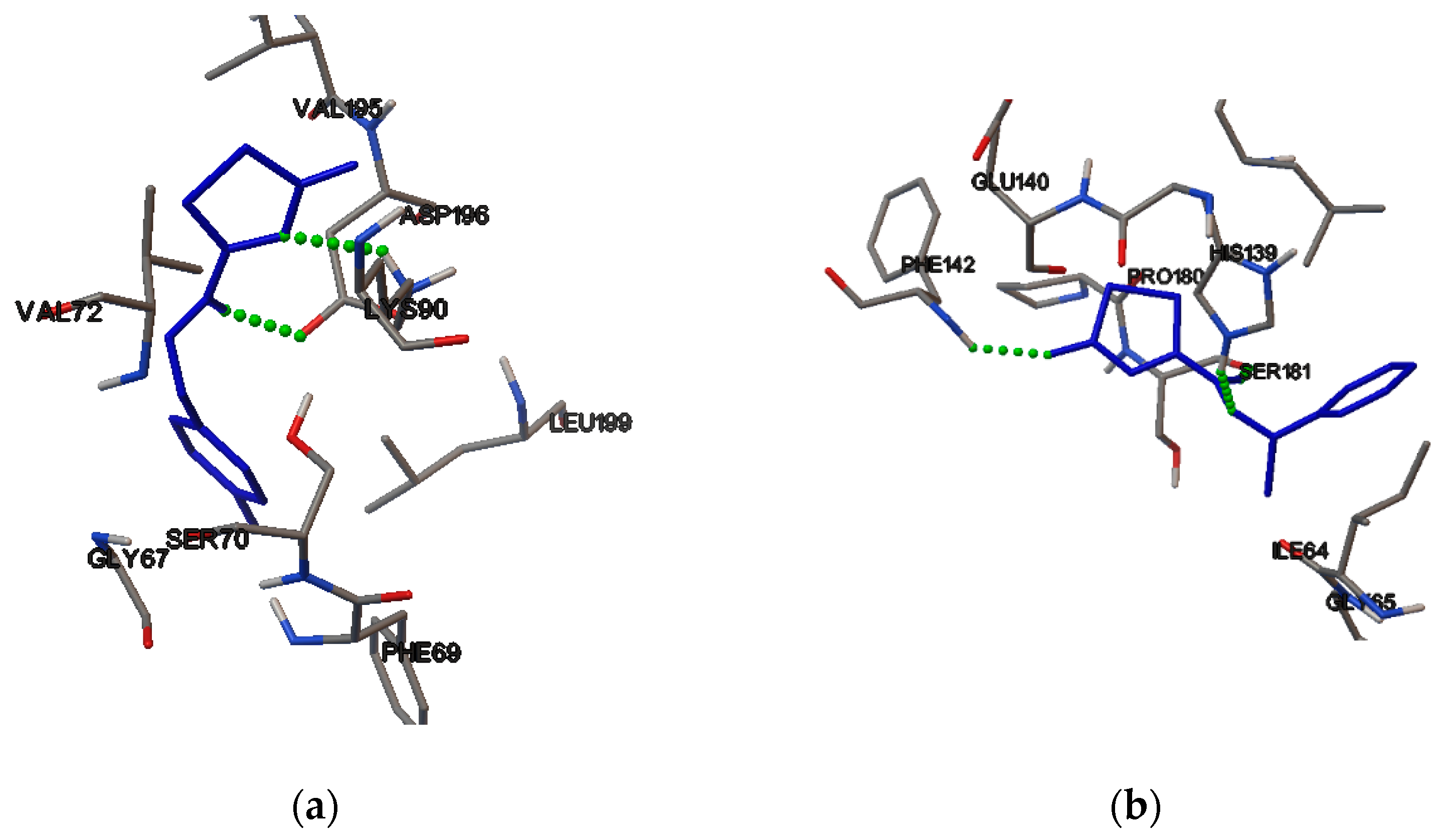
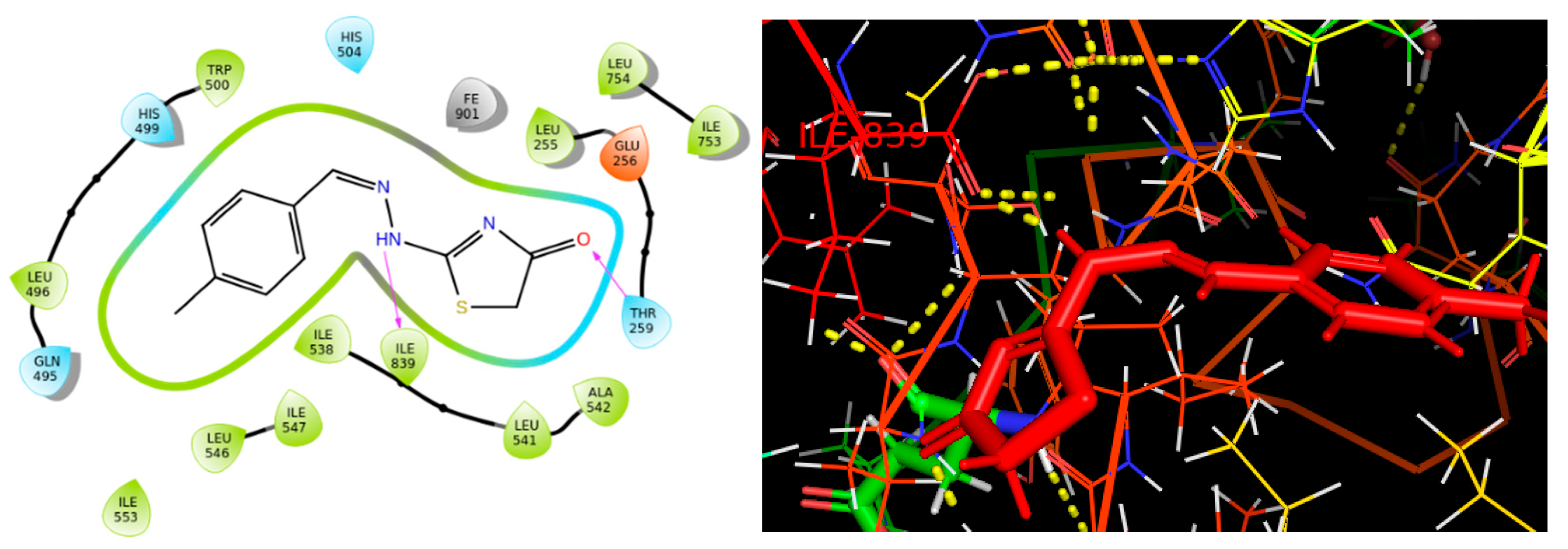
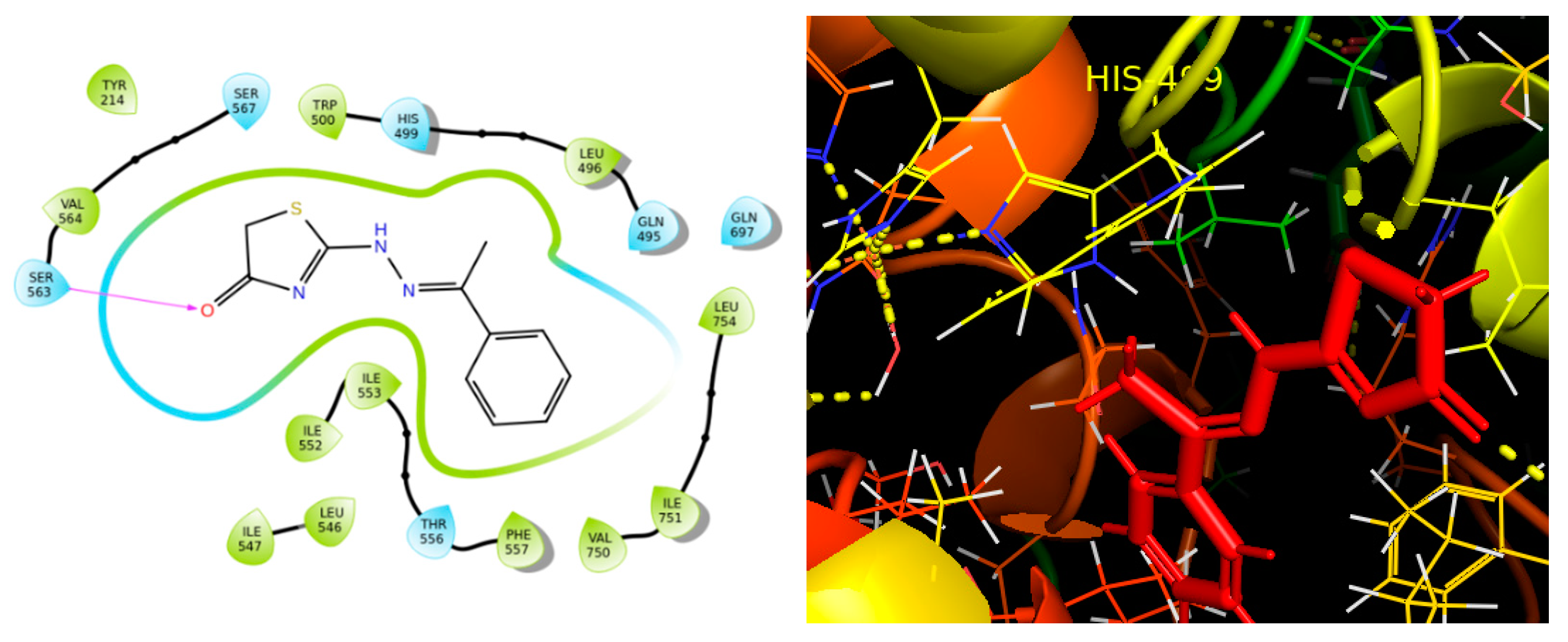
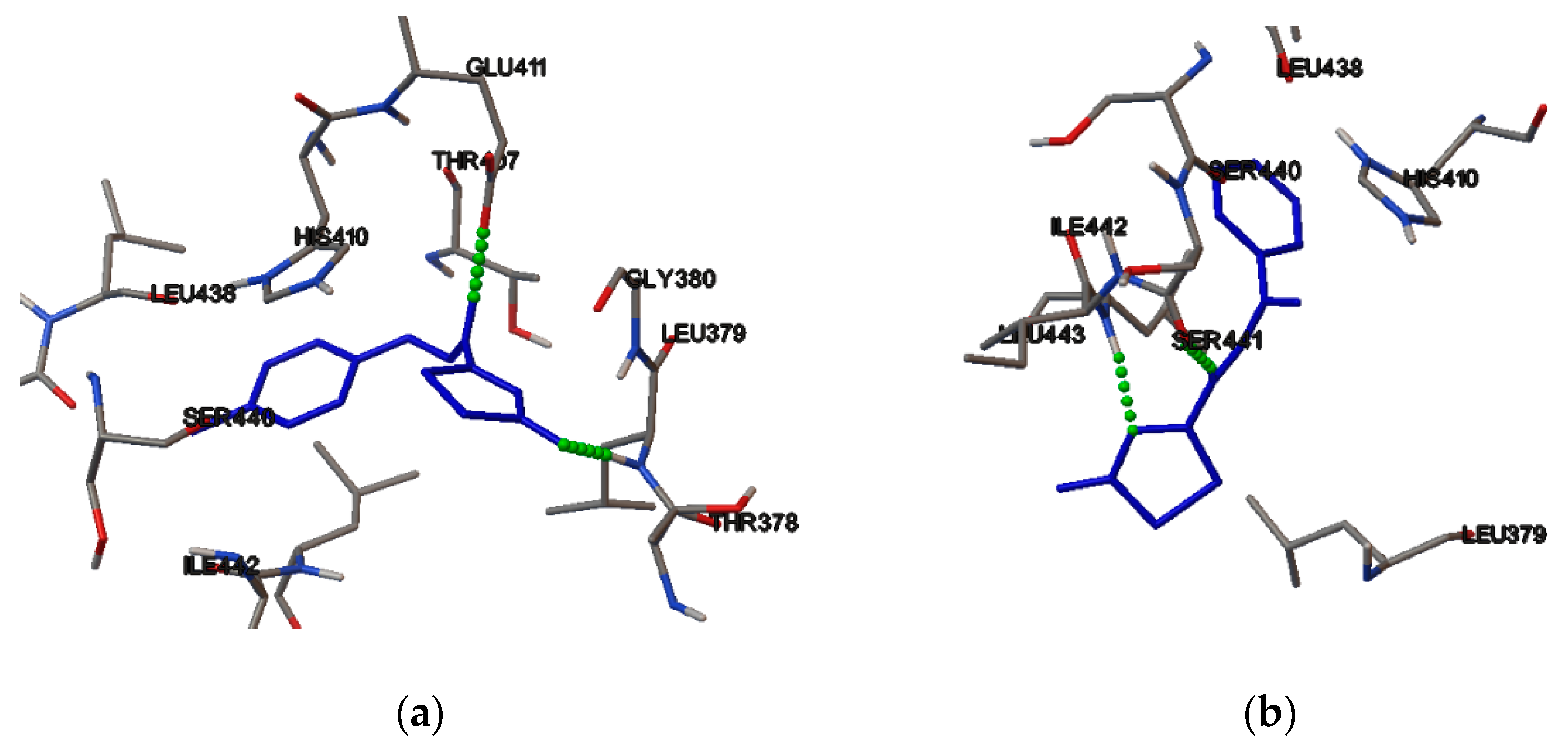
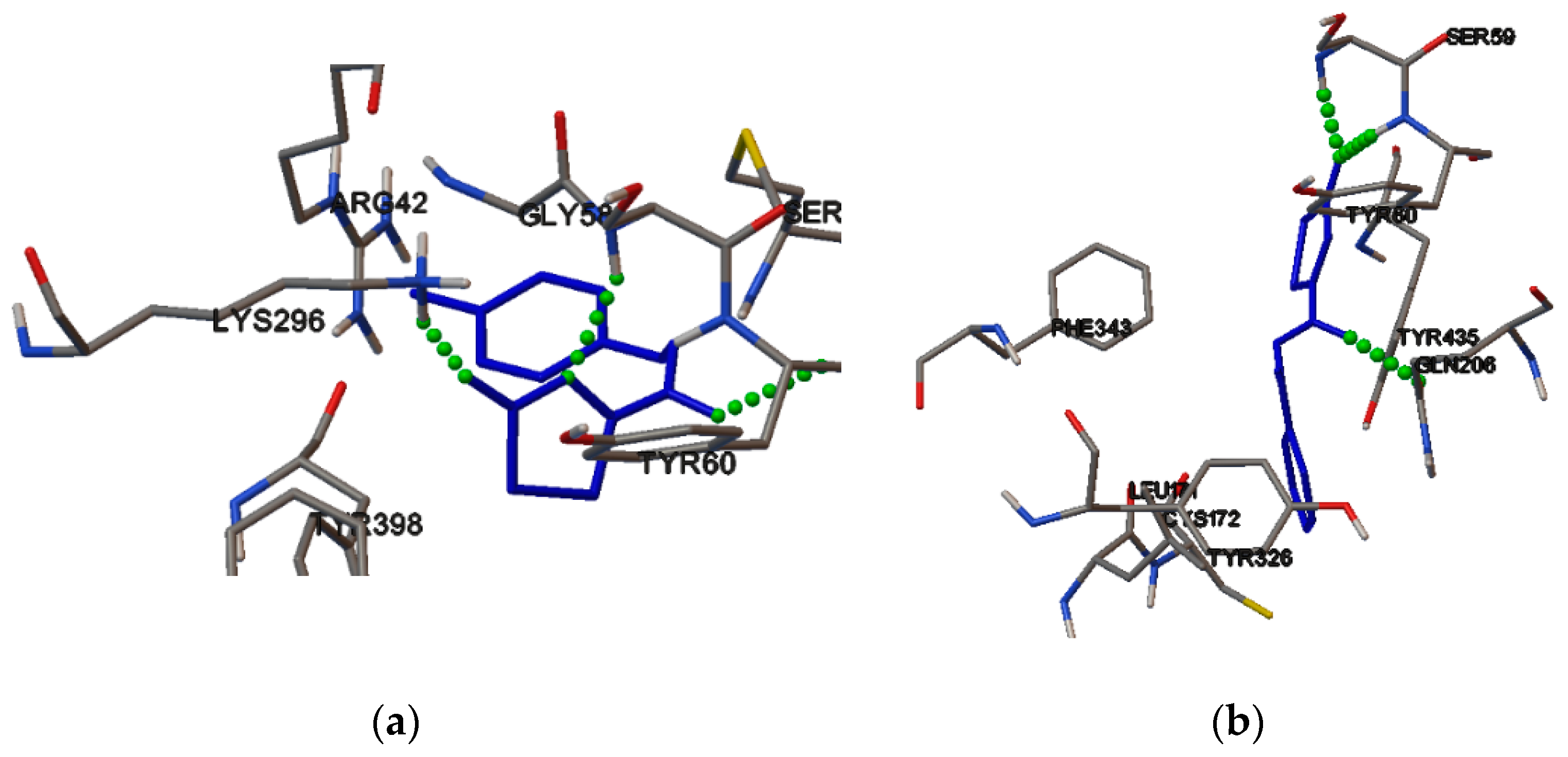
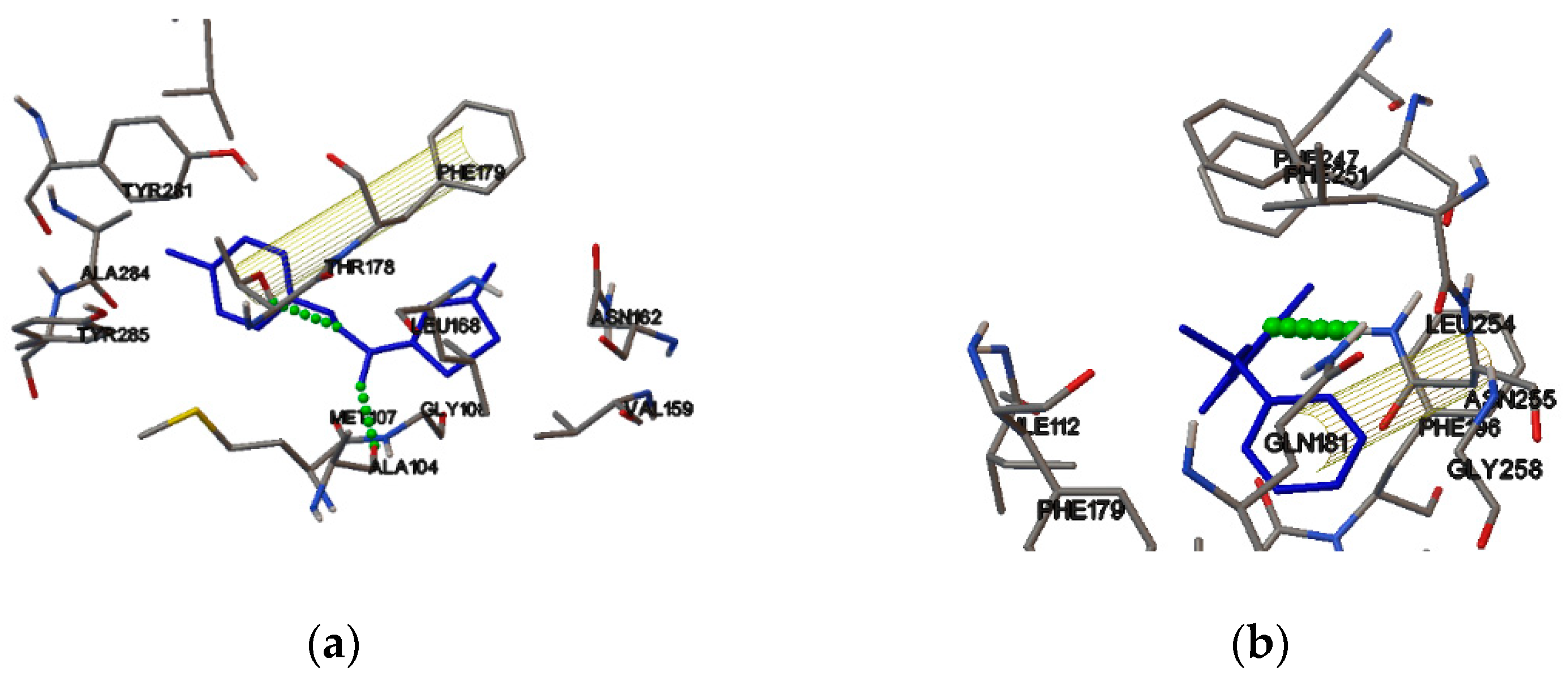
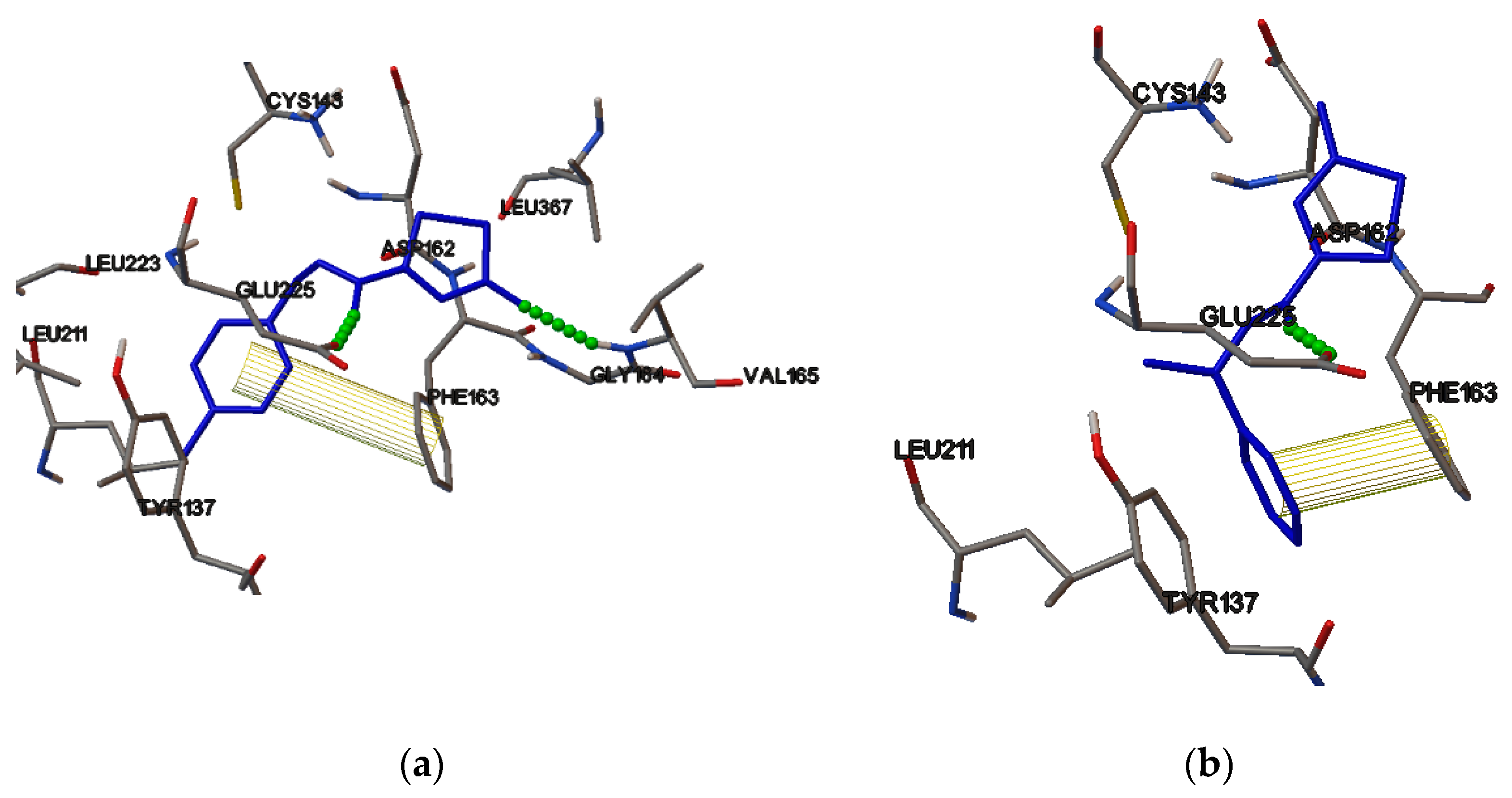
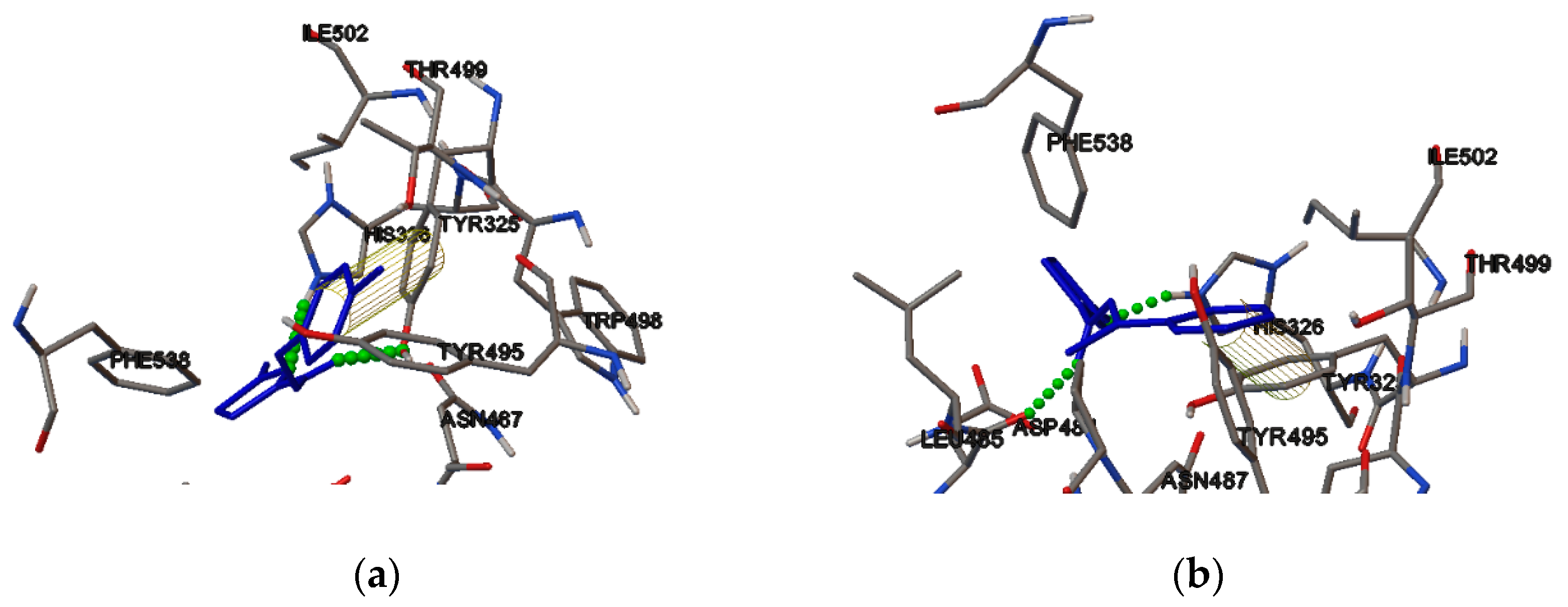
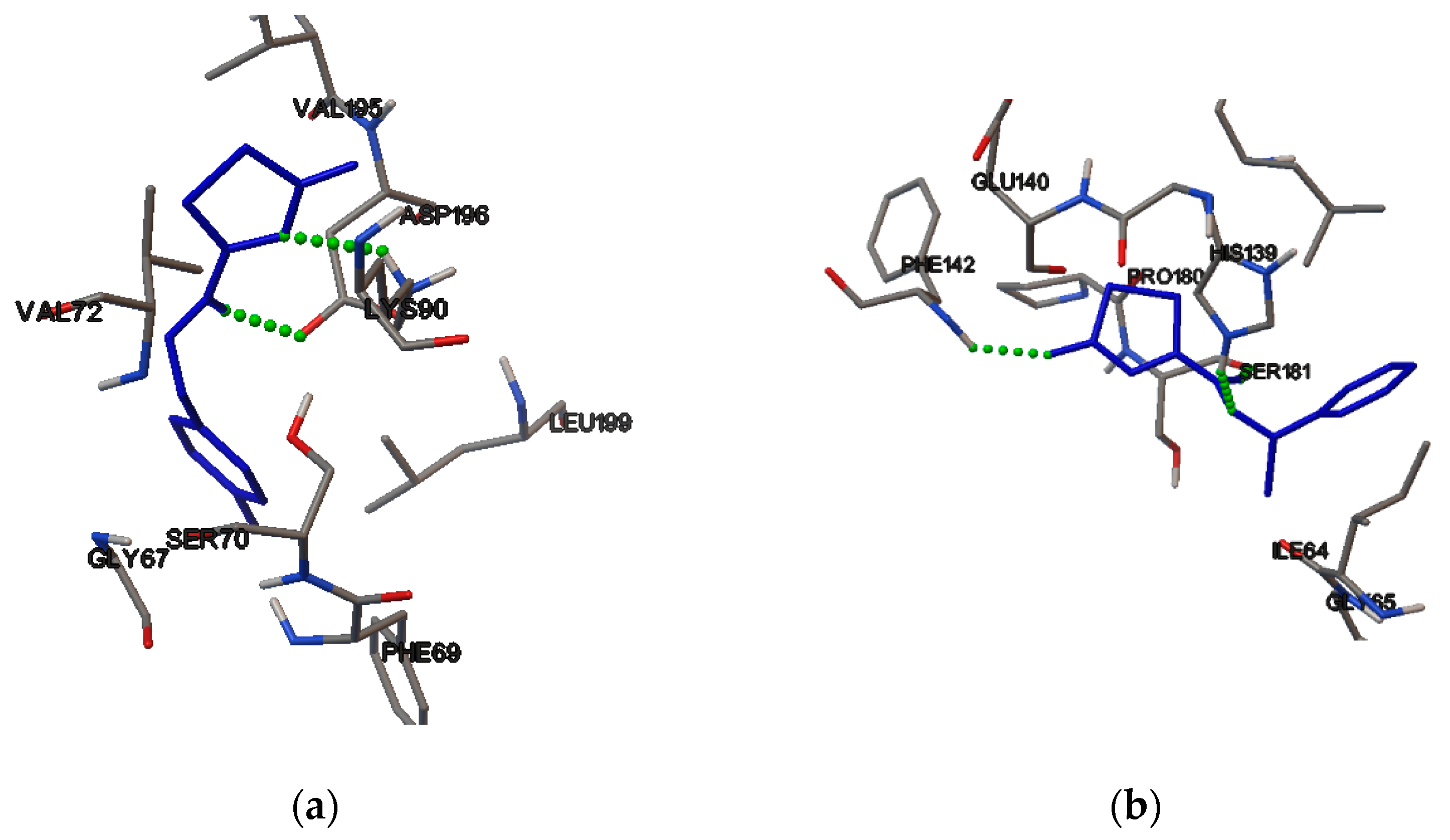
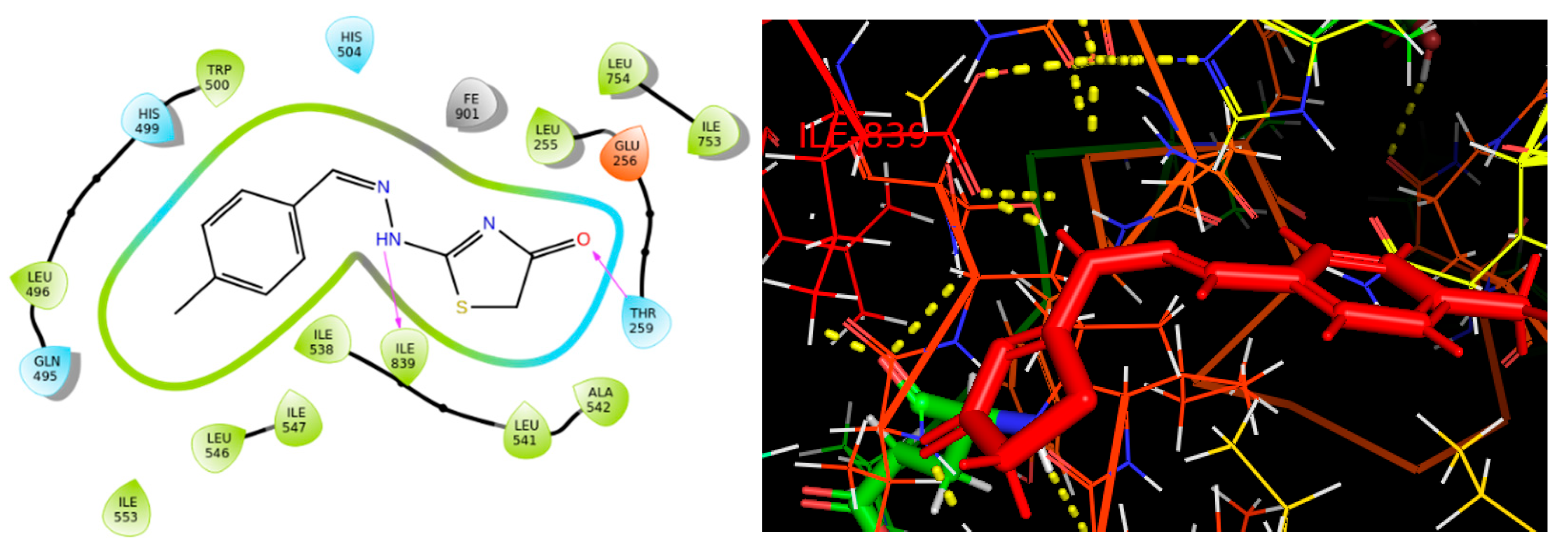
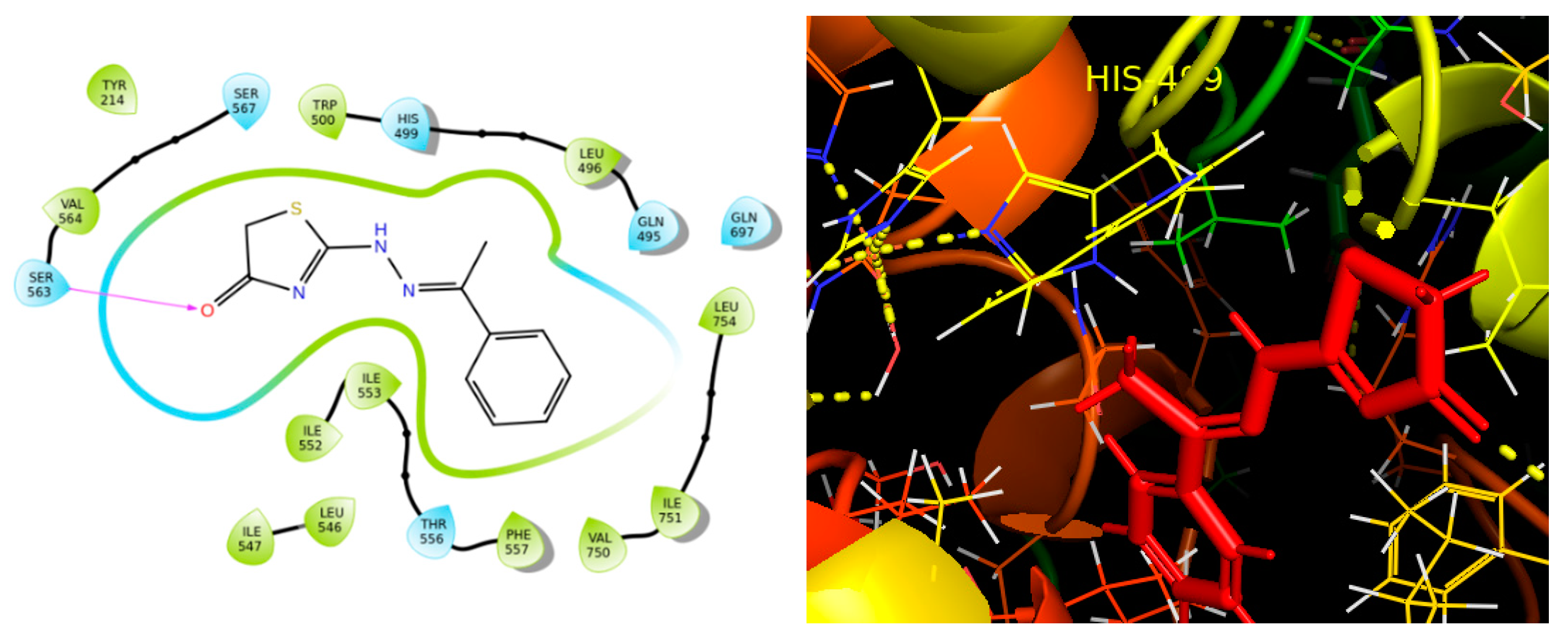
2.4. Molecular Binding
2.5. Results of the Pharmacokinetics and Toxicity Properties of the Two Compounds
3. Materials and Methods
3.1. Synthesis
3.2. Structure Assignment
3.3. Conformational Analysis
3.4. Reaction Mechanism
3.5. Molecular Binding
3.6. Induced Fit Docking
3.7. ADMET Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Parrilha, G.L.; dos Santos, R.G.; Beraldo, H. Applications of Radiocomplexes with Thiosemicarbazones and Bis(Thiosemicarbazones) in Diagnostic and Therapeutic Nuclear Medicine. Coord. Chem. Rev. 2022, 458, 214418. [Google Scholar] [CrossRef]
- Sevinçli, Z.Ş.; Duran, G.N.; Özbil, M.; Karalı, N. Synthesis, Molecular Modeling and Antiviral Activity of Novel 5-Fluoro-1H-Indole-2,3-Dione 3-Thiosemicarbazones. Bioorg. Chem. 2020, 104, 104202. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, O.; Singh-Pillay, A.; Koorbanally, N.A.; Singh, P. Antibacterial Evaluation and Molecular Docking Studies of Pyrazole–Thiosemicarbazones and Their Pyrazole–Thiazolidinone Conjugates. Mol. Divers. 2021, 25, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, V.; Haj Mohammad Ebrahim Tehrani, K.; Azerang, P.; Sardari, S.; Kobarfard, F. Synthesis, Antimycobacterial and Anticancer Activity of Novel Indole-Based Thiosemicarbazones. Arch. Pharm. Res. 2021, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, D.; Kryshchyshyn, A.; Lesyk, R. 5-Ene-4-Thiazolidinones—An Efficient Tool in Medicinal Chemistry. Eur. J. Med. Chem. 2017, 140, 542–594. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Roman, O.; Lesyk, R. Synthetic Approaches, Structure Activity Relationship and Biological Applications for Pharmacologically Attractive Pyrazole/Pyrazoline–Thiazolidine-Based Hybrids. Eur. J. Med. Chem. 2016, 113, 145–166. [Google Scholar] [CrossRef]
- Jain, V.S.; Vora, D.K.; Ramaa, C.S. Thiazolidine-2,4-Diones: Progress towards Multifarious Applications. Bioorg. Med. Chem. 2013, 21, 1599–1620. [Google Scholar] [CrossRef]
- Verma, A.; Saraf, S.K. 4-Thiazolidinone—A Biologically Active Scaffold. Eur. J. Med. Chem. 2008, 43, 897–905. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Gupta, S.J.; Fatima, G.N.; Sonar, P.K.; Verma, A.; Saraf, S.K. 4-Thiazolidinones: The Advances Continue…. Eur. J. Med. Chem. 2014, 72, 52–77. [Google Scholar] [CrossRef]
- Benmohammed, A.; Rekiba, N.; Sehanine, Y.; Louail, A.A.; Khoumeri, O.; Kadiri, M.; Djafri, A.; Terme, T.; Vanelle, P. Synthesis and Antimicrobial Activities of New Thiosemicarbazones and Thiazolidinones in Indole Series. Chem. Mon. 2021, 152, 977–986. [Google Scholar] [CrossRef]
- Nechak, R.; Bouzroura, S.A.; Benmalek, Y.; Salhi, L.; Martini, S.P.; Morizur, V.; Dunach, E.; Kolli, B.N. Synthesis and Antimicrobial Activity Evaluation of Novel 4-Thiazolidinones Containing a Pyrone Moiety. Synth. Commun. 2015, 45, 262–272. [Google Scholar] [CrossRef]
- Salem, M.A.; Abbas, S.Y.; El-Sharief MA, M.S.; Alzahrani, A.Y.; Helal, M.H.; Thabet, H.K. Synthesis and Antimicrobial Activity of 4-Methylthiazole and 4-Thiazolidinone Derivatives Derived from 5-(Aryldiazo)Salicylaldehyde Thiosemicarbazones. Synth. Commun. 2021, 51, 3325–3331. [Google Scholar] [CrossRef]
- Trotsko, N.; Bekier, A.; Paneth, A.; Wujec, M.; Dzitko, K. Synthesis and In Vitro Anti-Toxoplasma Gondii Activity of Novel Thiazolidin-4-One Derivatives. Molecules 2019, 24, 3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Qi, H.; Hao, Z.; Shao, X.; Liu, T.; Yang, Q.; Qian, X. Thiazolylhydrazone Dervatives as Inhibitors for Insect N-Acetyl-β-d-Hexosaminidase and Chitinase. Chin. Chem. Lett. 2020, 31, 1271–1275. [Google Scholar] [CrossRef]
- Hoque, M.J.; Ahsan, A.; Hossain, B. Molecular Docking, Pharmacokinetic, and DFT Calculation of Naproxen and Its Degradants. Biomed. J. Sci. Tech. Res. 2018, 9, 7360–7365. [Google Scholar] [CrossRef] [Green Version]
- De Proft, F.; Geerlings, P. Conceptual and Computational DFT in the Study of Aromaticity. Chem. Rev. 2001, 101, 1451–1464. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Li, M.; Hubalek, F.; Mattevi, A.; Edmondson, D.E. Structural and Mechanistic Studies of Arylalkylhydrazine Inhibition of Human Monoamine Oxidases A and B. Biochemistry 2008, 47, 5616–5625. [Google Scholar] [CrossRef]
- Stauch, B.; Johansson, L.C.; McCorvy, J.D.; Patel, N.; Han, G.W.; Huang, X.-P.; Gati, C.; Batyuk, A.; Slocum, S.T.; Ishchenko, A.; et al. Structural Basis of Ligand Recognition at the Human MT1 Melatonin Receptor. Nature 2019, 569, 284–288. [Google Scholar] [CrossRef]
- Dementiev, A.; Joachimiak, A.; Nguyen, H.; Gorelik, A.; Illes, K.; Shabani, S.; Gelsomino, M.; Ahn, E.-Y.E.; Nagar, B.; Doan, N. Molecular Mechanism of Inhibition of Acid Ceramidase by Carmofur. J. Med. Chem. 2019, 62, 987–992. [Google Scholar] [CrossRef]
- Foster, S.A.; Whalen, D.M.; Özen, A.; Wongchenko, M.J.; Yin, J.; Yen, I.; Schaefer, G.; Mayfield, J.D.; Chmielecki, J.; Stephens, P.J.; et al. Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer Cell 2016, 29, 477–493. [Google Scholar] [CrossRef] [Green Version]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of Phosphodiesterase 4D (PDE4D) Allosteric Modulators for Enhancing Cognition with Improved Safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Elustondo, F.; Di Fonzo, A.; Leroux, F.G.; Wong, A.C.; Snijders, A.P.; Matthews, S.J.; Cherepanov, P. Crystal Structure of Human CDC7 Kinase in Complex with Its Activator DBF4. Nat. Struct. Mol. Biol. 2012, 19, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Mosyak, L.; Georgiadis, K.; Shane, T.; Svenson, K.; Hebert, T.; McDonagh, T.; Mackie, S.; Olland, S.; Lin, L.; Zhong, X.; et al. Crystal Structures of the Two Major Aggrecan Degrading Enzymes, ADAMTS4 and ADAMTS5. Protein Sci. 2008, 17, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Guy, R.C. Ames Test. In Encyclopedia of Toxicology; Elsevier: Amsterdam, The Netherlands, 2005; pp. 88–91. [Google Scholar] [CrossRef]
- Pires DE, V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.; Davis, N.E.; Binning, R.C.; Radom, L. Extension of Gaussian-2 Theory to Molecules Containing Third-row Atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Tzeli, D.; Tsoungas, P.G.; Petsalakis, I.D.; Kozielewicz, P.; Zloh, M. Intramolecular Cyclization of β-Nitroso-o-Quinone Methides. A Theoretical Endoscopy of a Potentially Useful Innate ‘Reclusive’ Reaction. Tetrahedron 2015, 71, 359–369. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New Developments in the Polarizable Continuum Model for Quantum Mechanical and Classical Calculations on Molecules in Solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Rizvi SM, D.; Shakil, S.; Haneef, M. A Simple Click by Click Protocol to Perform Docking: Autodock 4.2 Made Easy for Non-Bioinformaticians. EXCLI J. 2013, 12, 830–857. [Google Scholar] [CrossRef]
- Schleinkofer, K.; Wang, T.; Wade, R.C. Molecular Docking. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin/Heidelberg, Germany, 2006; Volume 443, pp. 1149–1153. [Google Scholar] [CrossRef]
- Ghafary, S.; Ghobadian, R.; Mahdavi, M.; Nadri, H.; Moradi, A.; Akbarzadeh, T.; Najafi, Z.; Sharifzadeh, M.; Edraki, N.; Moghadam, F.H.; et al. Design, Synthesis, and Evaluation of Novel Cinnamic Acid-Tryptamine Hybrid for Inhibition of Acetylcholinesterase and Butyrylcholinesterase. DARU J. Pharm. Sci. 2020, 28, 463–477. [Google Scholar] [CrossRef]
- Peperidou, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Voulgari, E.; Avgoustakis, K. Multifunctional Cinnamic Acid Derivatives. Molecules 2017, 22, 1247. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, A.R.; Hu, S.; Poss, E.M.; Carr, C.A.M.; Scouras, A.D.; Prigozhin, D.M.; Iavarone, A.T.; Palla, A.; Alber, T.; Fraser, J.S.; et al. Hydrogen-Deuterium Exchange of Lipoxygenase Uncovers a Relationship between Distal, Solvent Exposed Protein Motions and the Thermal Activation Barrier for Catalytic Proton-Coupled Electron Tunneling. ACS Cent. Sci. 2017, 3, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacroModel, Version 10; Schrodinger, LLC: New York, NY, USA, 2013.
- MacroModel, Version 10.2; Schrodinger, LLC: New York, NY, USA, 2013.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1H | COSY | NOESY | HSQC | HMBC | 13C |
|---|---|---|---|---|---|---|
| 1 | 2.35 | - | H-3 | C-1 | C-2. C-3 | 21.51 |
| 2 | - | - | - | - | H-1/H-3 | 141.03 |
| 3 | 7.26 | H-4 | H-1/H-4 | C-3 | C-2/C-5 | 129.90 |
| 4 | 7.66 | H-3 | H-3/H-6 | C-4 | C-5/C-3/C-6 | 128.10 |
| 5 | - | - | - | - | H-3/H-6/H-4 | 131.98 |
| 6 | 8.36 | H-4 | C-6 | H-5/H-4 | 156.68 | |
| 7 | ||||||
| 8 | ||||||
| 9 | - | - | - | - | C-11/C-12/H-8 | 165.20 |
| 10 | ||||||
| 11 | - | - | - | - | H-12 | 174.62 |
| 12 | 3.89 | - | - | C-12 | C-11/C-9 | 33.45 |
| Position | 1H | COSY | NOESY | HSQC | HMBC | 13C |
| 1 | 7.47 | H-2 | H-2 | C-1 | C-2. C-3 | 121.87 |
| 2 | 7.47 | H-1/H-3 | H-1/H-3 | C-2 | H-1/H-3 | 121.87 |
| 3 | 7.86 | H-2 | H-2/H-6 | C-3 | C-2/C-5/C-4 | 126.84 |
| 4 | - | - | - | - | H-3/H-2/H-6 | 138.22 |
| 5 | - | - | - | - | H-3/H-6/H-4 | 160.83 |
| 6 | 2.37 | - | H-3 | C-6 | H-5/H-4 | 15.04 |
| 7 | ||||||
| 8 | ||||||
| 9 | - | - | - | - | H-12 | 164.53 |
| 10 | ||||||
| 11 | - | - | - | - | H-12 | 174.42 |
| 12 | 3.85 | - | - | C-12 | C-11/C-9 | 33.26 |
| Binding Energy | Inhibition Constant | |
|---|---|---|
| 2VRM (MAO B) | ||
| DKI21 | −7.64 ± 0.5 | 2.52 ± 0.5 μΜ/3.13 ± 0.5 μΜ |
| DKI24 | −7.55 ± 0.5 | 2.93 ± 0.5 μΜ |
| 6ME2 (Melatonin receptor MT1) | ||
| DKI21 | −6.81 ± 0.5 | 10.17 ± 0.5 μΜ |
| DKI24 | −7.38 ± 0.5 | 3.92 ± 0.5 μΜ |
| 6MHM (Human acid ceramidase) | ||
| DKI21 | −7.10 ± 0.5 | 6.29 ± 0.5 μΜ |
| DKI24 | −7.07 ± 0.5 | 6.58 ± 0.5 μΜ |
| 5HIE (Kinase domain) | ||
| DKI21 | −6.41 ± 0.5 | 19.9 ± 0.5 μΜ |
| DKI24 | −6.36 ± 0.5 | 21.89 ± 0.5 μΜ |
| 3G4L(Phosphodiesterase 4D) | ||
| DKI21 | −7.1 ± 0.5 | 6.3 ± 0.5 μΜ |
| DKI24 | −7.76 ± 0.5 | 2.05 ± 0.5 μΜ |
| 2RJQ (ADAMTS5) | ||
| DKI21 | −7.65 ± 0.5 | 2.48 ± 0.5 μΜ |
| DKI24 | −7.7 ± 0.5 | 4.47 ± 0.5 μΜ |
| CDC7/DBF4 (Cell division cycle 7-related protein kinase/Activator of S phase kinase) (4F9C) | ||
| DKI21 | −6.07 ± 0.5 | 35.5 ± 0.5 μΜ |
| DKI24 | −6.20 ± 0.5 | 28.43 ± 0.5 μΜ |
| Properties | Compound DK121 | Compound DΚΙ24 |
|---|---|---|
| Molecular Weight | 233.296 | 233.296 |
| LogP | 1.54792 | 1.6296 |
| Rotable bonds | 2 | 2 |
| Hydrogen Bond Acceptors | 4 | 4 |
| Hydrogen Bond Donors | 1 | 1 |
| Surface Area | 97.846 (Å2) | 97.846 (Å2) |
| Water solubility | −2.855 (logmolL−1) | −2.702 (logmolL−1) |
| Compound DKI21 | Compound DKI24 | |
|---|---|---|
| BBB | 0.121871 | 0.370766 |
| Buffer_solubility_mg_L | 101.478 | 11192.1 |
| Caco2 | 8.61083 | 7.52339 |
| CYP_2C19_inhibition | Non | Non |
| CYP_2C9_inhibition | Non | Non |
| CYP_2D6_inhibition | Non | Non |
| CYP_2D6_substrate | Non | Non |
| CYP_3A4_inhibition | Non | Non |
| CYP_3A4_substrate | Non | Weakly |
| HIA | 96.096918 | 96.096626 |
| MDCK | 23.4668 | 22.523 |
| Pgp_inhibition | Non | Non |
| Plasma_Protein_Binding | 73.336958 | 70.164297 |
| Pure_water_solubility_mg_L | 12.6628 | 20.7683 |
| Skin_Permeability | −3.1538 | −3.40889 |
| Properties | Compound DK121 | Compound DKI24 |
|---|---|---|
| Toxicity | ||
| AMES toxicity | Yes | No |
| Max. tolerated dose (human) | 0.399 (log mg/kg/day) | 0.269 (log mg/kg/day) |
| Herg I inhibitor | No | No |
| Herg II inhibitor | No | No |
| Oral Rat Acute Toxicity (LD50) | 2.699 (mol/kg) | 2.878 (mol/kg) |
| Oral Rat Chronic Toxicity | 1.417 (log mg/kg_bw/day) | 1.386 (log mg/kg_bw/day) |
| Hepatotoxicity | Yes | Yes |
| Skin Sensitization | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiou, N.; Cheilari, A.; Karta, D.; Chontzopoulou, E.; Plavec, J.; Tzeli, D.; Vassiliou, S.; Mavromoustakos, T. Conformational Properties and Putative Bioactive Targets for Novel Thiosemicarbazone Derivatives. Molecules 2022, 27, 4548. https://doi.org/10.3390/molecules27144548
Georgiou N, Cheilari A, Karta D, Chontzopoulou E, Plavec J, Tzeli D, Vassiliou S, Mavromoustakos T. Conformational Properties and Putative Bioactive Targets for Novel Thiosemicarbazone Derivatives. Molecules. 2022; 27(14):4548. https://doi.org/10.3390/molecules27144548
Chicago/Turabian StyleGeorgiou, Nikitas, Antigoni Cheilari, Danai Karta, Eleni Chontzopoulou, Janez Plavec, Demeter Tzeli, Stamatia Vassiliou, and Thomas Mavromoustakos. 2022. "Conformational Properties and Putative Bioactive Targets for Novel Thiosemicarbazone Derivatives" Molecules 27, no. 14: 4548. https://doi.org/10.3390/molecules27144548
APA StyleGeorgiou, N., Cheilari, A., Karta, D., Chontzopoulou, E., Plavec, J., Tzeli, D., Vassiliou, S., & Mavromoustakos, T. (2022). Conformational Properties and Putative Bioactive Targets for Novel Thiosemicarbazone Derivatives. Molecules, 27(14), 4548. https://doi.org/10.3390/molecules27144548